

Genomic Analysis of the Rare Slightly Halophilic Myxobacterium “Paraliomyxa miuraensis” SMH-27-4, the Producer of the Antibiotic Miuraenamide A


Abstract
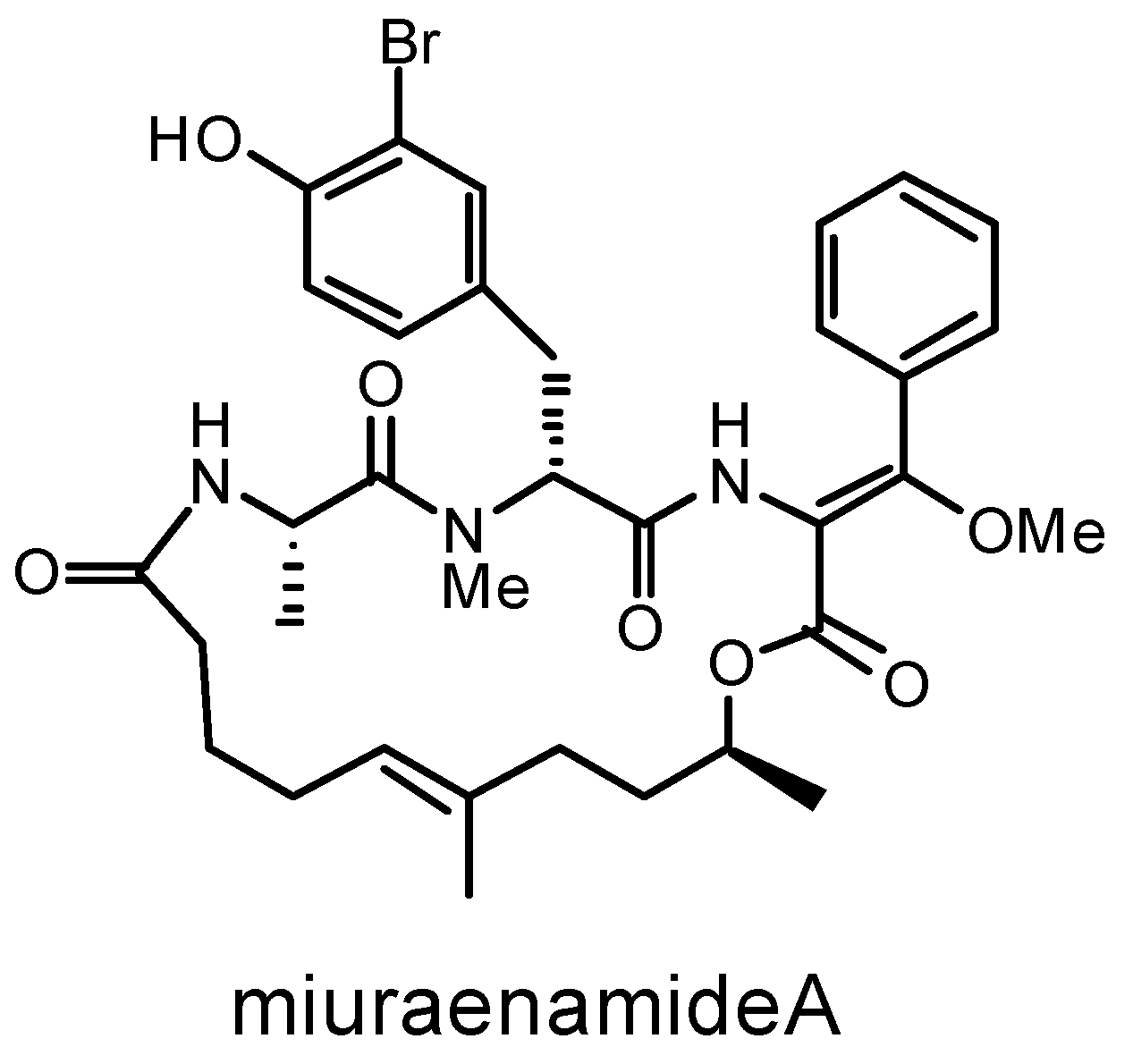
:1. Introduction
2. Materials and Methods
2.1. Cultivation and Genomic DNA Isolation
2.2. Draft Genome Sequencing, Assembly, and Annotation
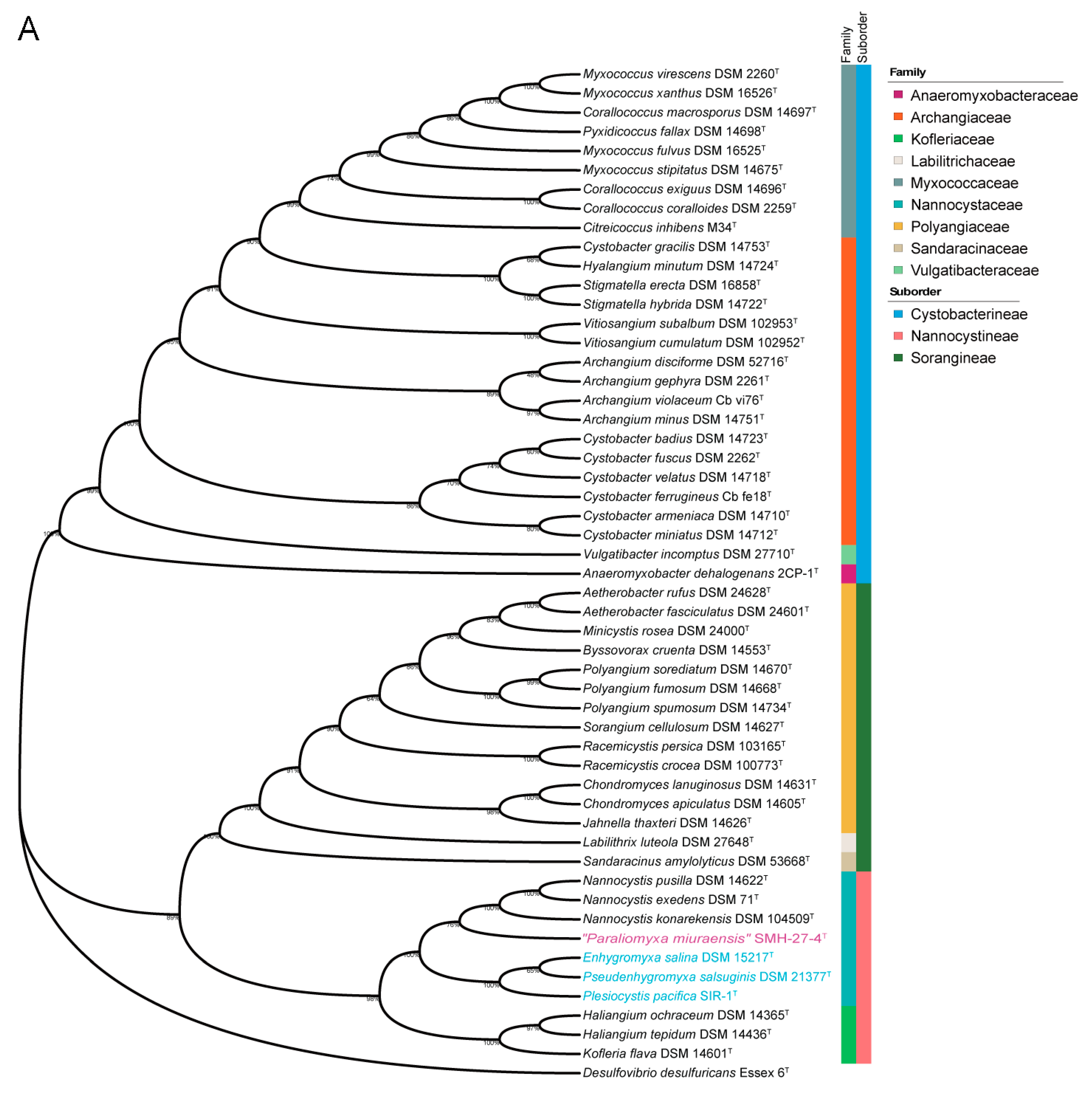
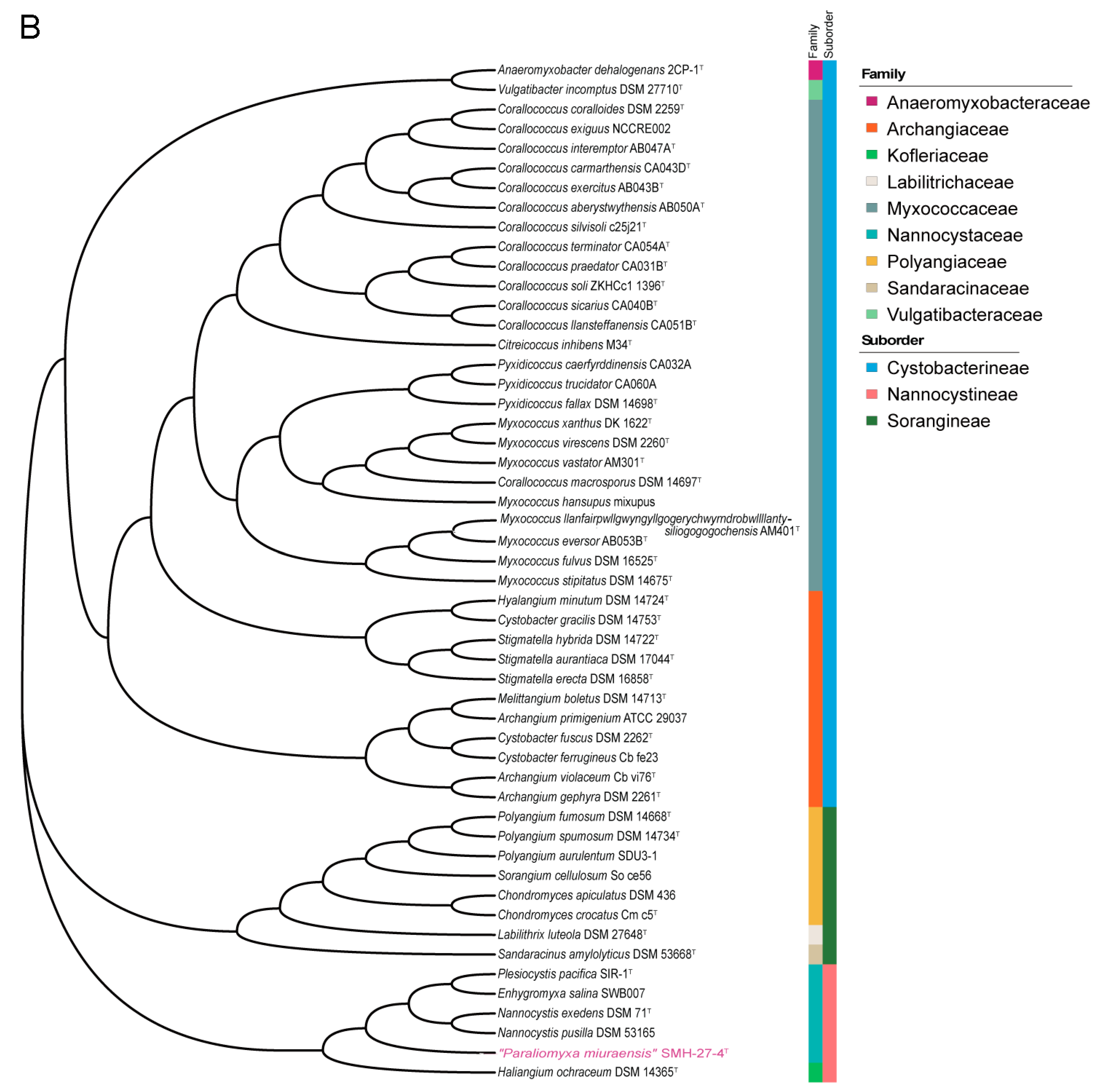
2.3. Phylogenetic Analysis
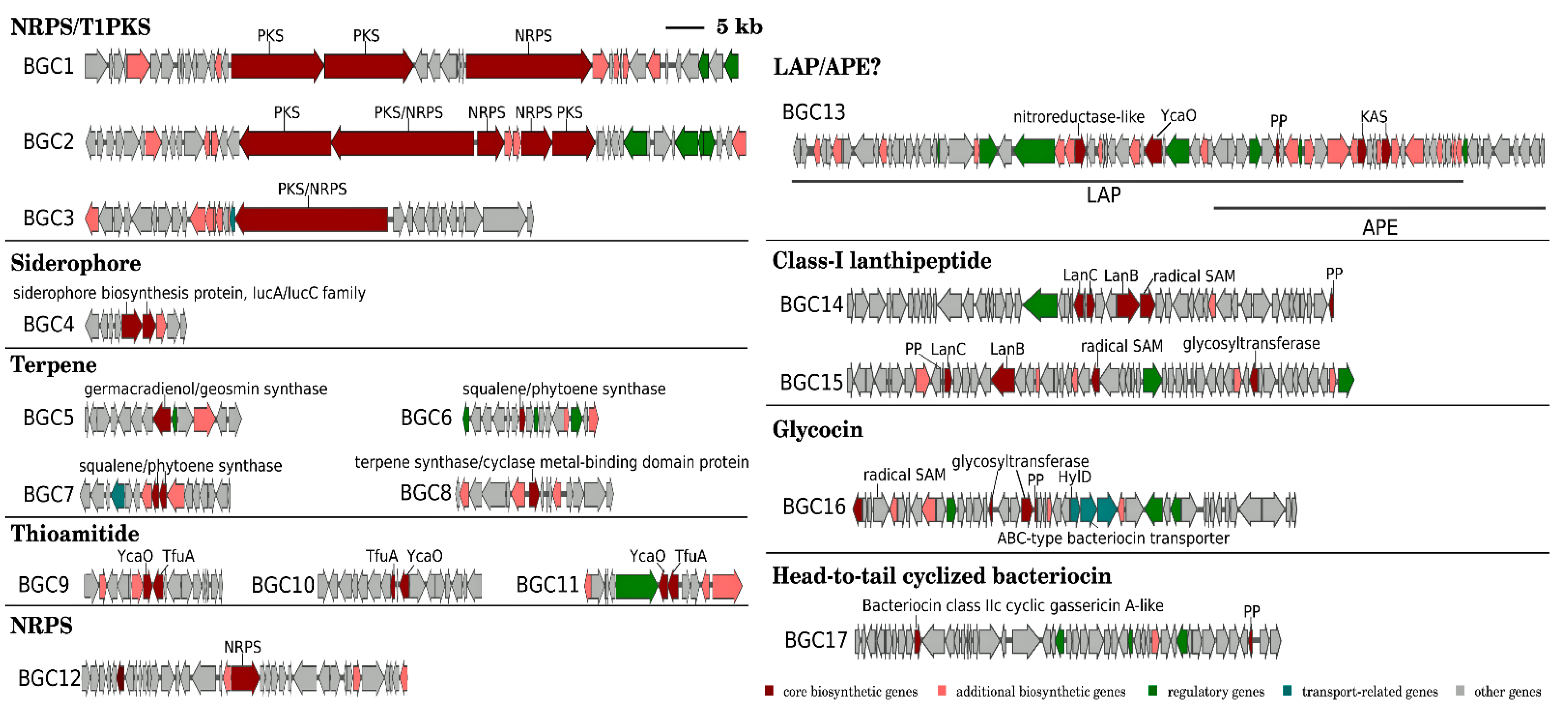
2.4. BGCs Prediction and Generation of Similarity Networks
2.5. Identification of Orthologous Proteins and Functional Categorization
2.6. Availability of Nucleotide Sequence Data
3. Results
3.1. Draft Genome Sequencing, Assembly, and Annotation
3.2. Phylogenetic Analysis
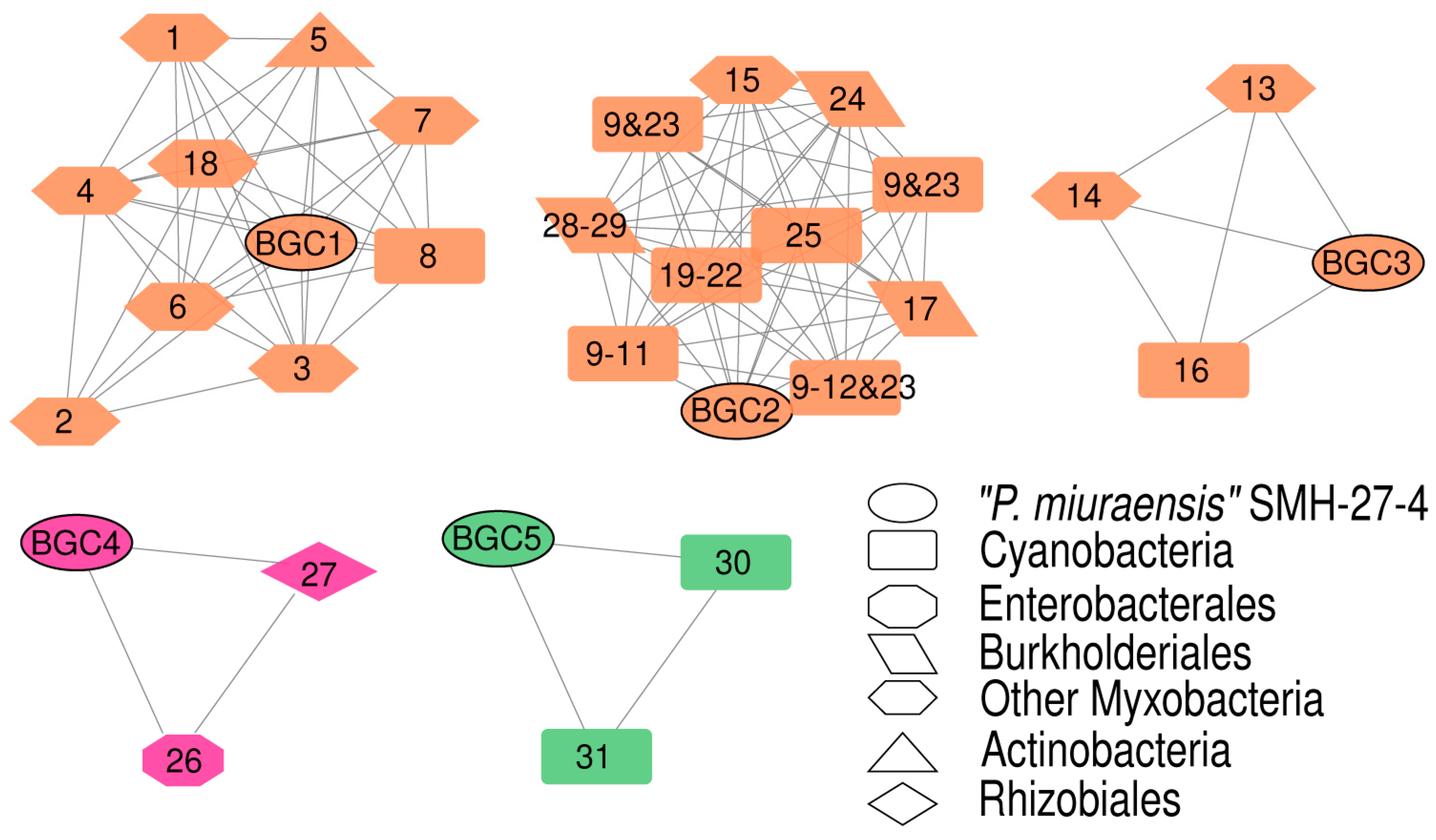
3.3. Biosynthetic Gene Clusters (BGCs)
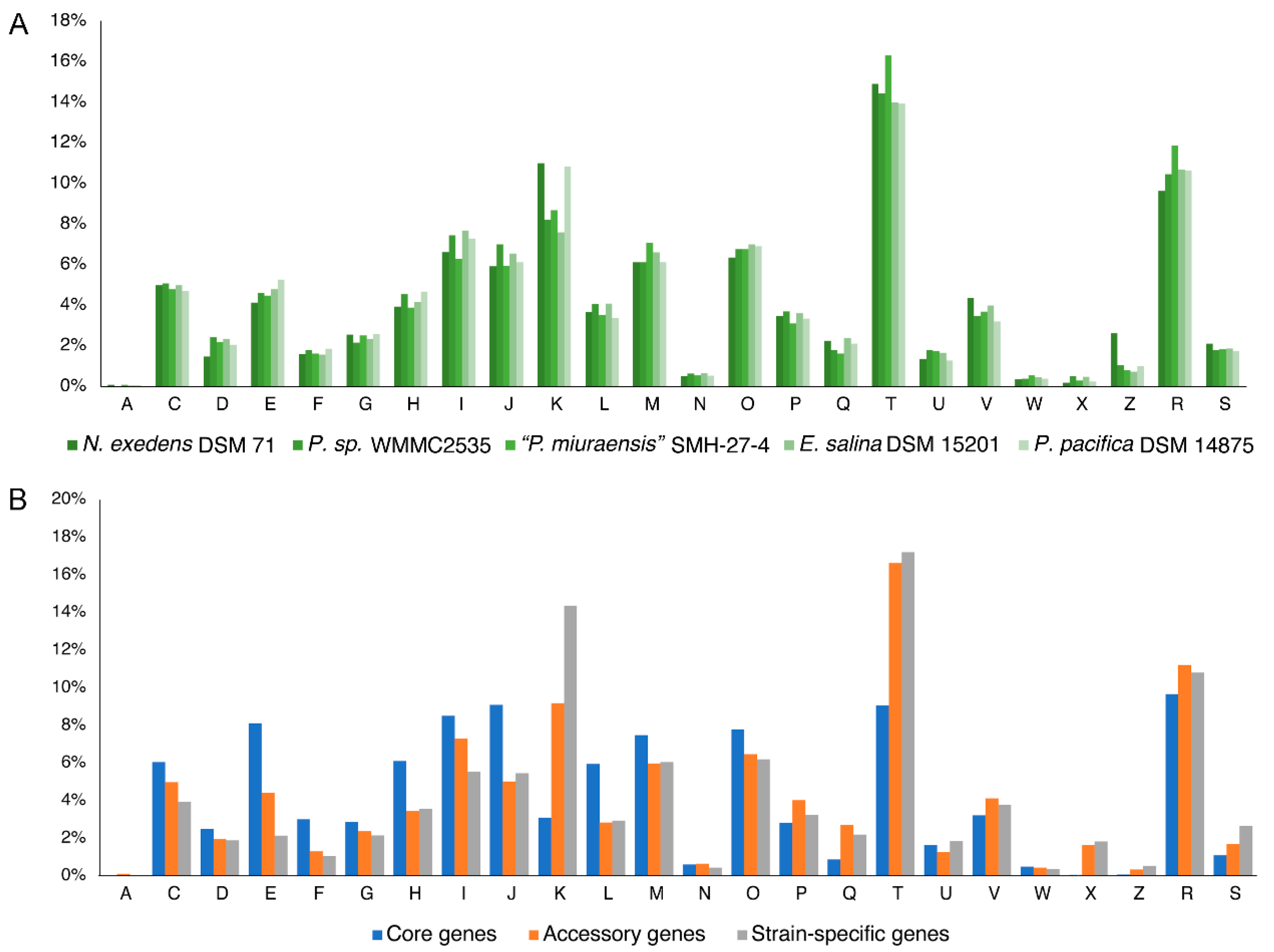
3.4. Distribution of Gene Fuctions of the Strains of the Family Nannocystaceae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Velicer, G.J.; Vos, M. Sociobiology of the myxobacteria. Annu. Rev. Microbiol. 2009, 63, 599–623. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D.; Robinson, M.; Kroos, L. Myxobacteria, polarity, and multicellular morphogenesis. Cold Spring Harb. Perspect. Biol. 2010, 2, a000380. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-dorado, J.; Marcos-torres, F.J.; García-bravo, E.; Moraleda-muñoz, A.; Pérez, J. Myxobacteria: Moving, killing, feeding, and surviving together. Front. Microbiol. 2016, 7, 781. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Müller, R. Myxobacteria—‘Microbial factories’ for the production of bioactive secondary metabolites. Mol. BioSyst. 2009, 5, 567–574. [Google Scholar] [CrossRef]
