
The Phylogeny and Metabolic Potentials of a Lignocellulosic Material-Degrading Aliiglaciecola Bacterium Isolated from Intertidal Seawater in East China Sea

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Description and Bacterial Isolation
2.2. Microbial Utilization of Carbohydrates and Peptones
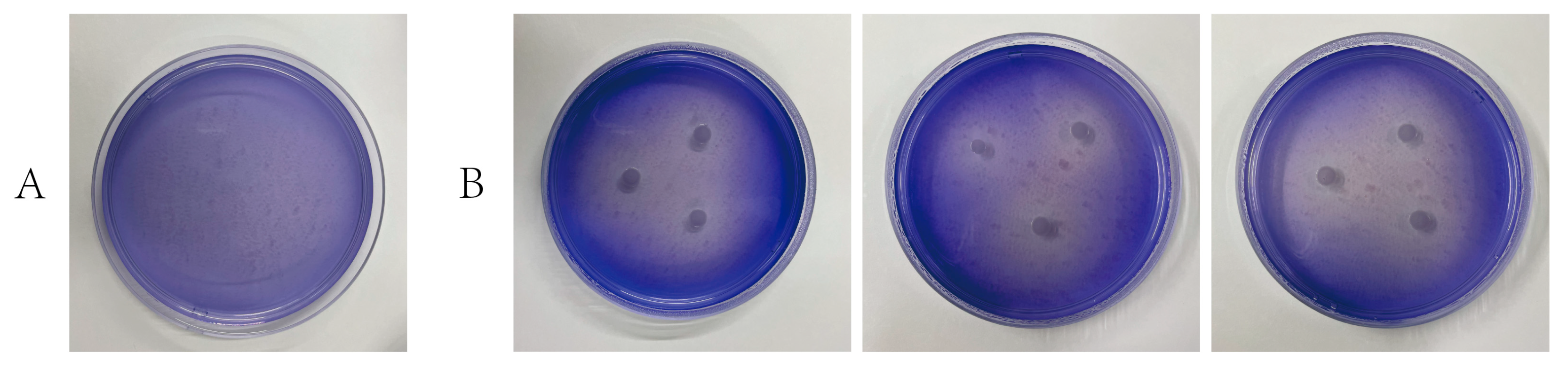
2.3. Microbial Degradation of Lignin
2.4. Genomic DNA Extraction, Sequencing and Assembly
2.5. Gene Annotation and Genomic Comparison
2.6. Phylogenetic Analysis
3. Results and Discussions
3.1. Description of Strain LCG003
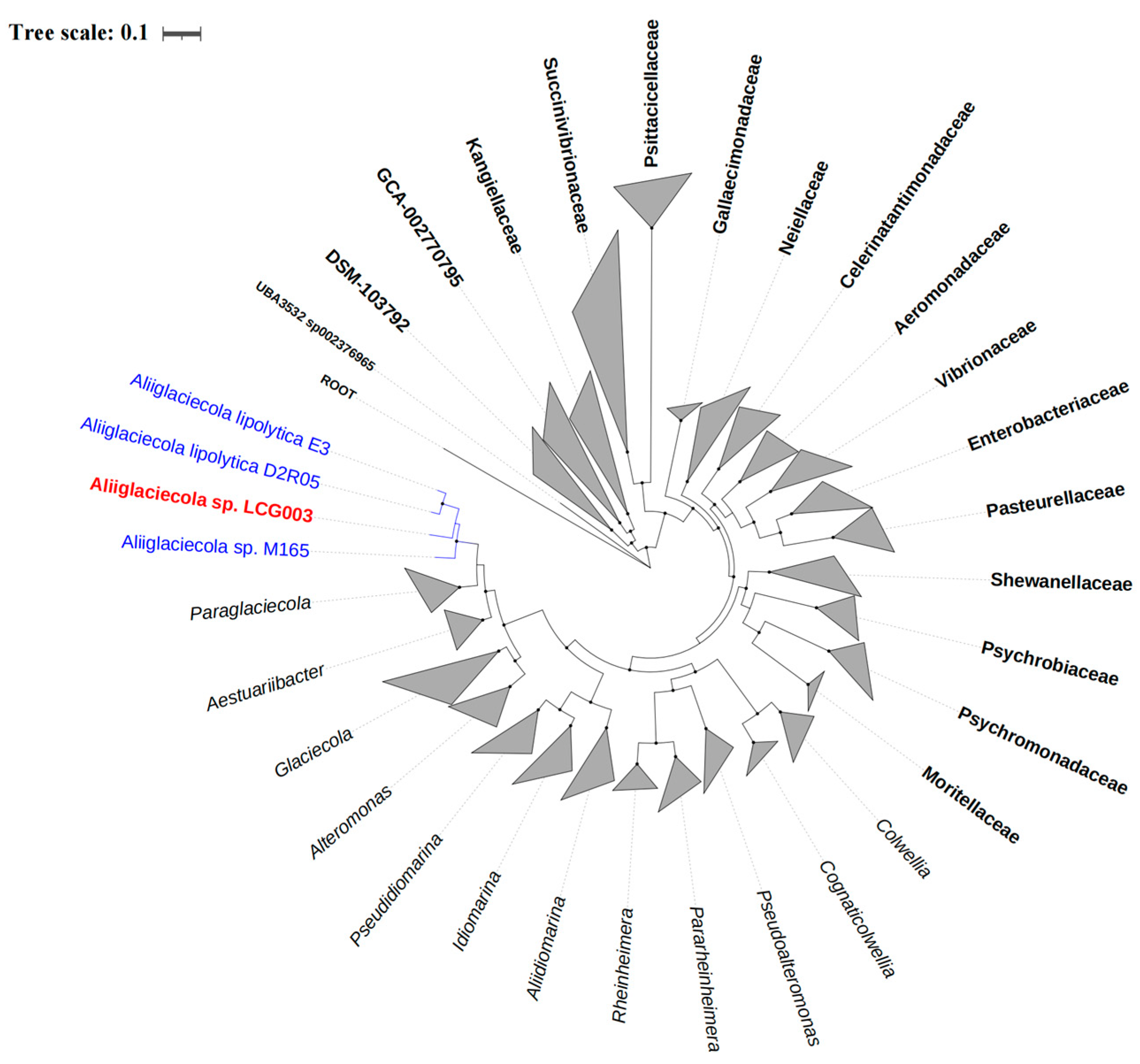
3.2. The Phylogeny of Strain LCG003
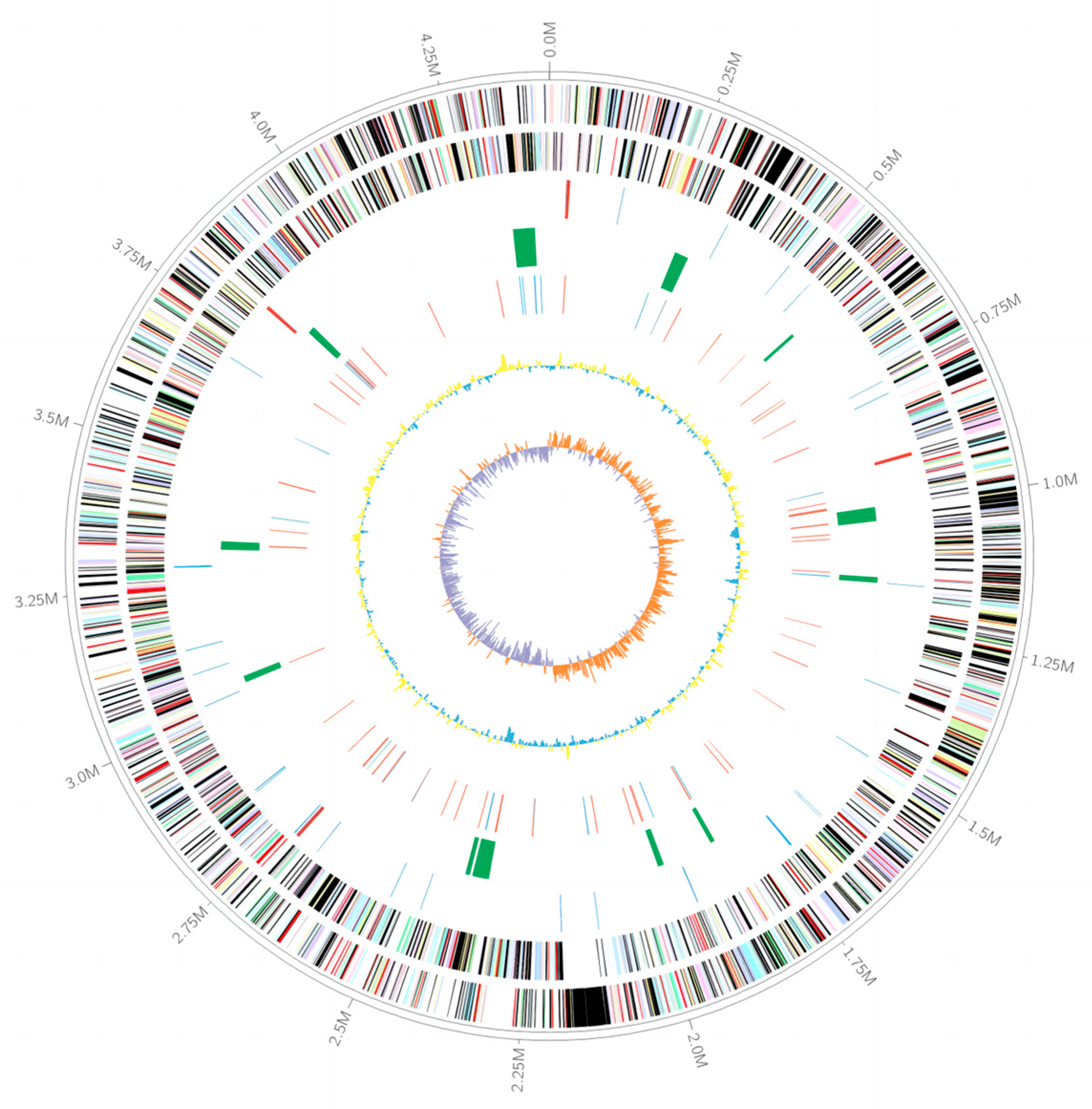
3.3. The Genomic Features of Strain LCG003
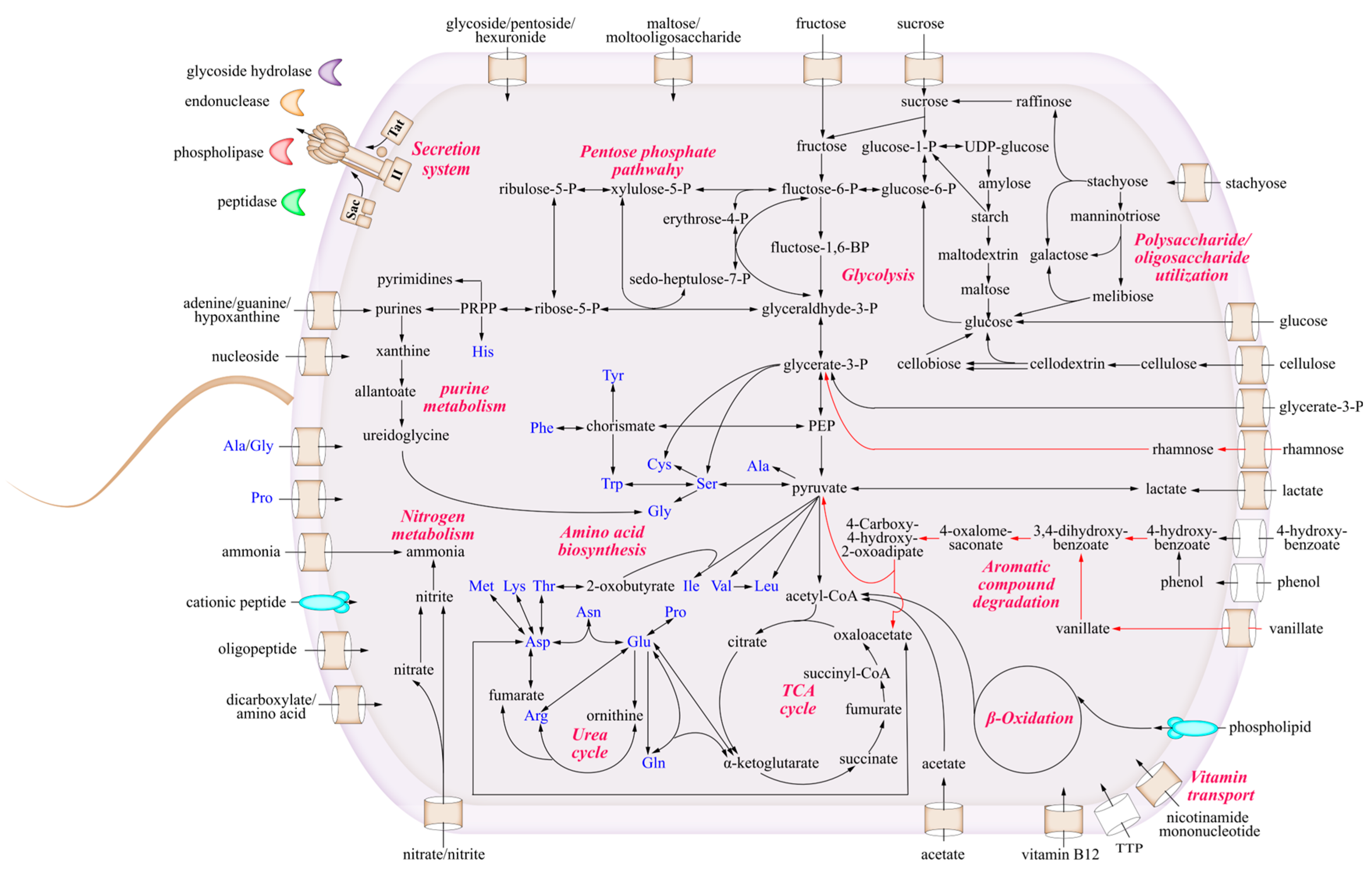
3.4. The Metabolic Characteristics of Strain LCG003
3.4.1. The Utilization of Carbohydrates and Carboxylic Acids
3.4.2. Metabolism of Amino Acids and Extracellular Proteins
3.4.3. Strain-Specific Degradation of Lignin and Aromatic Compounds
3.4.4. The Acquirement of Nitrogen and Phosphorus Sources
3.4.5. The Biosynthetic Deficiency of Thiamine and Cobalamin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Collins, M.N.; Nechifor, M.; Tanasa, F.; Zanoaga, M.; McLoughlin, A.; Strozyk, M.A.; Culebras, M.; Teaca, C.A. Valorization of lignin in polymer and composite systems for advanced engineering applications—A review. Int. J. Biol. Macromol. 2019, 131, 828–849. [Google Scholar] [CrossRef]
- Soltanian, S.; Aghbashlo, M.; Almasi, F.; Hosseinzadeh-Bandbafha, H.; Nizami, A.-S.; Ok, Y.S.; Lam, S.S.; Tabatabaei, M. A critical review of the effects of pretreatment methods on the exergetic aspects of lignocellulosic biofuels. Energy Convers. Manag. 2020, 212, 112792. [Google Scholar] [CrossRef]
- Isikgor, F.H.; Becer, C.R. Lignocellulosic biomass: A sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef]
