
Investigation of a Listeria monocytogenes Chromosomal Immigration Control Region Reveals Diverse Restriction Modification Systems with Complete Sequence Type Conservation

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Whole-Genome Sequence Analysis and Nucleotide Alignments
2.2. Generation of Minimum Spanning Trees and Gene Annotations
3. Results and Discussion
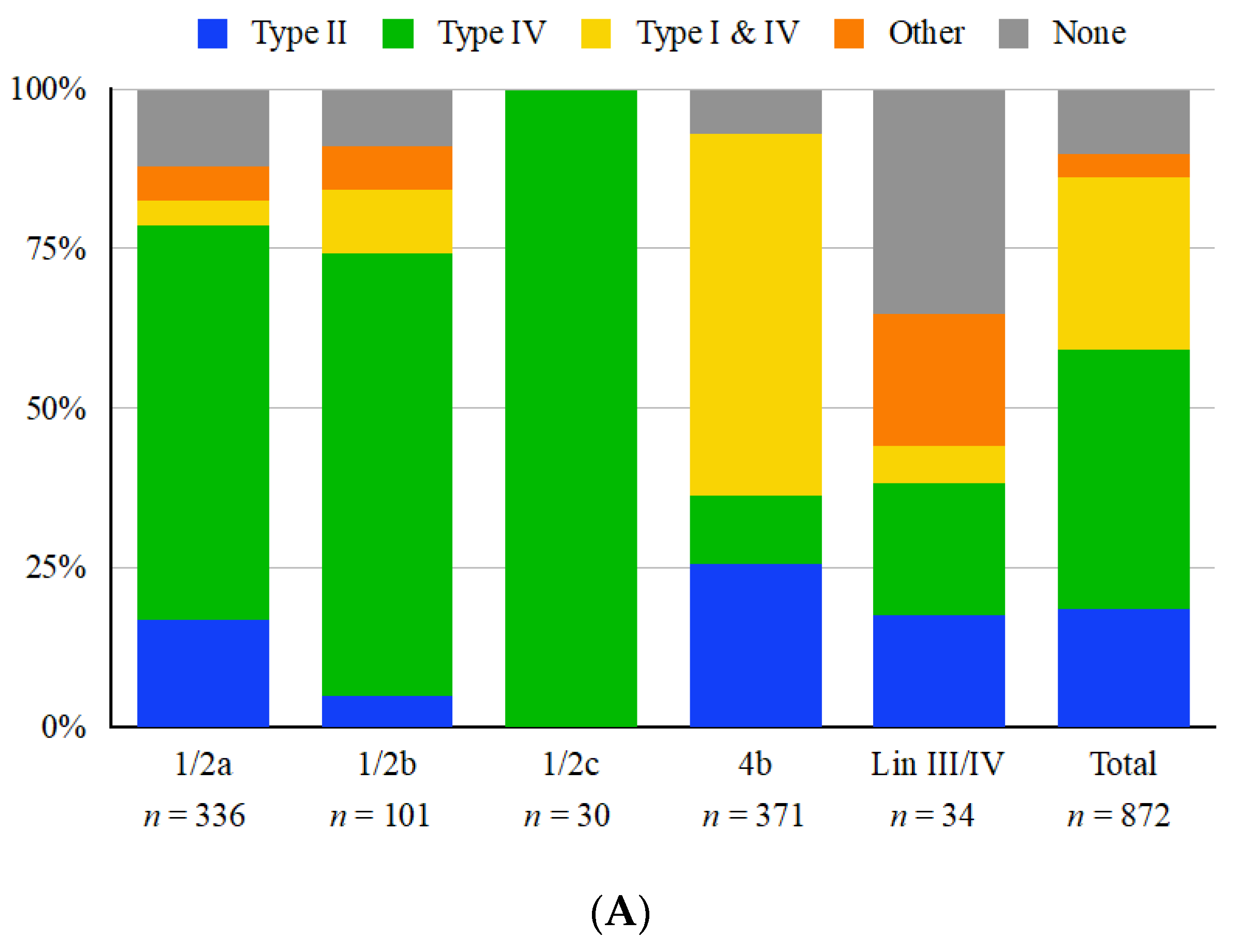
3.1. The Immigration Control Region Frequently Harbors Restriction Modification Systems That Exhibit Serotype-Dependent Trends
3.2. ICR Presence and Content Are Completely Conserved within Each Sequence Type
3.3. Serotype 4b Strains Commonly Harbor Paired Type I and IV Restriction Modification Systems
3.4. Type II Restriction Modification Systems Can Be Found Both inside and Flanking the ICR
3.5. DNA Helicase and Other Diverse Genes Are Occasionally Found in the Immigration Control Region
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- De Noordhout, C.M.; Devleesschauwer, B.; Angulo, F.J.; Verbeke, G.; Haagsma, J.; Kirk, M.; Havelaar, A.; Speybroeck, N. The global burden of listeriosis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 1073–1082. [Google Scholar] [CrossRef]
- Radoshevich, L.; Cossart, P. Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Painter, J.; Slutsker, L. Listeriosis in humans. In Listeriosis in Humans. Listeria, Listeriosis, and Food Safety, 3rd ed.; Ryser, E.T., Marth., E.H., Eds.; CRC Press: Boca Raton, FL, USA, 2007; pp. 85–110. [Google Scholar]
- Kathariou, S.; Evans, P.; Dutta, V. Strain-specific virulence differences in Listeria monocytogenes: Current perspectives in addressing an old and vexing issue. In Foodborne Pathogens. Food Microbiology and Food Safety; Gurtler, J., Doyle, M., Kornacki, J., Eds.; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Maury, M.M.; Bracq-Dieye, H.; Huang, L.; Vales, G.; Lavina, M.; Thouvenot, P.; Disson, O.; Leclercq, A.; Brisse, S.; Lecuit, M. Hypervirulent Listeria monocytogenes clones’ adaption to mammalian gut accounts for their association with dairy products. Nat. Commun. 2019, 10, 2488. [Google Scholar] [CrossRef] [PubMed]
- Merchel Piovesan Pereira, B.; Tagkopoulos, I. Benzalkonium chlorides: Uses, regulatory status, and microbial resistance. Appl. Environ. Microbiol. 2019, 85, e00377-19. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.A.; Nannapaneni, R.; Hagens, S. Reduction of Listeria monocytogenes on the surface of fresh channel catfish fillets by bacteriophage Listex P100. Foodborne Pathog. Dis. 2010, 7, 427–434. [Google Scholar] [CrossRef]