- Diez, J.; Martinez, J.P.; Mestres, J.; Sasse, F.; Frank, R.; Meyerhans, A. Myxobacteria: Natural pharmaceutical factories. Microb. Cell Fact. 2012, 11, 2–4. [Google Scholar] [CrossRef]
- Hug, J.J.; Müller, R. Host development for heterologous expression and biosynthetic studies of myxobacterial natural products. In Comprehensive Natural Products III, 3rd ed.; Liu, H., Begley, T.P., Eds.; Elsevier: San Diego, CA, USA, 2020; Volume 6, pp. 149–216. [Google Scholar]
- Thaxter, R. Contributions from the cryptogamic laboratory of Harvard University. XVIII. On the Myxobacteriaceae, a new order of Schizomycetes. Bot. Gaz. 1892, 12, 389–406. [Google Scholar] [CrossRef]
- Iizuka, T.; Jojima, Y.; Fudou, R.; Yamanaka, S. Isolation of myxobacteria from the marine environment. FEMS Microbiol. Lett. 1998, 169, 317–322. [Google Scholar] [CrossRef]
- Dávila-Céspedes, A.; Hufendiek, P.; Crüsemann, M.; Schäberle, T.F.; König, G.M. Marine-derived myxobacteria of the suborder Nannocystineae: An underexplored source of structurally intriguing and biologically active metabolites. Beilstein J. Org. Chem. 2016, 12, 969–984. [Google Scholar] [CrossRef]
- Albataineh, H.; Stevens, D.C. Marine myxobacteria: A few good halophiles. Mar. Drugs 2018, 16, 209. [Google Scholar] [CrossRef]