- Cesarino, I.; Araújo, P.; Júnior, A.P.D.; Mazzafera, P. An overview of lignin metabolism and its effect on biomass recalcitrance. Braz. J. Bot. 2012, 35, 303–311. [Google Scholar] [CrossRef]
- Sahoo, B.M.; Ravi Kumar, B.V.V.; Banik, B.K.; Borah, P. Polyaromatic Hydrocarbons (PAHs): Structures, Synthesis and their Biological Profile. Curr. Org. Synth. 2020, 17, 625–640. [Google Scholar] [CrossRef]
- Dhar, K.; Subashchandrabose, S.R.; Venkateswarlu, K.; Krishnan, K.; Megharaj, M. Anaerobic Microbial Degradation of Polycyclic Aromatic Hydrocarbons: A Comprehensive Review. Rev. Environ. Contam. Toxicol. 2020, 251, 25–108. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Wang, W.; Zhang, G.; Li, W.; Jiang, A.; Cao, M.; Zhang, X.; Xing, K.; Peng, X.; Yuan, B.; et al. Isolation and characterization marine bacteria capable of degrading lignin-derived compounds. PLoS ONE 2020, 15, e0240187. [Google Scholar] [CrossRef] [PubMed]
- Bleem, A.; Kato, R.; Kellermyer, Z.A.; Katahira, R.; Miyamoto, M.; Niinuma, K.; Kamimura, N.; Masai, E.; Beckham, G.T. Multiplexed fitness profiling by RB-TnSeq elucidates pathways for lignin-related aromatic catabolism in Sphingobium sp. SYK-6. Cell Rep. 2023, 42, 112847. [Google Scholar] [CrossRef]
- Li, J.; Dong, C.; Lai, Q.; Wang, G.; Shao, Z. Frequent Occurrence and Metabolic Versatility of Marinifilaceae Bacteria as Key Players in Organic Matter Mineralization in Global Deep Seas. mSystems 2022, 7, e0086422. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Dong, J.D.; Wang, Y.S.; Gu, J.D.; Yin, J.P.; Ahmad, M.; Ling, J. Comparative genomics reveals the evidence of aromatic hydrocarbons degradation potential in genus Roseovarius in marine environment. Int. Biodeterior. Biodegrad. 2022, 171, 105408. [Google Scholar] [CrossRef]
