


Helicobacter pylori and Its Role in Gastric Cancer

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. H. pylori and Associated Diseases
2.1. Gastric Cancer
2.2. Peptic Ulcer Disease
2.3. Mucosa-Associated Lymphoid Tissue (MALT) Lymphoma
2.4. Extragastric Manifestations of H. pylori
3. H. pylori Virulence Factors and Cancer Mechanisms
3.1. Urease
3.2. Adhesins
3.3. CagA and the Pathogenicity Island
3.4. Vacuolating Toxin (VacA)
3.5. H. pylori Neutrophil Activating Protein (HP-NAP)
3.6. H. pylori γ-Glutamyltranspeptidase (GGT)
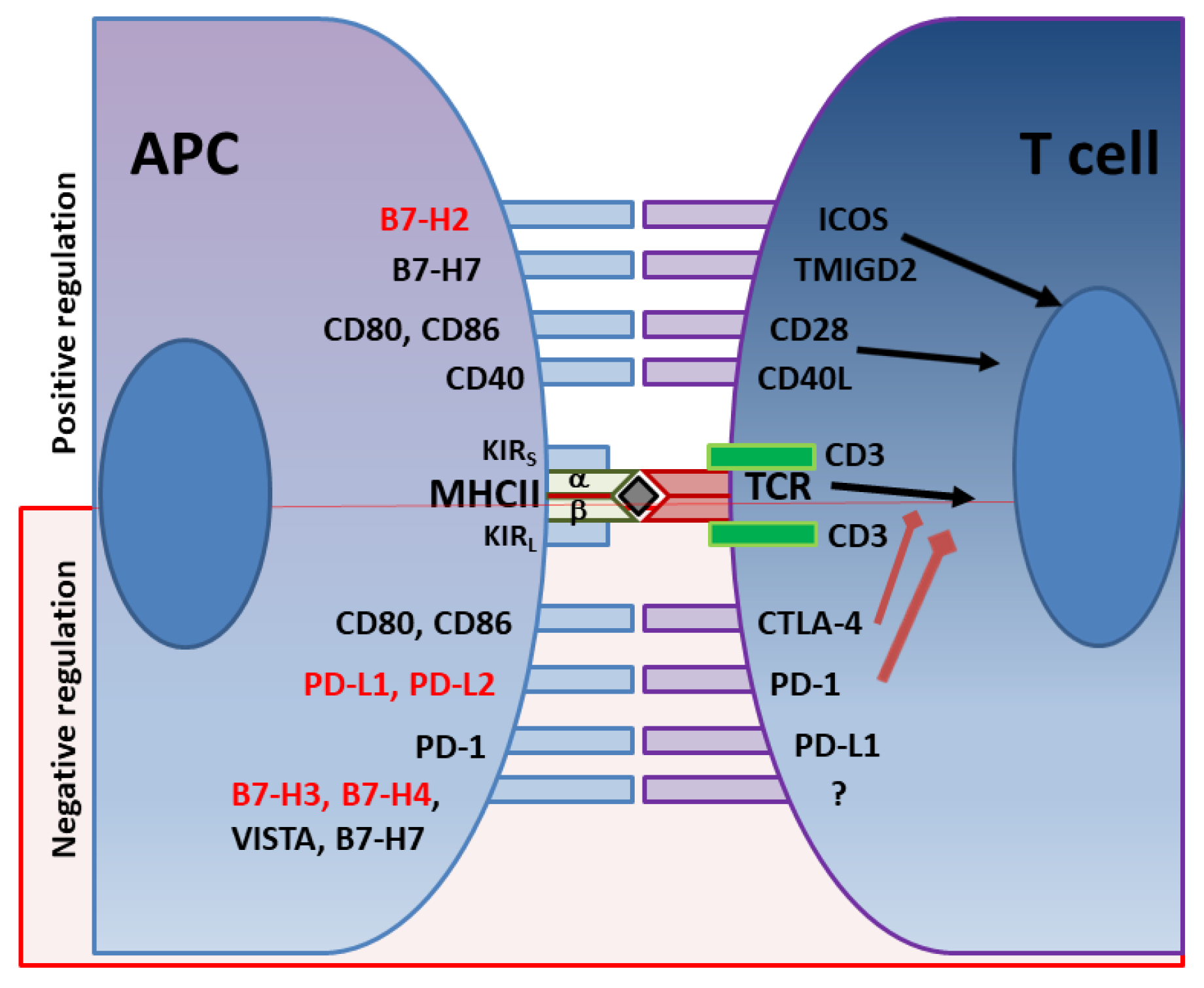
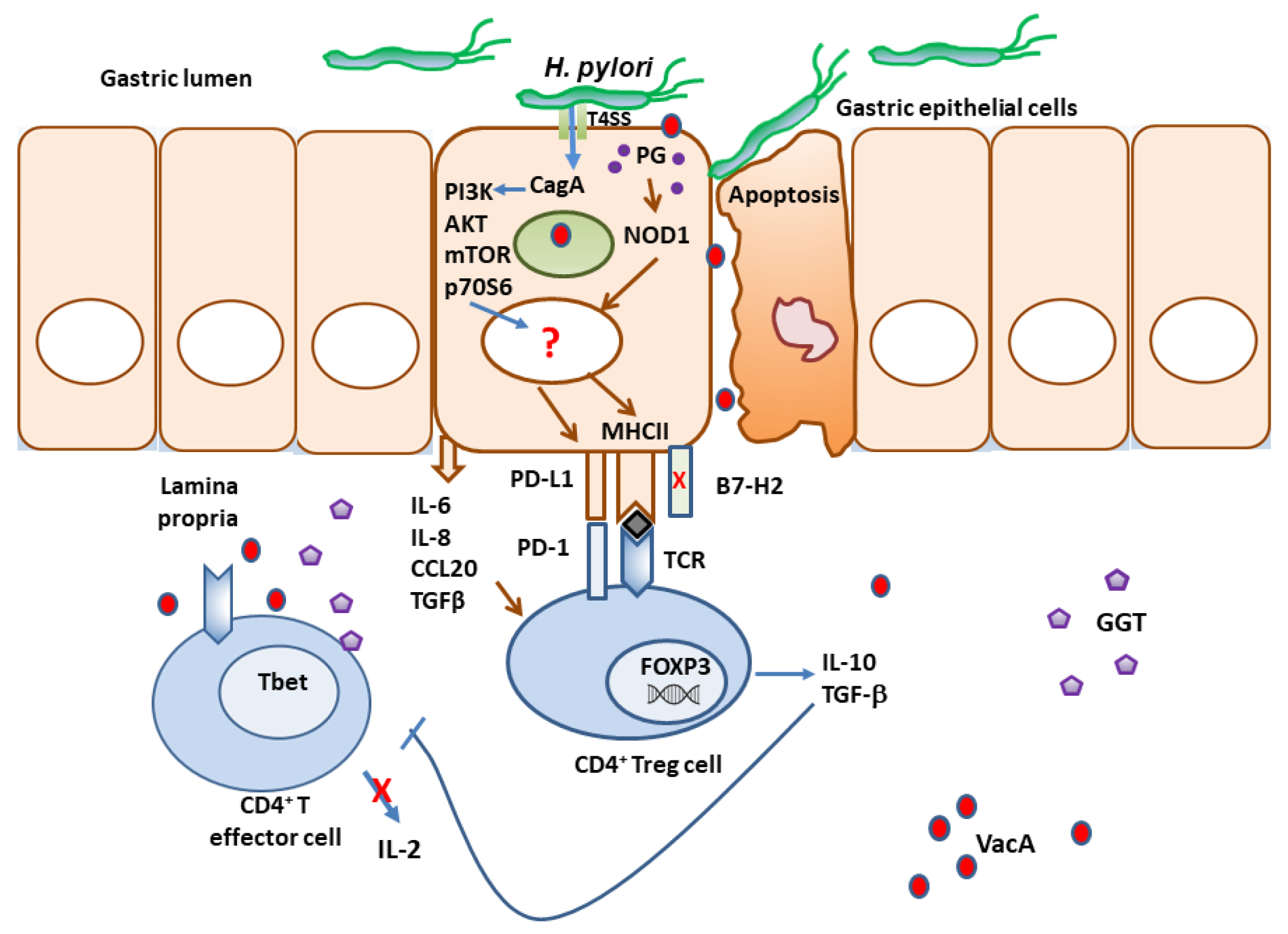
4. Immune Checkpoints in H. pylori Infection as Immune Escape Mechanisms
5. Conclusions and Future Directions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2023. [Google Scholar]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Infection with Helicobacter pylori. In Schistosomes, Liver Flukes and Helicobacter pylori; IARC: Lyon, France, 1994; Volume 61, pp. 177–240. [Google Scholar]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. A human model of gastric carcinogenesis. Cancer Res. 1988, 48, 3554–3560. [Google Scholar]
- Ma, J.; Shen, H.; Kapesa, L.; Zeng, S. Lauren classification and individualized chemotherapy in gastric cancer. Oncol. Lett. 2016, 11, 2959–2964. [Google Scholar] [CrossRef]
- Plummer, M.; Franceschi, S.; Vignat, J.; Forman, D.; De Martel, C. Global burden of gastric cancer attributable to Helicobacter pylori. Int. J. Cancer 2015, 136, 487–490. [Google Scholar] [CrossRef]
- Møller, H.; Heseltine, E.; Vainio, H. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Int. J. Cancer 1995, 60, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Holcombe, C. Helicobacter pylori: The African enigma. Gut 1992, 33, 429–431. [Google Scholar] [CrossRef]
- Agha, A.; Graham, D.Y. Evidence-based examination of the African enigma in relation to Helicobacter pylori infection. Scand. J. Gastroenterol. 2005, 40, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Bamford, K.B.; Fan, X.; Crowe, S.E.; Leary, J.F.; Gourley, W.K.; Luthra, G.K.; Brooks, E.G.; Graham, D.Y.; Reyes, V.E.; Ernst, P.B. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology 1998, 114, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Beck, P.; Dangler, C.A.; Whary, M.T.; Wang, T.C.; Shi, H.N.; Nagler-Anderson, C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat. Med. 2000, 6, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Feng, Y.; Muthupalani, S.; Eurell, L.L.; Taylor, N.S.; Whary, M.T.; Fox, J.G. Coinfection with enterohepatic helicobacter species can aeliorate or promote Helicobacter pylori-induced gastric pathology in C57BL/6 Mice. Infect. Immun. 2011, 79, 3861–3871. [Google Scholar] [CrossRef]
- Singh, K.; Ghoshal, U.C. Causal role of Helicobacter pylori infection in gastric cancer: An Asian enigma. World J. Gastroenterol. 2006, 12, 1346–1351. [Google Scholar] [CrossRef]
- Xia, Y.; Yamaoka, Y.; Zhu, Q.; Matha, I.; Gao, X. A comprehensive sequence and disease correlation analyses for the C-terminal region of CagA protein of Helicobacter pylori. PLoS ONE 2009, 4, e7736. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Kodama, T.; Kashima, K.; Graham, D.Y.; Sepulveda, A.R. Variants of the 3′ region of the cagA gene in Helicobacter pylori isolates from patients with different H. pylori-associated diseases. J. Clin. Microbiol. 1998, 36, 2258–2263. [Google Scholar] [CrossRef]
- Haenszel, W.; Duque, E.; Garcia, F.T.; Bolanos, O. Gastric cancer in colombia. iii. natural history of precursor lesions. J. Natl. Cancer Inst. 1976, 57, 1027–1035. [Google Scholar] [CrossRef]
- Chaturvedi, R.; de Sablet, T.; Asim, M.; Piazuelo, M.B.; Barry, D.P.; Verriere, T.G.; Sierra, J.C.; Hardbower, D.M.; Delgado, A.G.; Schneider, B.G.; et al. Increased Helicobacter pylori-associated gastric cancer risk in the Andean region of Colombia is mediated by spermine oxidase. Oncogene 2015, 34, 3429–3440. [Google Scholar] [CrossRef]
- Bravo, L.E.; Van Doorn, L.J.; Realpe, J.L.; Correa, P. Virulence-associated genotypes of Helicobacter pylori: Do they explain the African enigma? Am. J. Gastroenterol. 2002, 97, 2839–2842. [Google Scholar] [CrossRef]
- Kodaman, N.; Pazos, A.; Schneider, B.G.; Blanca Piazuelo, M.; Mera, R.; Sobota, R.S.; Sicinschi, L.A.; Shaffer, C.L.; Romero-Gallo, J.; De Sablet, T.; et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc. Natl. Acad. Sci. USA 2014, 111, 1455–1460. [Google Scholar] [CrossRef] [PubMed]
- Bakhti, S.Z.; Raei, N.; Latifi-Navid, S.; Zahri, S.; Yazdanbod, A. Inverse relationship between cagG-positive Helicobacter pylori status and risk of gastric ulcer. Br. J. Biomed. Sci. 2019, 76, 95–97. [Google Scholar] [CrossRef]
- Sverdén, E.; Agréus, L.; Dunn, J.M.; Lagergren, J. Peptic ulcer disease. BMJ 2019, 367, l5495. [Google Scholar] [CrossRef]
- Lau, J.Y.; Sung, J.; Hill, C.; Henderson, C.; Howden, C.W.; Metz, D.C. Systematic review of the epidemiology of complicated peptic ulcer disease: Incidence, recurrence, risk factors and mortality. Digestion 2011, 84, 102–113. [Google Scholar] [CrossRef]
- Lanas, A.; Chan, F.K.L. Peptic ulcer disease. Lancet 2017, 390, 613–624. [Google Scholar] [CrossRef]
- El-Omar, E.M.; Oien, K.; El-Nujumi, A.; Gillen, D.; Wirz, A.; Dahill, S.; Williams, C.; Ardill, J.E.S.; McColl, K.E.L. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology 1997, 113, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, P.G.; Du, M.Q. Gastrointestinal lymphoma: Where morphology meets molecular biology. J. Pathol. 2005, 205, 255–274. [Google Scholar] [CrossRef] [PubMed]
- D’Elios, M.M.; Amedei, A.; Manghetti, M.; Costa, F.; Baldari, C.T.; Quazi, A.S.; Telford, J.L.; Romagnani, S.; Del Prete, G. Impaired T-cell regulation of B-cell growth in Helicobacter pylori–related gastric low-grade MALT lymphoma. Gastroenterology 1999, 117, 1105–1112. [Google Scholar] [CrossRef]