- Gemperlein, K.; Zaburannyi, N.; Garcia, R.; La Clair, J.J.; Müller, R. Metabolic and biosynthetic diversity in marine myxobacteria. Mar. Drugs 2018, 16, 314. [Google Scholar] [CrossRef] [Green Version]
- Moghaddam, J.A.; Crüsemann, M.; Alanjary, M.; Harms, H.; Dávila-Céspedes, A.; Blom, J.; Poehlein, A.; Ziemert, N.; König, G.M.; Schäberle, T.F. Analysis of the genome and metabolome of marine myxobacteria reveals high potential for biosynthesis of novel specialized metabolites. Sci. Rep. 2018, 8, 16600. [Google Scholar] [CrossRef]
- Iizuka, T.; Fudou, R.; Jojima, Y.; Ogawa, S.; Yamanaka, S.; Inukai, Y.; Ojika, M. Miuraenamides A and B, novel antimicrobial cyclic depsipeptides from a new slightly halophilic myxobacterium: Taxonomy, production, and biological properties. J. Antibiot. 2006, 59, 385–391. [Google Scholar] [CrossRef]
- Iizuka, T. Isolation and Characterization of Novel Myxobacteria and Their Significance as Biomedical Resources. Ph.D. Thesis, Toyohashi University of Technology, Toyohashi, Japan, January 2016. [Google Scholar]
- Garcia, R.; Müller, R. The family Nannocystaceae. In The Prokaryotes—Deltaproteobacteria and Epsilonproteobacteria, 4th ed.; Rosenberg, E., Delong, E.F., Loy, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 10, pp. 213–229. [Google Scholar]
- Iizuka, T.; Jojima, Y.; Hayakawa, A.; Fujii, T.; Yamanaka, S.; Fudou, R. Pseudenhygromyxa salsuginis gen. nov., sp. nov., a myxobacterium isolated from an estuarine marsh. Int. J. Syst. Evol. Microbiol. 2013, 63, 1360–1369. [Google Scholar] [CrossRef]