- Perera, M.N.; Abuladze, T.; Li, M.; Woolston, J.; Sulakvelidze, A. Bacteriophage cocktail significantly reduces or eliminates Listeria monocytogenes contamination on lettuce, apples, cheese, smoked salmon and frozen foods. Food Microbiol. 2015, 52, 42–48. [Google Scholar] [CrossRef]
- Kim, J.W.; Siletzky, R.M.; Kathariou, S. Host ranges of Listeria-specific bacteriophages from the turkey processing plant environment in the United States. Appl. Environ. Micrbiol. 2008, 74, 6623–6630. [Google Scholar] [CrossRef]
- Bigot, B.; Lee, W.J.; McIntyre, L.; Wilson, T.; Hudson, J.A.; Billington, C.; Heinemann, J.A. Control of Listeria monocytogenes growth in a ready-to-eat poultry product using a bacteriophage. Food Microbiol. 2011, 28, 1448–1452. [Google Scholar] [CrossRef]
- Guenther, S.; Loessner, M.J. Bacteriophage biocontrol of Listeria monocytogenes on soft ripened white mold and red-smear cheeses. Bacteriophage 2011, 1, 94–100. [Google Scholar] [CrossRef]
- Vongkamjan, K.; Roof, S.; Stasiewicz, M.J.; Wiedmann, M. Persistent Listeria monocytogenes subtypes isolated from a smoked fish processing facility included both phage susceptible and resistant isolates. Food Microbiol. 2013, 35, 38–48. [Google Scholar] [CrossRef]
- Eugster, M.R.; Morax, L.S.; Hüls, V.J.; Huwiler, S.G.; Leclercq, A.; Lecuit, M.; Loessner, M.J. Bacteriophage predation promotes serovar diversification in Listeria monocytogenes. Mol. Microbiol. 2015, 97, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Chen, Y.; Parsons, C.; Brown, E.; Loessner, M.J.; Shen, Y.; Kathariou, S. Whole genome sequence analysis of phage-resistant Listeria monocytogenes serotype 1/2a strains from turkey processing plants. Pathogens 2021, 10, 199. [Google Scholar] [CrossRef] [PubMed]
- Promadej, N.; Fiedler, F.; Cossart, P.; Dramsi, S.; Kathariou, S. Cell wall teichoic acid glycosylation in Listeria monocytogenes serotype 4b requires gtcA, a novel, serogroup-specific gene. J. Bacteriol. 1999, 181, 418–425. [Google Scholar] [CrossRef]
- Cheng, Y.; Promadej, N.; Kim, J.W.; Kathariou, S. Teichoic acid glycosylation mediated by gtcA is required for phage adsorption and susceptibility of Listeria monocytogenes serotype 4b. Appl. Environ. Microbiol. 2008, 74, 1653–1655. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Lawrence, M.L.; Wiedmann, M.; Gorski, L.; Mandrell, R.E.; Ainsworth, A.J.; Austin, F.W. Listeria monocytogenes subgroups IIIA, IIIB, and IIIC delineate genetically distinct populations with varied pathogenic potential. J. Clin. Microbiol. 2006, 44, 4229–4233. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.J.; Ducey, T.F.; Usgaard, T.; Dunn, K.A.; Bielawski, J.P. Multilocus genotyping assays for single nucleotide polymorphism-based subtyping of Listeria monocytogenes isolates. Appl. Environ. Microbiol. 2008, 74, 7629–7642. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, J.; Fang, C.; Xia, Y.; Cheng, C.; Jiang, L.; Fang, W. Deciphering the biodiversity of Listeria monocytogenes lineage III strains by polyphasic approaches. J. Microbiol. 2011, 49, 759–767. [Google Scholar] [CrossRef]
- Kathariou, S. Listeria monocytogenes virulence and pathogenicity, a food safety perspective. J. Food Prot. 2002, 65, 1811–1829. [Google Scholar] [CrossRef]
- Swaminathan, B.; Gerner-Smidt, P. The epidemiology of human listeriosis. Microbes Infect. 2007, 9, 1236–1243. [Google Scholar] [CrossRef]
- Maury, M.M.; Tsai, Y.H.; Charlier, C.; Touchon, M.; Chenal-Francisque, V.; Leclercq, A.; Criscuolo, A.; Gaultier, C.; Roussel, S.; Brisabois, A.; et al. Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat. Genet. 2016, 48, 308–313. [Google Scholar] [CrossRef]
- Audurier, A.; Taylor, A.G.; Carbonnelle, B.; McLauchlin, J. A phage typing system for Listeria monocytogenes and its use in epidemiological studies. Clin. Investig. Med. 1984, 7, 229–232. [Google Scholar]
- Sumrall, E.T.; Shen, Y.; Keller, A.P.; Rismondo, J.; Pavlou, M.; Eugster, M.R.; Boulos, S.; Disson, O.; Thouvenot, P.; Kilcher, S.; et al. Phage resistance at the cost of virulence: Listeria monocytogenes serovar 4b requires galactosylated teichoic acids for InlB-mediated invasion. PLoS Pathog. 2019, 15, e1008032. [Google Scholar] [CrossRef] [PubMed]
- Hain, T.; Ghai, R.; Billion, A.; Kuenne, C.T.; Steinweg, C.; Izar, B.; Mohamed, W.; Mraheil, M.A.; Domann, E.; Schaffrath, S.; et al. Comparative genomics and transcriptomics of lineages I, II, and III strains of Listeria monocytogenes. BMC Genomics 2012, 13, 144. [Google Scholar] [CrossRef]
- Di, H.; Ye, L.; Yan, H.; Meng, H.; Yamasak, S.; Shi, L. Comparative analysis of CRISPR loci in different Listeria monocytogenes lineages. Biochem. Biophys. Res. Commun. 2014, 454, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Parsons, C.; Brown, P.; Kathariou, S. Use of bacteriophage amended with CRISPR-Cas systems to combat antimicrobial resistance in the bacterial foodborne pathogen Listeria monocytogenes. Antibiotics 2021, 10, 308. [Google Scholar] [CrossRef] [PubMed]
