
A Comparative Analysis of the Stomach, Gut, and Lung Microbiomes in Rattus norvegicus

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site and Sample Collection
2.2. DNA Extraction and Bacterial 16S rRNA Gene Amplification
2.3. Library Construction and Sequencing
2.4. Bioinformatics Analysis
3. Results
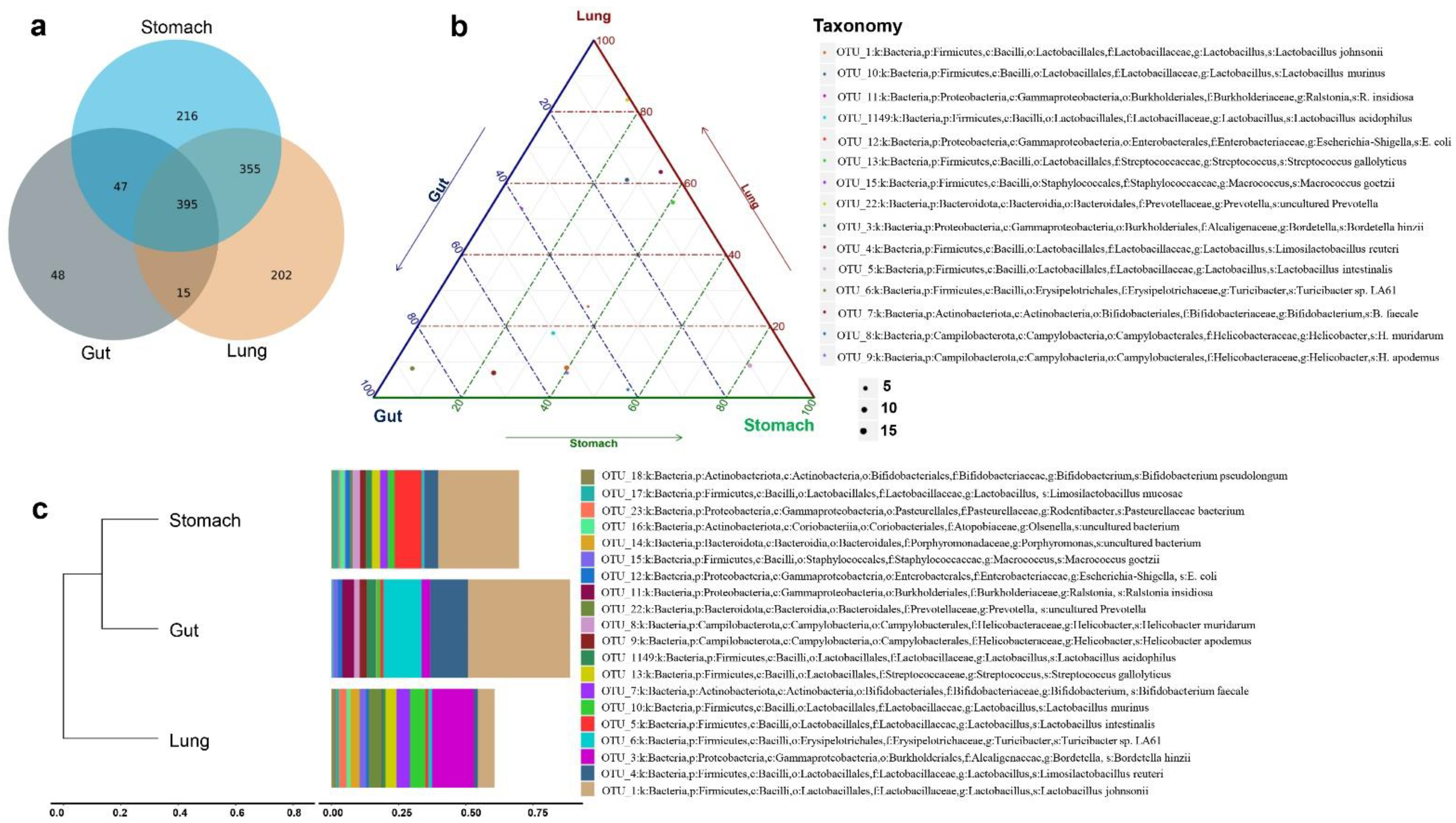
3.1. R. norvegicus Stomach, Gut, and Lung Microbial Compositions
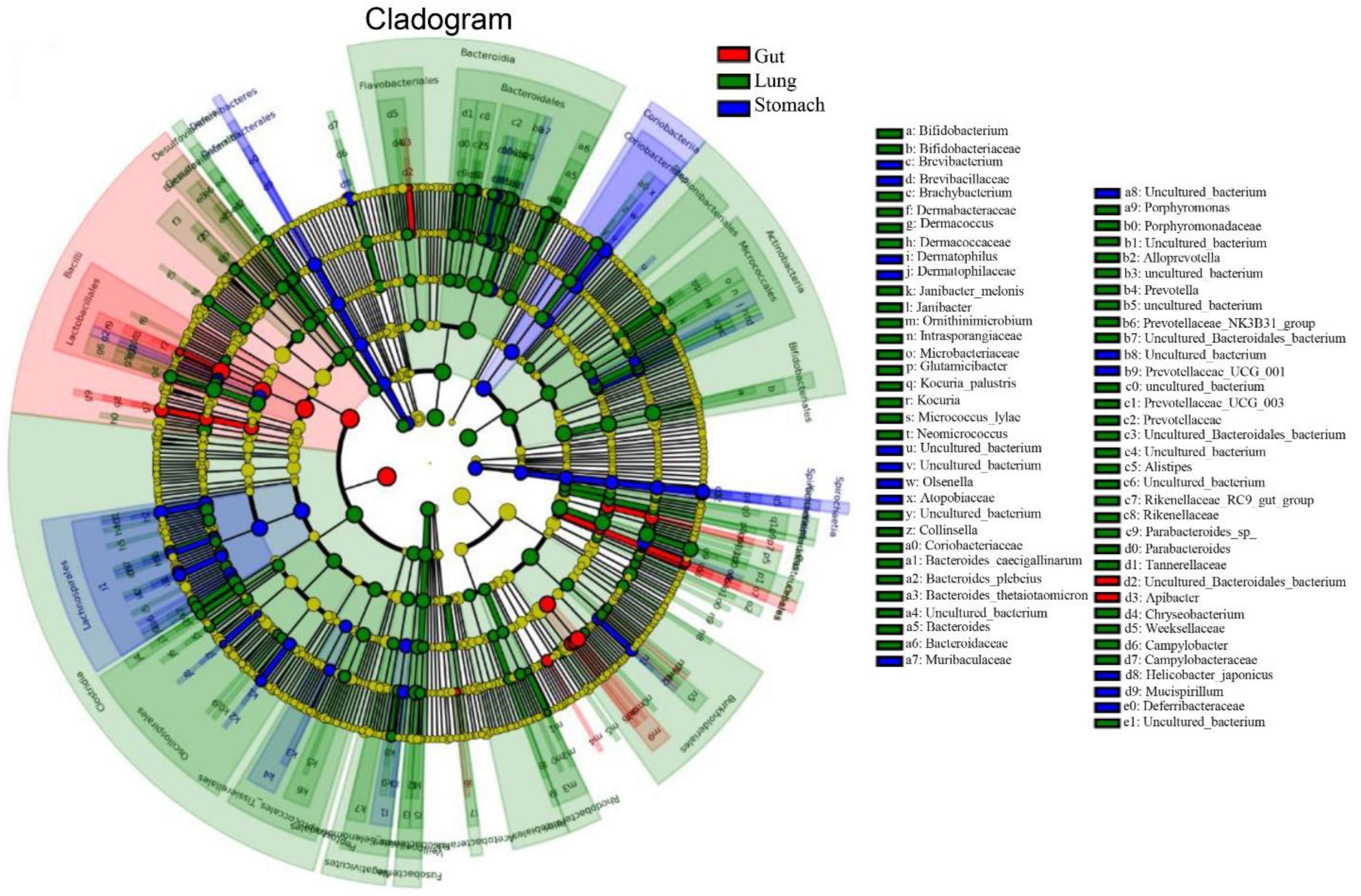
3.2. Comparing the Stomach, Gut, and Lung Microbiomes of R. norvegicus
3.3. R. norvegicus Stomach, Gut, and Lung Microbial Diversity
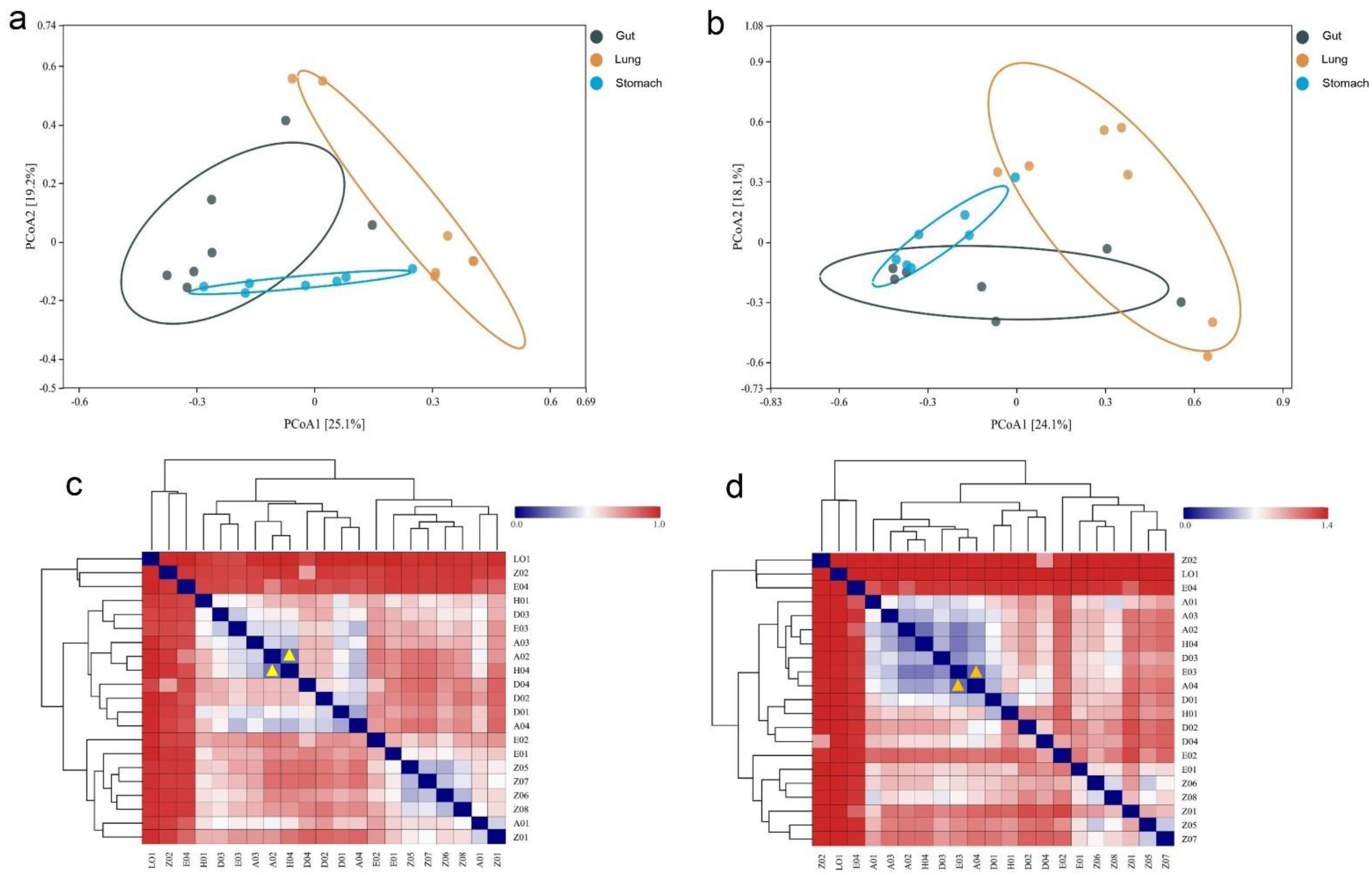
3.4. Variation of R. norvegicus Stomach, Gut, and Lung Microbial Communities
3.5. Evaluating Unique OTUs in Lungs
3.6. Evaluating Beneficial, Opportunistic, and Highly Pathogenic Bacteria at the Species Level
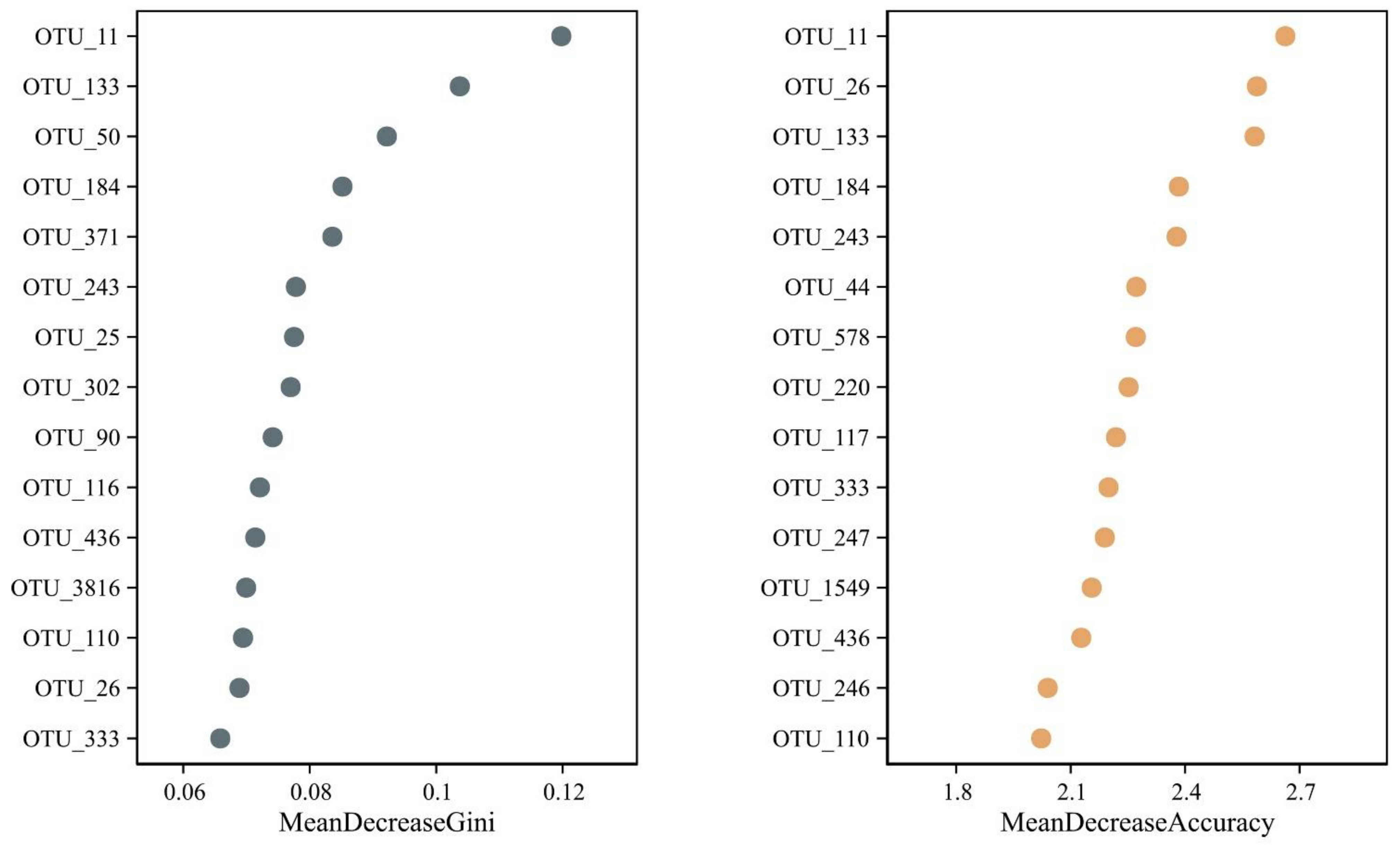
3.7. Identification of Potential Microbial OTU Biomarkers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Y.; Zhou, J.; Wang, L. Role and Mechanism of Gut Microbiota in Human Disease. Front. Cell Infect. Microbiol. 2021, 11, 625913. [Google Scholar] [CrossRef] [PubMed]
- Enaud, R.; Prevel, R.; Ciarlo, E.; Beaufils, F.; Wieërs, G.; Guery, B.; Delhaes, L. The Gut-Lung Axis in Health and Respiratory Diseases: A Place for Inter-Organ and Inter-Kingdom Crosstalks. Front. Cell Infect. Microbiol. 2020, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.A. Links between Natural Variation in the Microbiome and Host Fitness in Wild Mammals. Integr. Comp. Biol. 2017, 57, 756–769. [Google Scholar] [CrossRef]