- Hussell, T.; Isaacson, P.G.; Crabtkee, J.E.; Spencer, J.O. Helicobacter pylori-specific tumour-infiltrating T cells provide contact-dependent help for the growth of malignant B cells in low-grade gastric lymphoma of mucosa-associated lymphoid tissue. J. Pathol. 1996, 178, 122–127. [Google Scholar] [CrossRef]
- Parsonnet, J.; Hansen, S.; Rodriguez, L.; Gelb, A.B.; Warnke, R.A.; Jellum, E.; Orentreich, N.; Vogelman, J.H.; Friedman, G.D. Helicobacter pylori Infection and Gastric Lymphoma. N. Engl. J. Med. 1994, 330, 1267–1271. [Google Scholar] [CrossRef]
- Ruskoné-Fourmestraux, A.; Fischbach, W.; Aleman, B.M.P.; Boot, H.; Du, M.Q.; Megraud, F.; Montalban, C.; Raderer, M.; Savio, A.; Wotherspoon, A. EGILS consensus report. Gastric extranodal marginal zone B-cell lymphoma of MALT. Gut 2011, 60, 747–758. [Google Scholar] [CrossRef]
- Zullo, A.; Hassan, C.; Cristofari, F.; Andriani, A.; De Francesco, V.; Ierardi, E.; Tomao, S.; Stolte, M.; Morini, S.; Vaira, D. Effects of Helicobacter pylori Eradication on Early Stage Gastric Mucosa-Associated Lymphoid Tissue Lymphoma. Clin. Gastroenterol. Hepatol. 2010, 8, 105–110. [Google Scholar] [CrossRef]
- Li, J.Z.; Li, J.Y.; Wu, T.F.; Xu, J.H.; Huang, C.Z.; Cheng, D.; Chen, Q.K.; Yu, T. Helicobacter pylori Infection Is Associated with Type 2 Diabetes, Not Type 1 Diabetes: An Updated Meta-Analysis. Gastroenterol. Res. Pract. 2017, 2017, 5715403. [Google Scholar] [CrossRef] [PubMed]
- Upala, S.; Sanguankeo, A.; Saleem, S.A.; Jaruvongvanich, V. Effects of Helicobacter pylori eradication on insulin resistance and metabolic parameters: A systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 2017, 29, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F.; Shi, S. Helicobacter pylori Infection Increase the Risk of Myocardial Infarction: A Meta-Analysis of 26 Studies Involving more than 20,000 Participants. Helicobacter 2015, 20, 176–183. [Google Scholar] [CrossRef]
- Xu, M.Y.; Cao, B.; Yuan, B.S.; Yin, J.; Liu, L.; Lu, Q. Bin. Association of anaemia with Helicobacter pylori infection: A retrospective study. Sci. Rep. 2017, 7, 13434. [Google Scholar] [CrossRef]
- Satoh, T.; Pandey, J.P.; Okazaki, Y.; Asahi, A.; Kawakami, Y.; Ikeda, Y.; Kuwana, M. Single nucleotide polymorphism of interleukin-1beta associated with Helicobacter pylori infection in immune thrombocytopenic purpura. Tissue Antigens 2009, 73, 353–357. [Google Scholar] [CrossRef]
- Shen, X.; Yang, H.; Wu, Y.; Zhang, D.; Jiang, H. Meta-analysis: Association of Helicobacter pylori infection with Parkinson’s diseases. Helicobacter 2017, 22, e12398. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.J.; Lim, S.H.; Choi, J.; Kim, D.; Kim, Y.S.; Park, M.J.; Yim, J.Y.; Kim, J.S.; Cho, S.H.; Jung, H.C.; et al. Helicobacter pylori Serology Inversely Correlated with the Risk and Severity of Reflux Esophagitis in Helicobacter pylori Endemic Area: A Matched Case-Control Study of 5616 Health Check-Up Koreans. J. Neurogastroenterol. Motil. 2011, 17, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Y.; Hu, J.; Wang, X.; Ren, M.; Lu, G.; Lu, X.; Zhang, D.; He, S. The Effect of Helicobacter pylori Eradication in Patients with Gastroesophageal Reflux Disease: A Meta-Analysis of Randomized Controlled Studies. Dig. Dis. 2020, 38, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Islami, F.; Kamangar, F. Helicobacter pylori and Esophageal Cancer Risk—A Meta-Analysis. Cancer Prev. Res. 2008, 1, 329–338. [Google Scholar] [CrossRef]
- Chen, Y.; Blaser, M.J. Helicobacter pylori Colonization Is Inversely Associated with Childhood Asthma. J. Infect. Dis. 2008, 198, 553–560. [Google Scholar] [CrossRef]
- Amedei, A.; Codolo, G.; Del Prete, G.; de Bernard, M.; D’Elios, M.M. The effect of Helicobacter pylori on asthma and allergy. J. Asthma Allergy 2010, 3, 139–147. [Google Scholar] [PubMed]
- Elias, N.; Nasrallah, E.; Khoury, C.; Mansour, B.; Abu Zuher, L.; Asato, V.; Muhsen, K. Associations of Helicobacter pylori seropositivity and gastric inflammation with pediatric asthma. Pediatr. Pulmonol. 2020, 55, 2236–2245. [Google Scholar] [CrossRef] [PubMed]
- Karttunen, R.; Andersson, G.; Poikonen, K.; Kosunen, T.U.; Kartunen, T.; Juutinen, K.; Niemela, S. Helicobacter pylori induces lymphocyte activation in peripheral blood cultures. Clin. Exp. Immunol. 1990, 82, 485–488. [Google Scholar] [CrossRef]
- Amedei, A.; Cappon, A.; Codolo, G.; Cabrelle, A.; Polenghi, A.; Benagiano, M.; Tasca, E.; Azzurri, A.; D’Elios, M.M.; Del, P.G.; et al. The neutrophil-activating protein of Helicobacter pylori promotes Th1 immune responses. J. Clin. Investig. 2006, 116, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Reyes, V.E.; Beswick, E.J. Helicobacter pylori neutrophil activating protein’s potential as tool in therapeutic immune modulation. Expert Opin. Ther. Pat. 2007, 17, 1315–1320. [Google Scholar] [CrossRef]
- Lundgren, A.; Suri-Payer, E.; Enarsson, K.; Svennerholm, A.M.; Lundin, B.S. Helicobacter pylori-specific CD4 + CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect. Immun. 2003, 71, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Beswick, E.J.; Pinchuk, I.V.; Earley, R.B.; Schmitt, D.A.; Reyes, V.E. Role of gastric epithelial cell-derived transforming growth factor β in reduced CD4+ T cell proliferation and development of regulatory T cells during Helicobacter pylori infection. Infect. Immun. 2011, 79, 2737–2745. [Google Scholar] [CrossRef]
- Lundgren, A.; Stromberg, E.; Sjoling, A.; Lindholm, C.; Enarsson, K.; Edebo, A.; Johnsson, E.; Suri-Payer, E.; Larsson, P.; Rudin, A.; et al. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect. Immun. 2005, 73, 523–531. [Google Scholar] [CrossRef]