- Reichenbach, H. Nannocystis exedens gen. nov., spec. nov., a new myxobacterium of the family Sorangiaceae. Arch. Mikrobiol. 1970, 70, 119–138. [Google Scholar] [CrossRef]
- Sumiya, E.; Shimogawa, H.; Sasaki, H.; Tsutsumi, M.; Yoshita, K.; Ojika, M.; Suenaga, K.; Uesugi, M. Cell-morphology profiling of a natural product library identifies bisebromoamide and miuraenamide A as actin filament stabilizers. ACS Chem. Biol. 2011, 6, 425–431. [Google Scholar] [CrossRef]
- Karmann, L.; Schultz, K.; Herrmann, J.; Müller, R.; Kazmaier, U. Total syntheses and biological evaluation of miuraenamides. Angew. Chem. Int. Ed. 2015, 54, 4502–4507. [Google Scholar] [CrossRef]
- Ojima, D.; Yasui, A.; Tohyama, K.; Tokuzumi, K.; Toriihara, E.; Ito, K.; Iwasaki, A.; Tomura, T.; Ojika, M.; Suenaga, K. Total synthesis of miuraenamides A and D. J. Org. Chem. 2016, 81, 9886–9894. [Google Scholar] [CrossRef]
- Moser, C.; Rüdiger, D.; Förster, F.; von Blume, J.; Yu, P.; Kazmaier, U.; Vollmar, A.M.; Zahler, S. Persistent inhibition of pore-based cell migration by sub-toxic doses of miuraenamide, an actin filament stabilizer. Sci. Rep. 2017, 7, 16407. [Google Scholar] [CrossRef]
- Kappler, S.; Karmann, L.; Prudel, C.; Herrmann, J.; Caddeu, G.; Müller, R.; Vollmar, A.M.; Zahler, S.; Kazmaier, U. Synthesis and biological evaluation of modified miuraenamides. Eur. J. Org. Chem. 2018, 48, 6952–6965. [Google Scholar] [CrossRef]