- Pan, H.; Guo, R.; Zhu, J.; Wang, Q.; Ju, Y.; Xie, Y.; Zheng, Y.; Wang, Z.; Li, T.; Liu, Z.; et al. A gene catalogue of the Sprague-Dawley rat gut metagenome. Gigascience 2018, 7, giy055. [Google Scholar] [CrossRef]
- Niu, L.N.; Zhang, G.N.; Xuan, D.D.; Lin, C.; Lu, Z.; Cao, P.P.; Chen, S.W.; Zhang, Y.; Cui, X.J.; Hu, S.K. Comparative analysis of the gut microbiota of wild adult rats from nine district areas in Hainan, China. World J. Gastroenterol. 2023, 29, 3469–3481. [Google Scholar] [CrossRef] [PubMed]
- He, W.Q.; Xiong, Y.Q.; Ge, J.; Chen, Y.X.; Chen, X.J.; Zhong, X.S.; Ou, Z.J.; Gao, Y.H.; Cheng, M.J.; Mo, Y.; et al. Composition of gut and oropharynx bacterial communities in Rattus norvegicus and Suncus murinus in China. BMC Vet. Res. 2020, 16, 413. [Google Scholar] [CrossRef] [PubMed]
- Himsworth, C.G.; Parsons, K.L.; Jardine, C.; Patrick, D.M. Rats, cities, people, and pathogens: A systematic review and narrative synthesis of literature regarding the ecology of rat-associated zoonoses in urban centers. Vector Borne Zoonotic Dis. 2013, 13, 349–359. [Google Scholar] [CrossRef]
- Zhao, W.; Zhou, H.; Yang, L.; Ma, T.; Zhou, J.; Liu, H.; Lu, G.; Huang, H. Prevalence, genetic diversity and implications for public health of Enterocytozoon bieneusi in various rodents from Hainan Province, China. Parasit. Vectors 2020, 13, 438. [Google Scholar] [CrossRef]