- Wang, S.K.; Zhu, H.F.; He, B.S.; Zhang, Z.Y.; Chen, Z.T.; Wang, Z.Z.; Wu, G.L. CagA+ H. pylori infection is associated with polarization of T helper cell immune responses in gastric carcinogenesis. World J. Gastroenterol. 2007, 13, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Mestecky, J. The common mucosal immune system and current strategies for induction of immune responses in external secretions. J. Clin. Immunol. 1987, 7, 265–276. [Google Scholar] [CrossRef]
- Sayar, R.; Shirvani, J.S.; Hajian-Tilaki, K.; Vosough, Z.; Ranaei, M. The negative association between inflammatory bowel disease and Helicobacter pylori seropositivity. Casp. J. Intern. Med. 2019, 10, 217–222. [Google Scholar]
- Castanõ-Rodríguez, N.; Kaakoush, N.O.; Lee, W.S.; Mitchell, H.M. Dual role of Helicobacter and Campylobacter species in IBD: A systematic review and meta-analysis. Gut 2017, 66, 235–249. [Google Scholar] [CrossRef]
- Ewe, K.; Schwartz, S.; Petersen, S.; Press, A.G. Inflammation does not decrease intraluminal pH in chronic inflammatory bowel disease. Dig. Dis. Sci. 1999, 44, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Reyes, V.E.; Almanza, R.J.; Ye, G. Helicobacter pylori: A Trojan horse in the gastric environment? Mucosal Immunol. Update 1998, 5, 68–70. [Google Scholar]
- Beswick, E.J.; Suarez, G.; Reyes, V.E. H. pylori and host interactions that influence pathogenesis. World J. Gastroenterol. 2006, 12, 5599–5605. [Google Scholar] [CrossRef] [PubMed]
- Suarez, G.; Reyes, V.E.; Beswick, E.J. Immune response to H. pylori. World J. Gastroenterol. 2006, 12, 5593–5598. [Google Scholar] [CrossRef]
- Reyes, V.E.; Suárez, G.; Sierra, J.C.; Beswick, E.J. Helicobacter pylori. In Vaccines for Biodefense and Emerging and Neglected Diseases; Barrett, A.D.T., Stanberry, L., Eds.; Academic Press: Cambridge, MA, USA, 2009; pp. 983–1012. [Google Scholar]
- Alzahrani, S.; Lina, T.T.; Gonzalez, J.; Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Effect of Helicobacter pylori on gastric epithelial cells. World J. Gastroenterol. 2014, 20, 12767–12780. [Google Scholar] [CrossRef]
- Lina, T.T.; Alzahrani, S.; Gonzalez, J.; Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Immune evasion strategies used by Helicobacter pylori. World J. Gastroenterol. 2014, 20, 12753–12766. [Google Scholar] [CrossRef]
- Ha, N.C.; Oh, S.T.; Sung, J.Y.; Cha, K.A.; Lee, M.H.; Oh, B.H. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat. Struct. Biol. 2001, 8, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Mobley, H.L. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment. Pharmacol. Ther. 1996, 10, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.E.; Phadnis, S.H. Structure, function and localization of Helicobacter pylori urease. Yale J. Biol. Med. 1998, 71, 63–73. [Google Scholar] [PubMed]
- Dunn, B.E.; Vakil, N.B.; Schneider, B.G.; Miller, M.M.; Zitzer, J.B.; Peutz, T.; Phadnis, S.H. Localization of Helicobacter pylori urease and heat shock protein in human gastric biopsies. Infect. Immun. 1997, 65, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Phadnis, S.H.; Parlow, M.H.; Levy, M.; Ilver, D.; Caulkins, C.M.; Connors, J.B.; Dunn, B.E. Surface localization of Helicobacter pylori urease and a heat shock protein homolog requires bacterial autolysis. Infect. Immun. 1996, 64, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Gunasena, H.; Cheng, Z.; Espejo, R.; Crowe, S.E.; Ernst, P.B.; Reyes, V.E. Helicobacter pylori urease binds to class II MHC on gastric epithelial cells and induces their apoptosis. J. Immunol. 2000, 165, 1918–1924. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Crowe, S.E.; Behar, S.; Gunasena, H.; Ye, G.; Haeberle, H.; Van Houten, N.; Gourley, W.K.; Ernst, P.B.; Reyes, V.E. The effect of class II major histocompatibility complex expression on adherence of Helicobacter pylori and induction of apoptosis in gastric epithelial cells: A mechanism for T helper cell type 1-mediated damage. J. Exp. Med. 1998, 187, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Barrera, C.; Espejo, R.; Reyes, V.E. Differential glycosylation of MHC class II molecules on gastric epithelial cells: Implications in local immune responses. Hum. Immunol. 2002, 63, 384–393. [Google Scholar] [CrossRef]
- Fan, X.J.; Gunasena, H.; Gonzales, M.; Almanza, R.; Cheng, Z.; Ye, G.; Barrera, C.; Crowe, S.E.; Ernst, P.B.; Reyes, V.E. Class II MHC molecules on gastric epithelial cells act as receptors for Helicobacter pylori urease and signal apoptosis of the epithelium. FASEB J. 1998, 12, A1089. [Google Scholar]
- Beswick, E.J.; Pinchuk, I.V.; Minch, K.; Suarez, G.; Sierra, J.C.C.; Yamaoka, Y.; Reyes, V.E.E. The Helicobacter pylori Urease B Subunit Binds to CD74 on Gastric Epithelial Cells and Induces NF-{kappa}B Activation and Interleukin-8 Production. Infect. Immun. 2006, 74, 1148–1155. [Google Scholar] [CrossRef]
- Beswick, E.J.; Bland, D.A.; Suarez, G.; Barrera, C.A.; Fan, X.; Reyes, V.E. Helicobacter pylori binds to CD74 on gastric epithelial cells and stimulates interleukin-8 production. Infect. Immun. 2005, 73, 2736–2743. [Google Scholar] [CrossRef]
- Valenzuela-Valderrama, M.; Cerda-Opazo, P.; Backert, S.; González, M.F.; Carrasco-Véliz, N.; Jorquera-Cordero, C.; Wehinger, S.; Canales, J.; Bravo, D.; Quest, A.F.G. The Helicobacter pylori Urease Virulence Factor Is Required for the Induction of Hypoxia-Induced Factor-1α in Gastric Cells. Cancers 2019, 11, 799. [Google Scholar] [CrossRef]
- Kitajima, Y.; Miyazaki, K. The critical impact of HIF-1α on gastric cancer biology. Cancers 2013, 5, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Goda, N.; Ryan, H.E.; Khadivi, B.; McNulty, W.; Rickert, R.C.; Johnson, R.S. Hypoxia-Inducible Factor 1α Is Essential for Cell Cycle Arrest during Hypoxia. Mol. Cell Biol. 2003, 23, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sun, M.; Wang, L.; Jiao, B. HIFs, angiogenesis, and cancer. J. Cell Biochem. 2013, 114, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.N.; Hartung, M.L.; Urban, S.; Kyburz, A.; Bahlmann, A.S.; Lind, J.; Backert, S.; Taube, C.; Müller, A. Helicobacter urease-induced activation of the TLR2/NLRP3/IL-18 axis protects against asthma. J. Clin. Investig. 2015, 125, 3297–3302. [Google Scholar] [CrossRef]