- Gegenfurtner, F.A.; Zisis, T.; Al Danaf, N.; Schrimpf, W.; Kliesmete, Z.; Ziegenhain, C.; Enard, W.; Kazmaier, U.; Lamb, D.C.; Vollmar, A.M.; et al. Transcriptional effects of actin-binding compounds: The cytoplasm sets the tone. Cell. Mol. Life Sci. 2018, 75, 4539–4555. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Crevenna, A.H.; Ugur, I.; Marion, A.; Antes, I.; Kazmaier, U.; Hoyer, M.; Lamb, D.C.; Gegenfurtner, F.; Kliesmete, Z.; et al. Actin stabilizing compounds show specific biological effects due to their binding mode. Sci. Rep. 2019, 9, 9731. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meixner, M.; Yu, L.; Zhuo, L.; Karmann, L.; Kazmaier, U.; Vollmar, A.M.; Antes, I.; Zahler, S. Turning the actin nucleating compound miuraenamide into nucleation inhibitors. ACS Omega 2021, 6, 22165–22172. [Google Scholar] [CrossRef] [PubMed]
- Baltes, C.; Thalla, D.G.; Kazmaier, U.; Lautenschläger, F. Actin stabilization in cell migration. Front. Cell Dev. Biol. 2022, 10, 931880. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- Kajitani, R.; Toshimoto, K.; Noguchi, H.; Toyoda, A.; Ogura, Y.; Okuno, M.; Yabana, M.; Harada, M.; Nagayasu, E.; Maruyama, H.; et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014, 24, 1384–1395. [Google Scholar] [CrossRef]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Yamada, K.D.; Tomii, K.; Katoh, K. Application of the MAFFT sequence alignment program to large data—Reexamination of the usefulness of chained guide trees. Bioinformatics 2016, 32, 3246–3251. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (ITOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016, 4, e1900v1. [Google Scholar]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Agrawal, P.; Khater, S.; Gupta, M.; Sain, N.; Mohanty, D. RiPPMiner: A bioinformatics resource for deciphering chemical structures of RiPPs based on prediction of cleavage and cross-links. Nucleic Acids Res. 2017, 45, W80–W88. [Google Scholar] [CrossRef]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-) orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, J.C.; Rocha, M.; Alves, M.M.; Salvador, A.F. UPIMAPI, ReCOGnizer and KEGGCharter: Bioinformatics tools for functional annotation and visualization of (meta)-omics datasets. Comput. Struct. Biotechnol. J. 2022, 20, 1798–1810. [Google Scholar] [CrossRef]
- Liaimera, A.; Helfrichb, E.J.N.; Hinrichsc, K.; Guljamowc, A.; Ishidab, K.; Hertweck, C.; Dittmann, E. Nostopeptolide plays a governing role during cellular differentiation of the symbiotic cyanobacterium Nostoc punctiforme. Proc. Natl. Acad. Sci. USA 2015, 112, 1862–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.E.; Novak, J.; Austin, F.W.; Gu, G.; Ellis, D.; Kirk, M.; Wilson-Stanford, S.; Tonelli, M.; Smith, L. Occidiofungin, a unique antifungal glycopeptide produced by a strain of Burkholderia contaminans. Biochemistry 2009, 48, 8312–8321. [Google Scholar] [CrossRef] [PubMed]