- Wedgwood, S.; Warford, C.; Agvatisiri, S.R.; Thai, P.N.; Chiamvimonvat, N.; Kalanetra, K.M.; Lakshminrusimha, S.; Steinhorn, R.H.; Mills, D.A.; Underwood, M.A. The developing gut-lung axis: Postnatal growth restriction, intestinal dysbiosis, and pulmonary hypertension in a rodent model. Pediatr. Res. 2020, 87, 472–479. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Qian, G.; Jiang, W.; Zou, B.; Feng, J.; Cheng, X.; Gu, J.; Chu, T.; Niu, C.; He, R.; Chu, Y.; et al. LPS inactivation by a host lipase allows lung epithelial cell sensitization for allergic asthma. J. Exp. Med. 2018, 215, 2397–2412. [Google Scholar] [CrossRef]
- Madan, J.C.; Koestler, D.C.; Stanton, B.A.; Davidson, L.; Moulton, L.A.; Housman, M.L.; Moore, J.H.; Guill, M.F.; Morrison, H.G.; Sogin, M.L.; et al. Serial analysis of the gut and respiratory microbiome in cystic fibrosis in infancy: Interaction between intestinal and respiratory tracts and impact of nutritional exposures. mBio 2012, 3, e00251-00212. [Google Scholar]
- Xu, Q.; Ni, J.J.; Han, B.X.; Yan, S.S.; Wei, X.T.; Feng, G.J.; Zhang, H.; Zhang, L.; Li, B.; Pei, Y.F. Causal Relationship Between Gut Microbiota and Autoimmune Diseases: A Two-Sample Mendelian Randomization Study. Front. Immunol. 2021, 12, 746998. [Google Scholar] [CrossRef]