- Boren, T.; Falk, P.; Roth, K.A.; Larson, G.; Normark, S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 1993, 262, 1892–1895. [Google Scholar] [CrossRef]
- Sheu, B.S.; Sheu, S.M.; Yang, H.B.; Huang, A.H.; Wu, J.J. Host gastric Lewis expression determines the bacterial density of Helicobacter pylori in babA2 genopositive infection. Gut 2003, 52, 927–932. [Google Scholar] [CrossRef]
- Fujimoto, S.; Olaniyi Ojo, O.; Arnqvist, A.; Yih Wu, J.; Odenbreit, S.; Haas, R.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori BabA Expression, Gastric Mucosal Injury, and Clinical Outcome. Clin. Gastroenterol. Hepatol. 2007, 5, 49–58. [Google Scholar] [CrossRef]
- Sheu, S.M.; Sheu, B.S.; Chiang, W.C.; Kao, C.Y.; Wu, H.M.; Yang, H.B.; Wu, J.J. H. pylori clinical isolates have diverse babAB genotype distributions over different topographic sites of stomach with correlation to clinical disease outcomes. BMC Microbiol. 2012, 12, 89. [Google Scholar] [CrossRef]
- Backert, S.; Selbach, M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 2008, 10, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Javaheri, A.; Kruse, T.; Moonens, K.; Mejías-Luque, R.; Debraekeleer, A.; Asche, C.I.; Tegtmeyer, N.; Kalali, B.; Bach, N.C.; Sieber, S.A.; et al. Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat. Microbiol. 2016, 2, 16189. [Google Scholar] [CrossRef]
- Taxauer, K.; Hamway, Y.; Ralser, A.; Dietl, A.; Mink, K.; Vieth, M.; Singer, B.B.; Gerhard, M.; Mejías-Luque, R. Engagement of CEACAM1 by Helicobacter pylori HopQ is important for the activation of non-canonical NF-κB in gastric epithelial cells. Microorganisms 2021, 9, 1748. [Google Scholar] [CrossRef] [PubMed]
- Feige, M.H.; Sokolova, O.; Pickenhahn, A.; Maubach, G.; Naumann, M. HopQ impacts the integrin α5β1-independent NF-κB activation by Helicobacter pylori in CEACAM expressing cells. Int. J. Med. Microbiol. 2018, 308, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Kwon, D.H.; Graham, D.Y. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2000, 97, 7533–7538. [Google Scholar] [CrossRef]
- Lu, H.; Jeng, Y.W.; Beswick, E.J.; Ohno, T.; Odenbreit, S.; Haas, R.; Reyes, V.E.; Kita, M.; Graham, D.Y.; Yamaoka, Y. Functional and intracellular signaling differences associated with the Helicobacter pylori AlpAB adhesin from Western and East Asian strains. J. Biol. Chem. 2007, 282, 6242–6254. [Google Scholar] [CrossRef]
- Odenbreit, S.; Till, M.; Hofreuter, D.; Faller, G.; Haas, R. Genetic and functional characterization of the alpAB gene locus essential for the adhesion of Helicobacter pylori to human gastric tissue. Mol. Microbiol. 1999, 31, 1537–1548. [Google Scholar] [CrossRef]
- Odenbreit, S.; Faller, G.; Haas, R. Role of the alpAB proteins and lipopolysaccharide in adhesion of Helicobacter pylori to human gastric tissue. Int. J. Med. Microbiol. 2002, 292, 247–256. [Google Scholar] [CrossRef]
- Mahdavi, J.; Sondén, B.; Hurtig, M.; Olfat, F.O.; Forsberg, L.; Roche, N.; Ångström, J.; Larsson, T.; Teneberg, S.; Karlsson, K.A.; et al. Helicobacter pylori SabA Adhesin in Persistent Infection and Chronic Inflammation. Science 2002, 297, 573–578. [Google Scholar] [CrossRef]
- Walz, A.; Odenbreit, S.; Mahdavi, J.; Borén, T.; Ruhl, S. Identification and characterization of binding properties of Helicobacter pylori by glycoconjugate arrays. Glycobiology 2005, 15, 700–708. [Google Scholar] [CrossRef]
- Hirmo, S.; Kelm, S.; Schauer, R.; Nilsson, B.; Wadström, T. Adhesion of Helicobacter pylori strains to α-2,3-linked sialic acids. Glycoconj. J. 1996, 13, 1005–1011. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ishijima, N.; Suzuki, M.; Ashida, H.; Ichikawa, Y.; Kanegae, Y.; Saito, I.; Borén, T.; Haas, R.; Sasakawa, C.; Mimuro, H. BabA-mediated Adherence Is a Potentiator of the Helicobacter pylori Type IV Secretion System Activity. J. Biol. Chem. 2011, 286, 25256–25264. [Google Scholar] [CrossRef]
- Backert, S.; Tegtmeyer, N.; Fischer, W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol. 2015, 10, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Structure and function of Helicobacter pylori caga, the first-identified bacterial protein involved in human cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 196–219. [Google Scholar] [CrossRef]
- Kaplan-Türköz, B.; Jiménez-Soto, L.F.; Dian, C.; Ertl, C.; Remaut, H.; Louche, A.; Tosi, T.; Haas, R.; Terradot, L. Structural insights into Helicobacter pylori oncoprotein CagA interaction with β1 integrin. Proc. Natl. Acad. Sci. USA 2012, 109, 14640–14645. [Google Scholar] [CrossRef]
- Jiménez-Soto, L.F.; Kutter, S.; Sewald, X.; Ertl, C.; Weiss, E.; Kapp, U.; Rohde, M.; Pirch, T.; Jung, K.; Retta, S.F.; et al. Helicobacter pylori type IV secretion apparatus exploits β1 integrin in a novel RGD-independent manner. PLoS Pathog. 2009, 5, e1000684. [Google Scholar] [CrossRef]
- Hatakeyama, M. Helicobacter pylori CagA and gastric cancer: A paradigm for hit-and-run carcinogenesis. Cell Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef]
- Higashi, H.; Tsutsumi, R.; Fujita, A.; Yamazaki, S.; Asaka, M.; Azuma, T.; Hatakeyama, M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc. Natl. Acad. Sci. USA 2002, 99, 14428–14433. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.; Machado, J.C.; Yamaoka, Y. Pathogenesis of Helicobacter pylori Infection. Helicobacter 2005, 10, 14–20. [Google Scholar] [CrossRef]