- Raghukumar, C.; Chandramohan, D.; Michel, F.C.; Redd, C.A. Degradation of lignin and decolorization of paper mill bleach plant effluent (BPE) by marine fungi. Biotechnol. Lett. 1996, 18, 105–106. [Google Scholar] [CrossRef]
- Li, Y.Q.; Wang, M.J.; Luo, C.B. Highly efficient polyhydroxyalkanoate production from lignin using genetically engineered Halomonas sp. Y3. Bioresour. Technol. 2023, 370, 128526. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, L.; Zhou, J. Links among extracellular enzymes, lignin degradation and cell growth establish the models to identify marine lignin-utilizing bacteria. Environ. Microbiol. 2021, 23, 160–173. [Google Scholar] [CrossRef]
- Tang, H.; Wang, M.J.; Gan, X.F.; Li, Y.Q. Funneling lignin-derived compounds into polyhydroxyalkanoate by Halomonas sp. Y3. Bioresour. Technol. 2022, 362, 127837. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Yan, H.H.; Shen, X.H.; Zhang, Y.T.; Wang, Y.; Sun, S.S.; Jiang, H.Y.; Zang, H.L.; Zhao, X.Y.; Hou, N.; et al. Genome Functional Analysis of the Psychrotrophic Lignin-Degrading Bacterium Arthrobacter sp. C2 and the Role of DyP in Catalyzing Lignin Degradation. Front. Microbiol. 2022, 13, 921549. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Cheng, Y.; Zang, H.L.; Chen, X.; Wang, Y.; Zhang, Y.T.; Wang, J.M.; Shen, X.H.; Li, C.Y. Biodegradation of lignin and the associated degradation pathway by psychrotrophic Arthrobacter sp. C2 from the cold region of China. Cellulose 2020, 27, 1423–1440. [Google Scholar] [CrossRef]
- Woo, H.L.; O’Dell, K.B.; Utturkar, S.; McBride, K.R.; Huntemann, M.; Clum, A.; Pillay, M.; Palaniappan, K.; Varghese, N.; Mikhailova, N.; et al. Near-Complete Genome Sequence of Thalassospira sp. Strain KO164 Isolated from a Lignin-Enriched Marine Sediment Microcosm. Genome Announc. 2016, 4, e01297-16. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.F.; Shen, X.B.; Gang, L.P.; Xu, H.J.; Wu, F.F.; Sheng, L.Q. A novel lignin degradation bacteria-Bacillus amyloliquefaciens SL-7 used to degrade straw lignin efficiently. Bioresour. Technol. 2020, 310, 123445. [Google Scholar] [CrossRef]