- Loenen, W.A.; Dryden, D.T.; Raleigh, E.A.; Wilson, G.G.; Murray, N.E. Highlights of the DNA cutters: A short history of the restriction enzymes. Nucleic Acids Res. 2014, 42, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Loenen, W.A.; Raleigh, E.A. The other face of restriction: Modification-dependent enzymes. Nucleic Acids Res. 2014, 42, 56–69. [Google Scholar] [CrossRef]
- Yildirim, S.; Elhanafi, D.; Lin, W.; Hitchins, A.D.; Siletzky, R.M.; Kathariou, S. Conservation of genomic localization and sequence content of Sau3AI-like restriction-modification gene cassettes among Listeria monocytogenes epidemic clone I and selected strains of serotype 1/2a. Appl. Environ. Microbiol. 2010, 76, 5577–5584. [Google Scholar] [CrossRef]
- Kim, J.W.; Dutta, V.; Elhanafi, D.; Lee, S.; Osborne, J.A.; Kathariou, S. A novel restriction-modification system is responsible for temperature-dependent phage resistance in Listeria monocytogenes ECII. Appl. Environ. Microbiol. 2012, 78, 1995–2004. [Google Scholar] [CrossRef]
- Lee, S.; Ward, T.J.; Siletzky, R.M.; Kathariou, S. Two novel type II restriction-modification systems occupying genomically equivalent locations on the chromosomes of Listeria monocytogenes strains. Appl. Environ. Microbiol. 2012, 78, 2623–2630. [Google Scholar] [CrossRef]
- Kuenne, C.; Billion, A.; Mraheil, M.A.; Strittmatter, A.; Daniel, R.; Goesmann, A.; Barbuddhe, S.; Hain, T.; Chakraborty, T. Reassessment of the Listeria monocytogenes pan-genome reveals dynamic integration hotspots and mobile genetic elements as major components of the accessory genome. BMC Genomics 2013, 14, 47. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; den Bakker, H.C.; Korlach, J.; Kong, N.; Storey, D.B.; Paxinos, E.E.; Ashby, M.; Clark, T.; Luong, K.; Wiedmann, M.; et al. Comparative genomics reveals the diversity of restriction-modification systems and DNA methylation sites in Listeria monocytogenes. Appl. Environ. Microbiol. 2017, 83, e02091-e16. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, E.A. Organization and function of the mcrBC genes of Escherichia coli K-12. Mol. Microbiol. 1992, 6, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Picchi, S.C.; Vilas-Boas, L.A.; Ceresini, P.C.; de Macedo Lemos, E.G.; Lemos, M.V. Strain variability in the DNA immigration control region (ICR) of Xylella fastidiosa. Res. Microbiol. 2006, 157, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Lee, S. Characterization of the immigration control region of Listeria monocytogenes. Ph.D. Thesis, North Carolina State University, Raleigh, NC, USA, 2011. Available online: http://www.lib.ncsu.edu/resolver/1840.16/7955 (accessed on 4 March 2023).