- Churin, Y.; Al Ghoul, L.; Kepp, O.; Meyer, T.F.; Birchmeier, W.; Naumann, M. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J. Cell Biol. 2003, 161, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Mimuro, H.; Kiga, K.; Fukumatsu, M.; Ishijima, N.; Morikawa, H.; Nagai, S.; Koyasu, S.; Gilman, R.H.; Kersulyte, D.; et al. Helicobacter pylori CagA Phosphorylation-Independent Function in Epithelial Proliferation and Inflammation. Cell Host Microbe 2009, 5, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Mimuro, H.; Suzuki, T.; Tanaka, J.; Asahi, M.; Haas, R.; Sasakawa, C. Grb2 Is a Key Mediator of Helicobacter pylori CagA Protein Activities. Mol. Cell 2002, 10, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Johnston, E.; Krishna, U.; Yamaoka, Y.; Israel, D.A.; Nagy, T.A.; Wroblewski, L.E.; Piazuelo, M.B.; Correa, P.; Peek, R.M., Jr. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008, 68, 379–387. [Google Scholar] [CrossRef]
- Saadat, I.; Higashi, H.; Obuse, C.; Umeda, M.; Murata-Kamiya, N.; Saito, Y.; Lu, H.; Ohnishi, N.; Azuma, T.; Suzuki, A.; et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 2007, 447, 330–333. [Google Scholar] [CrossRef]
- Murata-Kamiya, N.; Kurashima, Y.; Teishikata, Y.; Yamahashi, Y.; Saito, Y.; Higashi, H.; Aburatani, H.; Akiyama, T.; Peek, R.M., Jr.; Azuma, T.; et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 2007, 26, 4617–4626. [Google Scholar] [CrossRef]
- Palrasu, M.; Zaika, E.; Paulrasu, K.; Gokulan, R.C.; Suarez, G.; Que, J.; El-Rifai, W.; Peek, R.M.; Garcia-Buitrago, M.; Zaika, A.I. Helicobacter pylori pathogen inhibits cellular responses to oncogenic stress and apoptosis. PLoS Pathog. 2022, 18, e1010628. [Google Scholar] [CrossRef]
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Viala, J.; Chaput, C.; Boneca, I.G.; Cardona, A.; Girardin, S.E.; Moran, A.P.; Athman, R.; Memet, S.; Huerre, M.R.; Coyle, A.J.; et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 2004, 5, 1166–1174. [Google Scholar] [CrossRef]
- Grubman, A.; Kaparakis, M.; Viala, J.; Allison, C.; Badea, L.; Karrar, A.; Boneca, I.G.; Le Bourhis, L.; Reeve, S.; Smith, I.A.; et al. The innate immune molecule, NOD1, regulates direct killing of Helicobacter pylori by antimicrobial peptides 94. Cell Microbiol. 2010, 12, 626–639. [Google Scholar] [CrossRef]
- Suarez, G.; Romero-Gallo, J.; Piazuelo, M.B.; Wang, G.; Maier, R.J.; Forsberg, L.S.; Azadi, P.; Gomez, M.A.; Correa, P.; Peek, R.M. Modification of Helicobacter pylori peptidoglycan enhances NOD1 activation and promotes cancer of the stomach. Cancer Res. Am. Assoc. Cancer Res. Inc. 2015, 75, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Sorbara, M.T.; Ellison, L.K.; Ramjeet, M.; Travassos, L.H.; Jones, N.L.; Girardin, S.E.; Philpott, D.J. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity 2013, 39, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.G.; Feng, Y.; Xu, S.; Taylor, N.S.; Kinsel, M.; Dewhirst, F.E.; Paster, B.J.; Greenwell, M.; Levine, G.; Rogers, A.; et al. Helicobacter cetorum sp. nov., a Urease-positive Helicobacter species isolated from dolphins and whales. J. Clin. Microbiol. 2002, 40, 4536–4543. [Google Scholar] [CrossRef]
- Reyrat, J.M.; Lanzavecchia, S.; Lupetti, P.; De Bernard, M.; Pagliaccia, C.; Pelicic, V.; Charrel, M.; Ulivieri, C.; Norais, N.; Ji, X.; et al. 3D imaging of the 58 kDa cell binding subunit of the Helicobacter pylori cytotoxin. J. Mol. Biol. 1999, 290, 459–470. [Google Scholar] [CrossRef]
- McClain, M.S.; Iwamoto, H.; Cao, P.; Vinion-Dubiel, A.D.; Li, Y.; Szabo, G.; Shao, Z.; Cover, T.L. Essential role of a GXXXG motif for membrane channel formation by Helicobacter pylori vacuolating toxin. J. Biol. Chem. 2003, 278, 12101–12108. [Google Scholar] [CrossRef]
- González-Rivera, C.; Gangwer, K.A.; McClain, M.S.; Eli, I.M.; Chambers, M.G.; Ohi, M.D.; Lacy, D.B.; Cover, T.L. Reconstitution of Helicobacter pylori VacA toxin from purified components. Biochemistry 2010, 49, 5743–5752. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.J.; Ivie, S.E.; McClain, M.S.; Cover, T.L. Functional properties of the p33 and p55 domains of the Helicobacter pylori vacuolating cytotoxin. J. Biol. Chem. 2005, 280, 21107–21114. [Google Scholar] [CrossRef]
- Gupta, V.R.; Patel, H.K.; Kostolansky, S.S.; Ballivian, R.A.; Eichberg, J.; Blanke, S.R. Sphingomyelin functions as a novel receptor for Helicobacter pylori VacA. PLoS Pathog. 2008, 4, e1000073. [Google Scholar] [CrossRef]
- Ricci, V.; Galmiche, A.; Doye, A.; Necchi, V.; Solcia, E.; Boquet, P. High cell sensitivity to Helicobacter pylori VacA toxin depends on a GPI-anchored protein and is not blocked by inhibition of the clathrin-mediated pathway of endocytosis. Mol. Biol. Cell 2000, 11, 3897–3909. [Google Scholar] [CrossRef]
- Oldani, A.; Cormont, M.; Hofman, V.; Chiozzi, V.; Oregioni, O.; Canonici, A.; Sciullo, A.; Sommi, P.; Fabbri, A.; Ricci, V.; et al. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 2009, 5, e1000603. [Google Scholar] [CrossRef]
- Gauthier, N.C.; Monzo, P.; Gonzalez, T.; Doye, A.; Oldani, A.; Gounon, P.; Ricci, V.; Cormont, M.; Boquet, P. Early endosomes associated with dynamic F-actin structures are required for late trafficking of H. pylori VacA toxin. J. Cell Biol. 2007, 177, 343–354. [Google Scholar] [CrossRef]
- Yamasaki, E.; Wada, A.; Kumatori, A.; Nakagawa, I.; Funao, J.; Nakayama, M.; Hisatsune, J.; Kimura, M.; Moss, J.; Hirayama, T. Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J. Biol. Chem. 2006, 281, 11250–11259. [Google Scholar] [CrossRef] [PubMed]
- Galmiche, A.; Rassow, J.; Doye, A.; Cagnol, S.; Chambard, J.C.; Contamin, S.; De Thillot, V.; Just, I.; Ricci, V.; Solcia, E.; et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000, 19, 6361–6370. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Luo, Z.Q.; Blanke, S.R. Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc. Natl. Acad. Sci. USA 2011, 108, 16032–16037. [Google Scholar] [CrossRef]