- Khan, S.I.; Zarin, A.; Ahmed, S.; Hasan, F.; Belduz, A.O.; Canakci, S.; Khan, S.; Badshah, M.; Farman, M.; Shah, A.A. Degradation of lignin by Bacillus altitudinis SL7 isolated from pulp and paper mill effluent. Water Sci. Technol. 2022, 85, 420–432. [Google Scholar] [CrossRef]
- Chang, Y.C.; Choi, D.; Takamizawa, K.; Kikuchi, S. Isolation of Bacillus sp. strains capable of decomposing alkali lignin and their application in combination with lactic acid bacteria for enhancing cellulase performance. Bioresour. Technol. 2014, 152, 429–436. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, J.; Jiang, J.C.; Xu, H.; Zhang, N.; Xie, J.C.; Wei, M. Isolation and Characterization of Bacillus Sp. Capable of Degradating Alkali Lignin. Front. Energy Res. 2021, 9, 807286. [Google Scholar] [CrossRef]
- He, X.; Kim, H.; Dong, T.G.; Gates, I.; Lu, Q.Y. Green synthesis of Ag/lignin nanoparticle-loaded cellulose aerogel for catalytic degradation and antimicrobial applications. Cellulose 2022, 29, 9341–9360. [Google Scholar] [CrossRef]
- Ley, Y.; Cheng, X.Y.; Ying, Z.Y.; Zhou, N.Y.; Xu, Y. Characterization of Two Marine Lignin-Degrading Consortia and the Potential Microbial Lignin Degradation Network in Nearshore Regions. Microbiol. Spectr. 2023, 11, e0442422. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, E.P.; Mikhaĭlov, V.V. A new family of Alteromonadaceae fam. nov., including the marine proteobacteria species Alteromonas, Pseudoalteromonas, Idiomarina and Colwellia. Mikrobiologiia 2001, 70, 15–23. [Google Scholar] [PubMed]