- Wah-Suárez, M.I.; Vázquez, M.A.M.; Bosques-Padilla, F.J. Inflammatory bowel disease: The role of commensal microbiome in immune regulation. Gastroenterol. Hepatol. 2022, 45, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.M.; Elfers, C.; Ghallab, A.; Schneider, C.V.; Galvez, E.J.C.; Mohs, A.; Gui, W.; Candels, L.S.; Wirtz, T.H.; Zuehlke, S.; et al. Intestinal Dysbiosis Amplifies Acetaminophen-Induced Acute Liver Injury. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 909–933. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Heikenwalder, M.; Ganal-Vonarburg, S.C. The Liver at the Nexus of Host-Microbial Interactions. Cell Host Microbe 2016, 20, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Musser, G.; Carleton, M. Mammal Species of the World: A Taxonomic and Geographic Reference; Wilson, D.E., Reeder, D.M., Eds.; Johns Hopkins University Press: Baltimore, MD, USA, 2005. [Google Scholar]
- Schmidt, E.; Mykytczuk, N.; Schulte-Hostedde, A.I. Effects of the captive and wild environment on diversity of the gut microbiome of deer mice (Peromyscus maniculatus). ISME J. 2019, 13, 1293–1305. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef]
- Yu, C.; Su, Z.; Li, Y.; Li, Y.; Liu, K.; Chu, F.; Liu, T.; Chen, R.; Ding, X. Dysbiosis of gut microbiota is associated with gastric carcinogenesis in rats. Biomed. Pharmacother. 2020, 126, 110036. [Google Scholar] [CrossRef]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef]