- Baldeweg, F.; Kage, H.; Schieferdecker, S.; Allen, C.; Hoffmeister, D.; Nett, M. Structure of ralsolamycin, the interkingdom morphogen from the crop plant pathogen Ralstonia solanacearum. Org. Lett. 2017, 19, 4868–4871. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.W.; Wyatt, M.A.; Li, X.; Ibrahim, A.; Shuster, J.; Southam, G.; Magarvey, N.A. Gold biomineralization by a metallophore from a gold-associated microbe. Nat. Chem. Biol. 2013, 9, 241–243. [Google Scholar] [CrossRef]
- Ishida, K.; Murakami, M. Kasumigamide, an antialgal peptide from the cyanobacterium Microcystis aeruginosa. J. Org. Chem. 2000, 65, 5898–5900. [Google Scholar] [CrossRef]
- Varadarajan, A.R.; Allan, R.N.; Valentin, J.D.P.; Castañeda Ocampo, O.E.; Somerville, V.; Pietsch, F.; Buhmann, M.T.; West, J.; Skipp, P.J.; van der Mei, H.C.; et al. An integrated model system to gain mechanistic insights into biofilm-associated antimicrobial resistance in Pseudomonas aeruginosa MPAO1. NPJ Biofilms Microbiomes 2020, 6, 46. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of contigs | 164 |
| GC Content (%) | 69.7 |
| Estimated genome size based on Kmer analysis | 11,832,550 bp |
| Assembled genome size | 11,849,290 bp (100.1%) |
| N50 (bp) | 398,768 |
| L50 | 11 |
| Genes (total) | 9280 |
| Pseudogenes (total) | 40 |
| Genes (RNA) | 84 |
| tRNAs | 77 |
| rRNAs | 1, 1, 1 (5S, 16S, 23S) |
| ncRNAs | 4 |
| Genes (coding) | 9156 |
| Coding density | 90.7% |
| Hypothetic proteins | 3508 (38.3%) |
| Percentage (%) of complete BUSCOs in the genome assembly | 93.0% |
| Percentage (%) of complete BUSCOs among the annotated genes | 92.6% |
| Strains | Protein-Coding Genes | Orthologous Genes | COG Annotated Genes | ||