- Jean, W.D.; Hsu, C.Y.; Huang, S.-P.; Chen, J.-S.; Lin, S.; Su, M.-H.; Shieh, W.Y. Reclassification of [Glaciecola] lipolytica and [Aestuariibacter] litoralis in Aliiglaciecola gen. nov., as Aliiglaciecola lipolytica comb. nov. and Aliiglaciecola litoralis comb. nov., respectively. Int. J. Syst. Evol. Microbiol. 2013, 63 Pt 8, 2859–2864. [Google Scholar] [CrossRef]
- Gago, J.F.; Viver, T.; Urdiain, M.; Pastor, S.; Kampfer, P.; Ferreira, E.; Rossello-Mora, R. Description of three new Alteromonas species Alteromonas antoniana sp. nov., Alteromonas lipotrueae sp. nov. and Alteromonas lipotrueiana sp. nov. isolated from marine environments, and proposal for reclassification of the genus Salinimonas as Alteromonas. Syst. Appl. Microbiol. 2021, 44, 126226. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Sharma, G.; Srinivas, T.N.; Anil Kumar, P. Aliiglaciecola coringensis sp. nov., isolated from a water sample collected from mangrove forest in Coringa, Andhra Pradesh, India. Antonie Van. Leeuwenhoek 2014, 106, 1097–1103. [Google Scholar] [CrossRef]
- Jin, H.M.; Jeong, H.I.; Jeon, C.O. Aliiglaciecola aliphaticivorans sp. nov., an aliphatic hydrocarbon-degrading bacterium, isolated from a sea-tidal flat and emended description of the genus Aliiglaciecola Jean et al. 2013. Int. J. Syst. Evol. Microbiol. 2015, 65, 1550–1555. [Google Scholar] [CrossRef]
- Fang, J.; Kato, C.; Runko, G.M.; Nogi, Y.; Hori, T.; Li, J.; Morono, Y.; Inagaki, F. Predominance of Viable Spore-Forming Piezophilic Bacteria in High-Pressure Enrichment Cultures from ~1.5 to 2.4 km-Deep Coal-Bearing Sediments below the Ocean Floor. Front. Microbiol. 2017, 8, 137. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Vera Alvarez, R.; Landsman, D.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49, D274–D281. [Google Scholar] [CrossRef] [PubMed]