- Moura, A.; Criscuolo, A.; Pouseele, H.; Maury, M.M.; Leclercq, A.; Tarr, C.; Björkman, J.T.; Dallman, T.; Reimer, A.; Enouf, V.; et al. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbiol. 2016, 2, 16185. [Google Scholar] [CrossRef]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genomics 2008, 9, 75. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2015, 43, D298–D299. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Kathariou, S. Host-mediated modification of Sau3AI restriction in Listeria monocytogenes: Prevalence in epidemic-associated strains. Appl. Environ. Microbiol. 1997, 63, 3085–3089. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chen, Y.; Gorski, L.; Ward, T.J.; Osborne, J.; Kathariou, S. Listeria monocytogenes source distribution analysis indicates regional heterogeneity and ecological niche preference among serotype 4b clones. mBio 2018, 9, e00396-18. [Google Scholar] [CrossRef] [PubMed]
- Gorski, L.; Cooley, M.B.; Oryang, D.; Carychao, D.; Nguyen, K.; Luo, Y.; Weinstein, L.; Brown, E.; Allard, M.; Mandrell, R.E.; et al. Prevalence and clonal diversity of over 1,200 Listeria monocytogenes isolates collected from public access waters near produce production areas on the central California coast during 2011 to 2016. Appl. Environ. Microbiol. 2022, 88, e0035722. [Google Scholar] [CrossRef] [PubMed]
- Iwu, C.D.; Okoh, A.I. Characterization of antibiogram fingerprints in Listeria monocytogenes recovered from irrigation water and agricultural soil samples. PLoS ONE 2020, 15, e0228956. [Google Scholar] [CrossRef] [PubMed]
- Carlin, C.R.; Liao, J.; Weller, D.L.; Guo, X.; Orsi, R.; Wiedmann, M. Listeria cossartiae sp. nov., Listeria farberi sp. nov., Listeria immobilis sp. nov., Listeria portnoyi sp. nov. and Listeria rustica sp. nov., isolated from agricultural water and natural environments. Int. J. Syst. Evol. Microbiol. 2021, 71, 004795. [Google Scholar] [CrossRef] [PubMed]
- Orsi, R.H.; den Bakker, H.C.; Wiedmann, M. Listeria monocytogenes lineages: Genomics, evolution, ecology, and phenotypic characteristics. Int. J. Med. Microbiol. 2011, 301, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Glaser, P.; Frangeul, L.; Buchrieser, C.; Rusniok, C.; Amend, A.; Baquero, F.; Berche, P.; Bloecker, H.; Brandt, P.; Chakraborty, T.; et al. Comparative genomics of Listeria species. Science 2001, 294, 849–852. [Google Scholar] [CrossRef]
- Yang, H.; Hoffmann, M.; Allard, M.W.; Brown, E.W.; Chen, Y. Microevolution and gain or loss of mobile genetic elements of outbreak-related Listeria monocytogenes in food processing environments identified by whole genome sequencing analysis. Front. Microbiol. 2020, 11, 866. [Google Scholar] [CrossRef] [PubMed]
- Bullas, L.R.; Colson, C.; van Pel, A. DNA restriction and modification systems in Salmonella. SQ, a new system derived by recombination between the SB system of Salmonella typhimurium and the SP system of Salmonella potsdam. J. Gen. Microbiol. 1976, 95, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Loenen, W.A.; Dryden, D.T.; Raleigh, E.A.; Wilson, G.G. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014, 42, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Ward, T.J.; Graves, L.M.; Wolf, L.A.; Sperry, K.; Siletzky, R.M.; Kathariou, S. Atypical Listeria monocytogenes serotype 4b strains harboring a lineage II-specific gene cassette. Appl. Environ. Microbiol. 2012, 78, 660–667. [Google Scholar] [CrossRef]
- Ueno, T.; Ito, H.; Kimizuka, F.; Kotani, H.; Nakajima, K. Gene structure and expression of the MboI restriction–modification system. Nucleic Acids Res. 1993, 21, 2309–2313. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.N.; Dryden, D.T.; Bheemanaik, S. Type III restriction-modification enzymes: A historical perspective. Nucleic Acids Res. 2014, 42, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Snir, S.; Koonin, E.V. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 2011, 193, 6039–6056. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 2013, 41, 4360–4377. [Google Scholar] [CrossRef]