|---|---|---|---|---|---|
| Core Genes | Accessory Genes | Strain-Specific Genes | |||
| Pm | 9145 (100%) | 1460 (16%) | 2500 (27%) | 5185 (57%) | 3891 (43%) |
| Ne | 9295 (100%) | 1469 (16%) | 2347 (25%) | 5479 (59%) | 4020 (43%) |
| Ps | 7726 (100%) | 1475 (19%) | 2847 (37%) | 3404 (44%) | 3389 (44%) |
| Pp | 8182 (100%) | 1448 (18%) | 2859 (35%) | 3875 (47%) | 3516 (43%) |
| Es | 8079 (100%) | 1448 (18%) | 2788 (35%) | 3843 (48%) | 3390 (42%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Ojika, M. Genomic Analysis of the Rare Slightly Halophilic Myxobacterium “Paraliomyxa miuraensis” SMH-27-4, the Producer of the Antibiotic Miuraenamide A. Microorganisms 2023, 11, 371. https://doi.org/10.3390/microorganisms11020371
Liu Y, Ojika M. Genomic Analysis of the Rare Slightly Halophilic Myxobacterium “Paraliomyxa miuraensis” SMH-27-4, the Producer of the Antibiotic Miuraenamide A. Microorganisms. 2023; 11(2):371. https://doi.org/10.3390/microorganisms11020371
Chicago/Turabian StyleLiu, Ying, and Makoto Ojika. 2023. "Genomic Analysis of the Rare Slightly Halophilic Myxobacterium “Paraliomyxa miuraensis” SMH-27-4, the Producer of the Antibiotic Miuraenamide A" Microorganisms 11, no. 2: 371. https://doi.org/10.3390/microorganisms11020371
APA StyleLiu, Y., & Ojika, M. (2023). Genomic Analysis of the Rare Slightly Halophilic Myxobacterium “Paraliomyxa miuraensis” SMH-27-4, the Producer of the Antibiotic Miuraenamide A. Microorganisms, 11(2), 371. https://doi.org/10.3390/microorganisms11020371