- Elbourne, L.D.; Tetu, S.G.; Hassan, K.A.; Paulsen, I.T. TransportDB 2.0: A database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 2017, 45, D320–D324. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; McGinnis, S.; Madden, T.L. BLAST: Improvements for better sequence analysis. Nucleic Acids Res. 2006, 34, W6–W9. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. The Clustal Omega Multiple Alignment Package. Methods Mol. Biol. 2021, 2231, 3–16. [Google Scholar] [CrossRef]
- Tang, X.; Yu, L.; Yi, Y.; Wang, J.; Wang, S.; Meng, C.; Liu, S.; Hao, Y.; Zhang, Y.; Cao, X.; et al. Phylogenomic analysis reveals a two-stage process of the evolutionary transition of Shewanella from the upper ocean to the hadal zone. Environ. Microbiol. 2021, 23, 744–756. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Nethery, M.A.; Barrangou, R. Functional characterization of diverse type I-F CRISPR-associated transposons. Nucleic Acids Res. 2022, 50, 11670–11681. [Google Scholar] [CrossRef]
- Fülöp, V.; Jones, D.T. Beta propellers: Structural rigidity and functional diversity. Curr. Opin. Struct. Biol. 1999, 9, 715–721. [Google Scholar] [CrossRef]
- Moss, C.X.; Brown, E.; Hamilton, A.; Van der Veken, P.; Augustyns, K.; Mottram, J.C. An essential signal peptide peptidase identified in an RNAi screen of serine peptidases of Trypanosoma brucei. PLoS ONE 2015, 10, e0123241. [Google Scholar] [CrossRef]
- Eisenhaber, B.; Eisenhaber, S.; Kwang, T.Y.; Gruber, G.; Eisenhaber, F. Transamidase subunit GAA1/GPAA1 is a M28 family metallo-peptide-synthetase that catalyzes the peptide bond formation between the substrate protein’s omega-site and the GPI lipid anchor’s phosphoethanolamine. Cell Cycle 2014, 13, 1912–1917. [Google Scholar] [CrossRef]