- Floyd, J.L.; Smith, K.P.; Kumar, S.H.; Floyd, J.T.; Varela, M.F. LmrS is a multidrug efflux pump of the major facilitator superfamily from Staphylococcus aureus. Antimicrob. Agents Chemother. 2010, 54, 5406–5412. [Google Scholar] [CrossRef]
- Jiang, X.; Yu, T.; Xu, Y.; Wang, H.; Korkeala, H.; Shi, L. MdrL, a major facilitator superfamily efflux pump of Listeria monocytogenes involved in tolerance to benzalkonium chloride. Appl. Microbiol. Biotechnol. 2019, 103, 1339–1350. [Google Scholar] [CrossRef]
- Drew, D.; North, R.A.; Nagarathinam, K.; Tanabe, M. Structures and general transport mechanisms by the major facilitator superfamily (MFS). Chem. Rev. 2021, 121, 5289–5335. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Restriction Modification System 1 | Location | Incidence | Number of Strains | Conservation (nt Identity %) 2 | GC Content (%) |
|---|---|---|---|---|---|
| Type I: EcoKI-like (AACN6GTGC) | lmo0301-lmo0305 | 1/2a, 1/2b, 4b, lineage III | 235 | hdsR: ≥98.0 hdsM: ≥96.8 hdsS: ≥28.5 | 35 |
| Type I: EcoKI-like (AACN6GTGC) | lmo0293-lmo0294 | 1/2a | 33 | hdsR: 100 hdsM: 100 hdsS: 100 | 33 |
| Type II: HaeIII-like (GGCC) | lmo0305-lmo0314 | 1/2b | 9 | 100 | 27 |
| Type II: LmoJ2-like (GCWGC) | lmo0301-lmo0305 | 1/2a, lineage III | 15 | ≥99.6 | 32 |
| Type II: LmoJ3-like (GCNGC) | lmo0301-lmo0305 | 1/2a, 4b | 59 | ≥99.0 | 30 |
| Type II: MboI-like (GATC) | lmo0295-lmo0296 | 1/2b, 4b | 57 | 100 | 30 |
| Type II: NgoPII-like (GGCC) | lmo0305-lmo0314 | 1/2a, lineage III | 8 | ≥97.6 | 31 |
| Type II: SalI-like (GTCGAC) | lmo0318-lmo0319 | 1/2b, 4b | 35 | 99.9 | 31 |
| Type II: Sau3AI-like (GATC) | lmo0301-lmo0305 | 1/2a, 4b, lineage III | 89 | ≥98.0 | 31 |
| Type II: Sau3AI-like (GATC) | lmo0305-lmo0314 | 1/2a, 4b, lineage III, lineage IV | 53 | ≥48.8 | 31 |
| Type III: StyLTI-like (CAGAG) | lmo0293-lmo0294 | 4b | 1 | NA | 31 |
| Type IV Mrr: AspBHI-like (YSCNS) | lmo0301-lmo0305 | 1/2a, 1/2b, 4b | 100 | ≥97.0 | 31 |
| Type IV mcrB-like (RmC) | lmo0301-lmo0305 | 1/2a, 1/2b, 1/2c, 4b, lineage III | 540 | ≥95.0 | 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, P.; Lee, S.; Elhanafi, D.; Tham, W.; Danielsson-Tham, M.-L.; Lopez-Valladares, G.; Chen, Y.; Ivanova, M.; Leekitcharoenphon, P.; Kathariou, S. Investigation of a Listeria monocytogenes Chromosomal Immigration Control Region Reveals Diverse Restriction Modification Systems with Complete Sequence Type Conservation. Microorganisms 2023, 11, 699. https://doi.org/10.3390/microorganisms11030699
Brown P, Lee S, Elhanafi D, Tham W, Danielsson-Tham M-L, Lopez-Valladares G, Chen Y, Ivanova M, Leekitcharoenphon P, Kathariou S. Investigation of a Listeria monocytogenes Chromosomal Immigration Control Region Reveals Diverse Restriction Modification Systems with Complete Sequence Type Conservation. Microorganisms. 2023; 11(3):699. https://doi.org/10.3390/microorganisms11030699
Chicago/Turabian StyleBrown, Phillip, Sangmi Lee, Driss Elhanafi, Wilhelm Tham, Marie-Louise Danielsson-Tham, Gloria Lopez-Valladares, Yi Chen, Mirena Ivanova, Pimlapas Leekitcharoenphon, and Sophia Kathariou. 2023. "Investigation of a Listeria monocytogenes Chromosomal Immigration Control Region Reveals Diverse Restriction Modification Systems with Complete Sequence Type Conservation" Microorganisms 11, no. 3: 699. https://doi.org/10.3390/microorganisms11030699
APA StyleBrown, P., Lee, S., Elhanafi, D., Tham, W., Danielsson-Tham, M. -L., Lopez-Valladares, G., Chen, Y., Ivanova, M., Leekitcharoenphon, P., & Kathariou, S. (2023). Investigation of a Listeria monocytogenes Chromosomal Immigration Control Region Reveals Diverse Restriction Modification Systems with Complete Sequence Type Conservation. Microorganisms, 11(3), 699. https://doi.org/10.3390/microorganisms11030699