- Chew, S.Y.; Cheah, Y.K.; Seow, H.F.; Sandai, D.; Than, L.T. Probiotic Lactobacillus rhamnosus GR-1 and Lactobacillus reuteri RC-14 exhibit strong antifungal effects against vulvovaginal candidiasis-causing Candida glabrata isolates. J. Appl. Microbiol. 2015, 118, 1180–1190. [Google Scholar] [CrossRef]
- Wang, R.; Chen, S.; Jin, J.; Ren, F.; Li, Y.; Qiao, Z.; Wang, Y.; Zhao, L. Survival of Lactobacillus casei strain Shirota in the intestines of healthy Chinese adults. Microbiol. Immunol. 2015, 59, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, M.; Nomoto, R.; Mizuno, M.; Osawa, R. An in vitro investigation of immunomodulatory properties of Lactobacillus plantarum and L. delbrueckii cells and their extracellular polysaccharides. Biosci. Microbiota Food Health 2017, 36, 101–110. [Google Scholar] [CrossRef]
- Azad, M.A.K.; Sarker, M.; Li, T.; Yin, J. Probiotic Species in the Modulation of Gut Microbiota: An Overview. Biomed. Res. Int. 2018, 2018, 9478630. [Google Scholar] [CrossRef] [PubMed]
- Darnaud, M.; Dos Santos, A.; Gonzalez, P.; Augui, S.; Lacoste, C.; Desterke, C.; De Hertogh, G.; Valentino, E.; Braun, E.; Zheng, J.; et al. Enteric Delivery of Regenerating Family Member 3 alpha Alters the Intestinal Microbiota and Controls Inflammation in Mice With Colitis. Gastroenterology 2018, 154, 1009–1023.e14. [Google Scholar] [CrossRef]