- Wang, Y.-K.; Zhao, G.-Y.; Li, Y.; Chen, X.-L.; Xie, B.-B.; Su, H.-N.; Lv, Y.-H.; He, H.-L.; Liu, H.; Hu, J.; et al. Mechanistic Insight into the Function of the C-terminal PKD Domain of the Collagenolytic Serine Protease Deseasin MCP-01 from Deep Sea Pseudoalteromonas sp. SM9913: BINDING OF THE PKD DOMAIN TO COLLAGEN RESULTS IN COLLAGEN SWELLING BUT DOES NOT UNWIND THE COLLAGEN TRIPLE HELIX*. J. Biol. Chem. 2010, 285, 14285–14291. [Google Scholar] [CrossRef]
- Xiong, Y.I.; Zhao, Y.; Ni, K.; Shi, Y.; Xu, Q. Characterization of Ligninolytic Bacteria and Analysis of Alkali-Lignin Biodegradation Products. Pol. J. Microbiol. 2020, 69, 339–347. [Google Scholar] [CrossRef]
- Casciello, C.; Tonin, F.; Berini, F.; Fasoli, E.; Marinelli, F.; Pollegioni, L.; Rosini, E. A valuable peroxidase activity from the novel species Nonomuraea gerenzanensis growing on alkali lignin. Biotechnol. Rep. 2017, 13, 49–57. [Google Scholar] [CrossRef] [PubMed]
- de Gonzalo, G.; Colpa, D.I.; Habib, M.H.; Fraaije, M.W. Bacterial enzymes involved in lignin degradation. J. Biotechnol. 2016, 236, 110–119. [Google Scholar] [CrossRef]
- Lin, L.; Wang, X.; Cao, L.; Xu, M. Lignin catabolic pathways reveal unique characteristics of dye-decolorizing peroxidases in Pseudomonas putida. Environ. Microbiol. 2019, 21, 1847–1863. [Google Scholar] [CrossRef]
- Colpa, D.I.; Fraaije, M.W.; van Bloois, E. DyP-type peroxidases: A promising and versatile class of enzymes. J. Ind. Microbiol. Biotechnol. 2014, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Catucci, G.; Valetti, F.; Sadeghi, S.J.; Gilardi, G. Biochemical features of dye-decolorizing peroxidases: Current impact on lignin degradation. Biotechnol. Appl. Biochem. 2020, 67, 751–759. [Google Scholar] [CrossRef]