- Atherton, J.C.; Cao, P.; Peek, R.M.; Tummuru, M.K.R.; Blaser, M.J.; Cover, T.L. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J. Biol. Chem. 1995, 270, 17771–17777. [Google Scholar] [CrossRef]
- Bakhti, S.Z.; Latifi-Navid, S.; Mohammadi, S.; Zahri, S.; Bakhti, F.S.; Feizi, F.; Yazdanbod, A.; Siavoshi, F. Relevance of Helicobacter pylori vacA 3′-end Region Polymorphism to Gastric Cancer. Helicobacter 2016, 21, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Soyfoo, D.M.; Doomah, Y.H.; Xu, D.; Zhang, C.; Sang, H.M.; Liu, Y.Y.; Zhang, G.X.; Jiang, J.X.; Xu, S.F. New genotypes of Helicobacter pylori VacA d-region identified from global strains. BMC Mol. Cell Biol. 2021, 22, 4. [Google Scholar] [CrossRef]
- McClain, M.S.; Cao, P.; Iwamoto, H.; Vinion-Dubiel, A.D.; Szabo, G.; Shao, Z.; Cover, T.L. A 12-amino-acid segment, present in type s2 but not type s1 Helicobacter pylori VacA proteins, abolishes cytotoxin activity and alters membrane channel formation. J. Bacteriol. 2001, 183, 6499–6508. [Google Scholar] [CrossRef]
- Wang, H.J.; Kuo, C.H.; Yeh, A.A.M.; Chang, P.C.L.; Wang, W.C. Vacuolating toxin production in clinical isolates of Helicobacter pylori with different vacA genotypes. J. Infect. Dis. 1998, 178, 207–212. [Google Scholar] [CrossRef]
- Basso, D.; Zambon, C.F.; Letley, D.P.; Stranges, A.; Marchet, A.; Rhead, J.L.; Schiavon, S.; Guariso, G.; Ceroti, M.; Nitti, D.; et al. Clinical Relevance of Helicobacter pylori cagA and vacA Gene Polymorphisms. Gastroenterology 2008, 135, 91–99. [Google Scholar] [CrossRef]
- Nogueira, C.; Figueiredo, C.; Carneiro, F.; Gomes, A.T.; Barreira, R.; Figueira, P.; Salgado, C.; Belo, L.; Peixoto, A.; Bravo, J.C.; et al. Helicobacter pylori genotypes may determine gastric histopathology. Am. J. Pathol. 2001, 158, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Zali, M.R.; Yamaoka, Y. The association of vacA genotypes and Helicobacter pylori-related gastroduodenal diseases in the Middle East. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.L.; Yeh, Y.C.; Sheu, B.S. The impacts of H. pylori virulence factors on the development of gastroduodenal diseases. J. Biomed. Sci. 2018, 25, 68. [Google Scholar] [CrossRef]
- Chung, C.; Olivares, A.; Torres, E.; Yilmaz, O.; Cohen, H.; Perez-Perez, G. Diversity of VacA intermediate region among Helicobacter pylori strains from several regions of the world 1. J. Clin. Microbiol. 2010, 48, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Fernandez, T.; Burroni, D.; Pagliaccia, C.; Atherton, J.C.; Reyrat, J.M.; Rappuoli, R.; Telford, J.L. Cell specificity of Helicobacter pylori cytotoxin is determined by a short region in the polymorphic midregion. Infect. Immun. 2000, 68, 3754–3757. [Google Scholar] [CrossRef]
- Weiss, G.; Forster, S.; Irving, A.; Tate, M.; Ferrero, R.L.; Hertzog, P.; Frøkiær, H.; Kaparakis-Liaskos, M. Helicobacter pylori VacA Suppresses Lactobacillus acidophilus-Induced Interferon Beta Signaling in Macrophages via Alterations in the Endocytic Pathway. mBio 2013, 4, e00609-12. [Google Scholar] [CrossRef]
- Zheng, P.Y.; Jones, N.L. Helicobacter pylori strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO (coronin 1) protein. Cell Microbiol. 2003, 5, 25–40. [Google Scholar] [CrossRef]
- Jung, M.K.; Joo, S.K.; Jin, Y.L.; Kim, Y.J.; Youn, H.J.; In, Y.K.; Young, J.C.; Oh, Y.K.; Kim, N.; Hyun, C.J.; et al. Vacuolating Cytotoxin in Helicobacter pylori Water-Soluble Proteins Upregulates Chemokine Expression in Human Eosinophils via Ca2+ Influx, Mitochondrial Reactive Oxygen Intermediates, and NF-κB Activation. Infect. Immun. 2007, 75, 3373–3381. [Google Scholar]
- Supajatura, V.; Ushio, H.; Wada, A.; Yahiro, K.; Okumura, K.; Ogawa, H.; Hirayama, T.; Ra, C. Cutting edge: VacA, a vacuolating cytotoxin of Helicobacter pylori, directly activates mast cells for migration and production of proinflammatory cytokines. J. Immunol. 2002, 168, 2603–2607. [Google Scholar] [CrossRef]
- Oertli, M.; Noben, M.; Engler, D.B.; Semper, R.P.; Reuter, S.; Maxeiner, J.; Gerhard, M.; Taube, C.; Muller, A. Helicobacter pylori gamma-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc. Natl. Acad. Sci. USA 2013, 110, 3047–3052. [Google Scholar] [CrossRef]
- Boncristiano, M.; Paccani, S.R.; Barone, S.; Ulivieri, C.; Patrussi, L.; Ilver, D.; Amedei, A.; D’Elios, M.M.; Telford, J.L.; Baldari, C.T. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J. Exp. Med. 2003, 198, 1887–1897. [Google Scholar] [CrossRef]
- Gebert, B.; Fischer, W.; Weiss, E.; Hoffmann, R.; Haas, R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 2003, 301, 1099–1102. [Google Scholar] [CrossRef]
- Molinari, M.; Salio, M.; Galli, C.; Norais, N.; Rappuoli, R.; Lanzavecchia, A.; Montecucco, C. Selective inhibition of Ii-dependent antigen presentation by Helicobacter pylori toxin vacA. J. Exp. Med. 1998, 187, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.A.; Schlesinger, L.S.; Kang, B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J. Exp. Med. 2000, 191, 115–128. [Google Scholar] [CrossRef]
- Zanotti, G.; Papinutto, E.; Dundon, W.G.; Battistutta, R.; Seveso, M.; Giudice, G.; Del Rappuoli, R.; Montecucco, C. Structure of the neutrophil-activating protein from Helicobacter pylori. J. Mol. Biol. 2002, 323, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Satin, B.; Del, G.G.; Della, V.B.; Dusi, S.; Laudanna, C.; Tonello, F.; Kelleher, D.; Rappuoli, R.; Montecucco, C.; Rossi, F. The neutrophil-activating protein (HP-NAP) of Helicobacter pylori is a protective antigen and a major virulence factor. J. Exp. Med. 2000, 191, 1467–1476. [Google Scholar] [CrossRef]