- Boyle, R.J.; Robins-Browne, R.M.; Tang, M.L. Probiotic use in clinical practice: What are the risks? Am. J. Clin. Nutr. 2006, 83, 1256–1264, quiz 1446-7. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.P.; Lee, T.A.; Bolanos, J.T.; Danziger, L.H. Pathogenic relevance of Lactobacillus: A retrospective review of over 200 cases. Eur. J. Clin. Microbiol. Infect. Dis. 2005, 24, 31–40. [Google Scholar] [CrossRef]
- Chee, W.J.Y.; Chew, S.Y.; Than, L.T.L. Vaginal microbiota and the potential of Lactobacillus derivatives in maintaining vaginal health. Microb. Cell Fact. 2020, 19, 203. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Eun, C.S.; Kim, B.K.; Han, D.S.; Kim, S.Y.; Kim, K.M.; Choi, B.Y.; Song, K.S.; Kim, Y.S.; Kim, J.F. Differences in gastric mucosal microbiota profiling in patients with chronic gastritis, intestinal metaplasia, and gastric cancer using pyrosequencing methods. Helicobacter 2014, 19, 407–416. [Google Scholar] [CrossRef]
- Cotta, M.; Forster, R. The family Lachnospiraceae, including the genera Butyrivibrio, Lachnospira and Roseburia. Prokaryotes 2006, 4, 1002–1021. [Google Scholar]
- Anderson, M.; Sansonetti, P.J.; Marteyn, B.S. Shigella Diversity and Changing Landscape: Insights for the Twenty-First Century. Front. Cell Infect. Microbiol. 2016, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K. Strategies to promote abundance of Akkermansia muciniphila, an emerging probiotics in the gut, evidence from dietary intervention studies. J. Funct. Foods 2017, 33, 194–201. [Google Scholar] [CrossRef]
- Lennon, J.T.; Muscarella, M.E.; Placella, S.A.; Lehmkuhl, B.K. How, When, and Where Relic DNA Affects Microbial Diversity. mBio 2018, 9, e00637-18. [Google Scholar] [CrossRef]
- Sun, D.L.; Jiang, X.; Wu, Q.L.; Zhou, N.Y. Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl. Env. Microbiol. 2013, 79, 5962–5969. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | No. of OTUs | No. of Seq |
|---|---|---|
| Gut | 301 | 99,055 |
| Lung | 804 | 85,523 |
| Stomach | 701 | 99,395 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, T.; Wang, Y.; Wang, Y.; Li, Q.; Zhou, J.; Hou, Y.; Wang, B.; Xia, X. A Comparative Analysis of the Stomach, Gut, and Lung Microbiomes in Rattus norvegicus. Microorganisms 2023, 11, 2359. https://doi.org/10.3390/microorganisms11092359
Shah T, Wang Y, Wang Y, Li Q, Zhou J, Hou Y, Wang B, Xia X. A Comparative Analysis of the Stomach, Gut, and Lung Microbiomes in Rattus norvegicus. Microorganisms. 2023; 11(9):2359. https://doi.org/10.3390/microorganisms11092359
Chicago/Turabian StyleShah, Taif, Yuhan Wang, Yixuan Wang, Qian Li, Jiuxuan Zhou, Yutong Hou, Binghui Wang, and Xueshan Xia. 2023. "A Comparative Analysis of the Stomach, Gut, and Lung Microbiomes in Rattus norvegicus" Microorganisms 11, no. 9: 2359. https://doi.org/10.3390/microorganisms11092359
APA StyleShah, T., Wang, Y., Wang, Y., Li, Q., Zhou, J., Hou, Y., Wang, B., & Xia, X. (2023). A Comparative Analysis of the Stomach, Gut, and Lung Microbiomes in Rattus norvegicus. Microorganisms, 11(9), 2359. https://doi.org/10.3390/microorganisms11092359