- Evans, D.J., Jr.; Evans, D.G.; Takemura, T.; Nakano, H.; Lampert, H.C.; Graham, D.Y.; Granger, D.N.; Kvietys, P.R. Characterization of a Helicobacter pylori neutrophil-activating protein. Infect. Immun. 1995, 63, 2213–2220. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.W. Helicobacter pylori neutrophil-activating protein: From molecular pathogenesis to clinical applications. World J. Gastroenterol. 2014, 20, 5294–5301. [Google Scholar] [CrossRef]
- Montemurro, P.; Nishioka, H.; Dundon, W.G.; de Bernard, M.; Del, G.G.; Rappuoli, R.; Montecucco, C. The neutrophil-activating protein (HP-NAP) of Helicobacter pylori is a potent stimulant of mast cells. Eur. J. Immunol. 2002, 32, 671–676. [Google Scholar] [CrossRef]
- Polenghi, A.; Bossi, F.; Fischetti, F.; Durigutto, P.; Cabrelle, A.; Tamassia, N.; Cassatella, M.A.; Montecucco, C.; Tedesco, F.; de Bernard, M. The neutrophil-activating protein of Helicobacter pylori crosses endothelia to promote neutrophil adhesion in vivo. J. Immunol. 2007, 178, 1312–1320. [Google Scholar] [CrossRef]
- Wen, S.H.; Hong, Z.W.; Chen, C.C.; Chang, H.W.; Fu, H.W. Helicobacter pylori neutrophil-activating protein directly interacts with and activates toll-like receptor 2 to induce the secretion of interleukin-8 from neutrophils and atra-induced differentiated hl-60 cells. Int. J. Mol. Sci. 2021, 22, 11560. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood Content Repos. 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Oleastro, M.; Ménard, A. The role of Helicobacter pylori outer membrane proteins in adherence and pathogenesis. Biology 2013, 2, 1110–1134. [Google Scholar] [CrossRef]
- Huang, T.; Zhou, F.; Yuan, X.; Yang, T.; Liang, X.; Wang, Y.; Tu, H.; Chang, J.; Nan, K.; Wei, Y. Reactive Oxygen Species Are Involved in the Development of Gastric Cancer and Gastric Cancer-Related Depression through ABL1-Mediated Inflammation Signaling Pathway. Oxidative Med. Cell. Longev. 2019, 2019, 5813985. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, M.; Jin, C.; Yu, D.; Eriksson, F.; Essand, M. Vector-Encoded Helicobacter pylori Neutrophil-Activating Protein Promotes Maturation of Dendritic Cells with Th1 Polarization and Improved Migration. J. Immunol. 2014, 193, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Bolz, C.; Revez, J.; Javed, S.; El-Najjar, N.; Anderl, F.; Hyytiäinen, H.; Vuorela, P.; Gerhard, M.; Hänninen, M.L. Evidence for conserved function of γ-glutamyltranspeptidase in Helicobacter genus. PLoS ONE 2012, 7, e30543. [Google Scholar] [CrossRef]
- Saini, M.; Kashyap, A.; Bindal, S.; Saini, K.; Gupta, R. Bacterial Gamma-Glutamyl Transpeptidase, an Emerging Biocatalyst: Insights Into Structure–Function Relationship and Its Biotechnological Applications. Front. Microbiol. 2021, 2021, 641251. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, K.; Wachino, J.I.; Arakawa, Y.; Saidijam, M.; Rutherford, N.G.; Henderson, P.J.F. Metabolism of glutamine and glutathione via gamma-glutamyltranspeptidase and glutamate transport in Helicobacter pylori: Possible significance in the pathophysiology of the organism. Mol. Microbiol. 2007, 64, 396–406. [Google Scholar] [CrossRef]
- McGovern, K.J.; Blanchard, T.G.; Gutierrez, J.A.; Czinn, S.J.; Krakowka, S.; Youngman, P. γ-glutamyltransferase is a Helicobacter pylori virulence factor but is not essential for colonization. Infect. Immun. 2001, 69, 4168–4173. [Google Scholar] [CrossRef]
- Chevalier, C.; Thiberge, J.M.; Ferrero, R.L.; Labigne, A. Essential role of Helicobacter pylori gamma-glutamyltranspeptidase for the colonization of the gastric mucosa of mice. Mol. Microbiol. 1999, 31, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Lee, S.G.; Park, M.G.; Song, J.Y.; Kang, H.L.; Lee, W.K.; Cho, M.J.; Rhee, K.H.; Youn, H.S.; Baik, S.C. Gamma-glutamyltranspeptidase of Helicobacter pylori induces mitochondria-mediated apoptosis in AGS cells. Biochem. Biophys. Res. Commun. 2007, 355, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, M.; Bravo, D.; Canales, J.; Sanhueza, C.; Díaz, N.; Almarza, O.; Toledo, H.; Quest, A.F.G. Helicobacter pylori-induced loss of survivin and gastric cell viability is attributable to secreted bacterial gamma-glutamyl transpeptidase activity. J. Infect. Dis. 2013, 208, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Lee, S.G.; Kim, J.M.; Kim, D.S.; Song, J.Y.; Kang, H.L.; Lee, W.K.; Cho, M.J.; Rhee, K.H.; Youn, H.S.; et al. Helicobacter pylori γ-glutamyltranspeptidase induces cell cycle arrest at the G1-S phase transition. J. Microbiol. 2010, 48, 372–377. [Google Scholar] [CrossRef]
- Gong, M.; Ling, S.S.M.; Lui, S.Y.; Yeoh, K.G.; Ho, B. Helicobacter pylori γ-glutamyl transpeptidase is a pathogenic factor in the development of peptic ulcer disease. Gastroenterology 2010, 139, 564–573. [Google Scholar] [CrossRef]
- Busiello, I.; Acquaviva, R.; Di Popolo, A.; Blanchard, T.G.; Ricci, V.; Romano, M.; Zarrilli, R. Helicobacter pylori γ-glutamyltranspeptidase upregulates COX-2 and EGF-related peptide expression in human gastric cells. Cell Microbiol. 2004, 6, 255–267. [Google Scholar] [CrossRef]
- Nagy, T.A.; Frey, M.R.; Yan, F.; Israel, D.A.; Polk, D.B.; Peek, R.M. Helicobacter pylori regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J. Infect. Dis. 2009, 199, 641–651. [Google Scholar] [CrossRef]
- Murayama, Y.; Miyagawa, J.I.; Shinomura, Y.; Kanayama, S.; Isozaki, K.; Yamamori, K.; Mizuno, H.; Ishiguro, S.; Kiyohara, T.; Miyazaki, Y.; et al. Significance of the association between heparin-binding epidermal growth factor-like growth factor and CD9 in human gastric cancer. Int. J. Cancer 2002, 98, 505–513. [Google Scholar] [CrossRef]
- Yin, F.; Grabowska, A.M.; Clarke, P.A.; Whelband, E.; Robinson, K.; Argent, R.H.; Tobias, A.; Kumari, R.; Atherton, J.C.; Watson, S.A. Helicobacter pylori potentiates epithelial:mesenchymal transition in gastric cancer: Links to soluble HB-EGF, gastrin and matrix metalloproteinase-7. Gut 2010, 59, 1037–1045. [Google Scholar] [CrossRef]
- Shi, H.; Qi, C.; Meng, L.; Yao, H.; Jiang, C.; Fan, M.; Zhang, Q.; Hou, X.; Lin, R. Bone marrow-derived mesenchymal stem cells promote Helicobacter pylori-associated gastric cancer progression by secreting thrombospondin-2. Cell Prolif. 2021, 54, e13114. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Shi, H.; Meng, L.; Jiang, X.; Pang, S.; Fan, M.; Lin, R. Gamma-glutamyltransferase of Helicobacter pylori alters the proliferation, migration, and pluripotency of mesenchymal stem cells by affecting metabolism and methylation status. J. Microbiol. 2022, 60, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Shi, H.; Wang, Z.; Fan, M.; Pang, S.; Lin, R. The Gamma-glutamyltransferase gene of Helicobacter pylori can promote gastric carcinogenesis by activating Wnt signal pathway through up-regulating TET1. Life Sci. 2021, 267, 118921. [Google Scholar] [CrossRef] [PubMed]
- Schmees, C.; Prinz, C.; Treptau, T.; Rad, R.; Hengst, L.; Voland, P.; Bauer, S.; Brenner, L.; Schmid, R.M.; Gerhard, M. Inhibition of T-cell proliferation by Helicobacter pylori gamma-glutamyl transpeptidase. Gastroenterology 2007, 132, 1820–1833. [Google Scholar] [CrossRef]
- Gerhard, M.; Schmees, C.; Voland, P.; Endres, N.; Sander, M.; Reindl, W.; Rad, R.; Oelsner, M.; Decker, T.; Mempel, M.; et al. A secreted low-molecular-weight protein from Helicobacter pylori induces cell-cycle arrest of T cells. Gastroenterology 2005, 128, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Fassi Fehri, L.; Koch, M.; Belogolova, E.; Khalil, H.; Bolz, C.; Kalali, B.; Mollenkopf, H.J.; Beigier-Bompadre, M.; Karlas, A.; Schneider, T.; et al. Helicobacter pylori induces miR-155 in T cells in a cAMP-Foxp3-dependent manner. PLoS ONE 2010, 5, e9500. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Kandulski, A.; Wex, T.; Kuester, D.; Peitz, U.; Gebert, I.; Roessner, A.; Malfertheiner, P. Naturally occurring regulatory T cells (CD4+, CD25high, FOXP3+) in the antrum and cardia are associated with higher H. pylori colonization and increased gene expression of TGF-beta1. Helicobacter 2008, 13, 295–303. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Lina, T.T.; Alzahrani, S.; House, J.; Yamaoka, Y.; Sharpe, A.H.; Rampy, B.A.; Pinchuk, I.V.; Reyes, V.E. Helicobacter pylori cag pathogenicity island’s role in B7-H1 induction and immune evasion. PLoS ONE 2015, 10, e0121841. [Google Scholar] [CrossRef]
- Das, S.; Suarez, G.; Beswick, E.J.; Sierra, J.C.; Graham, D.Y.; Reyes, V.E. Expression of B7-H1 on gastric epithelial cells: Its potential role in regulating T cells during Helicobacter pylori infection. J. Immunol. 2006, 176, 3000–3009. [Google Scholar] [CrossRef]
- Beswick, E.J.; Pinchuk, I.V.; Das, S.; Powell, D.W.W.; Reyes, V.E.E. B7-H1 Expression on Gastric Epithelial Cells after Helicobacter pylori Exposure Promotes the Development of CD4+ CD25+ FoxP3+ Regulatory T Cells. Infect. Immun. 2007, 75, 4334–4341. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Li, W.; Li, R.; Wu, K.; Zhao, E.; Zhang, Y.; Zhang, P.; Shi, L.; Wang, D.; Yin, Y.; et al. Helicobacter pylori Promote B7-H1 Expression by Suppressing miR-152 and miR-200b in Gastric Cancer Cells. PLoS ONE 2017, 12, e0168822. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhu, X.; Guo, X.; Yang, Z.; Cai, Q.; Gu, D.; Luo, W.; Yuan, C.; Xiang, Y. Helicobacter urease suppresses cytotoxic CD8+ T-cell responses through activating Myh9-dependent induction of PD-L1. Int. Immunol. 2021, 33, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Lina, T.T.; Pinchuk, I.V.; House, J.; Yamaoka, Y.; Graham, D.Y.; Beswick, E.J.; Reyes, V.E. CagA-dependent downregulation of B7-H2 expression on gastric mucosa and inhibition of Th17 responses during Helicobacter pylori infection. J. Immunol. 2013, 191, 3838–3846. [Google Scholar] [CrossRef]
- Hou, J.; Yu, Z.; Xiang, R.; Li, C.; Wang, L.; Chen, S.; Li, Q.; Chen, M.; Wang, L. Correlation between infiltration of FOXP3+ regulatory T cells and expression of B7-H1 in the tumor tissues of gastric cancer. Exp. Mol. Pathol. 2014, 96, 284–291. [Google Scholar] [CrossRef]
- Shigemori, T.; Toiyama, Y.; Okugawa, Y.; Yamamoto, A.; Yin, C.; Narumi, A.; Ichikawa, T.; Ide, S.; Shimura, T.; Fujikawa, H.; et al. Soluble PD-L1 Expression in Circulation as a Predictive Marker for Recurrence and Prognosis in Gastric Cancer: Direct Comparison of the Clinical Burden Between Tissue and Serum PD-L1 Expression. Ann. Surg. Oncol. 2019, 26, 876–883. [Google Scholar] [CrossRef]
- Böger, C.; Behrens, H.M.; Krüger, S.; Röcken, C.; Boger, C.; Behrens, H.M.; Kruger, S.; Rocken, C. The novel negative checkpoint regulator VISTA is expressed in gastric carcinoma and associated with PD-L1/PD-1: A future perspective for a combined gastric cancer therapy? Oncoimmunology 2017, 6, e1293215. [Google Scholar] [CrossRef]
- Lina, T.T.; Gonzalez, J.; Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Helicobacter pylori Elicits B7-H3 Expression on Gastric Epithelial Cells: Implications in Local T Cell Regulation and Subset Development During Infection. Clin. Oncol. Res. 2019, 2, 1–12. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes, V.E. Helicobacter pylori and Its Role in Gastric Cancer. Microorganisms 2023, 11, 1312. https://doi.org/10.3390/microorganisms11051312
Reyes VE. Helicobacter pylori and Its Role in Gastric Cancer. Microorganisms. 2023; 11(5):1312. https://doi.org/10.3390/microorganisms11051312
Chicago/Turabian StyleReyes, Victor E. 2023. "Helicobacter pylori and Its Role in Gastric Cancer" Microorganisms 11, no. 5: 1312. https://doi.org/10.3390/microorganisms11051312
APA StyleReyes, V. E. (2023). Helicobacter pylori and Its Role in Gastric Cancer. Microorganisms, 11(5), 1312. https://doi.org/10.3390/microorganisms11051312