- Chen, L.P.; Xu, H.Y.; Fu, S.Z.; Fan, H.X.; Liu, Y.H.; Liu, S.J.; Liu, Z.P. Glaciecola lipolytica sp. nov., isolated from seawater near Tianjin city, China. Int. J. Syst. Evol. Microbiol. 2009, 59, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Gruber, K.; Puffer, B.; Kräutler, B. Vitamin B12-derivatives-enzyme cofactors and ligands of proteins and nucleic acids. Chem. Soc. Rev. 2011, 40, 4346–4363. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Description |
|---|---|
| Size (bp) | 4,411,932 |
| G + C content (%) | 42.5 |
| Coding sequence (%) | 87.51 |
| Total genes | 3871 |
| Protein-coding genes | 3771 |
| Genes assigned to COG | 2732 |
| rRNA operons | 4 |
| tRNA genes | 58 |
| ncRNA genes | 4 |
| Pseudogene | 26 |
| Gene islands | 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Wang, Z.; Yu, X.; Cao, J.; Bao, T.; Liu, J.; Sun, C.; Wang, J.; Fang, J. The Phylogeny and Metabolic Potentials of a Lignocellulosic Material-Degrading Aliiglaciecola Bacterium Isolated from Intertidal Seawater in East China Sea. Microorganisms 2024, 12, 144. https://doi.org/10.3390/microorganisms12010144
Zhang H, Wang Z, Yu X, Cao J, Bao T, Liu J, Sun C, Wang J, Fang J. The Phylogeny and Metabolic Potentials of a Lignocellulosic Material-Degrading Aliiglaciecola Bacterium Isolated from Intertidal Seawater in East China Sea. Microorganisms. 2024; 12(1):144. https://doi.org/10.3390/microorganisms12010144
Chicago/Turabian StyleZhang, Hongcai, Zekai Wang, Xi Yu, Junwei Cao, Tianqiang Bao, Jie Liu, Chengwen Sun, Jiahua Wang, and Jiasong Fang. 2024. "The Phylogeny and Metabolic Potentials of a Lignocellulosic Material-Degrading Aliiglaciecola Bacterium Isolated from Intertidal Seawater in East China Sea" Microorganisms 12, no. 1: 144. https://doi.org/10.3390/microorganisms12010144
APA StyleZhang, H., Wang, Z., Yu, X., Cao, J., Bao, T., Liu, J., Sun, C., Wang, J., & Fang, J. (2024). The Phylogeny and Metabolic Potentials of a Lignocellulosic Material-Degrading Aliiglaciecola Bacterium Isolated from Intertidal Seawater in East China Sea. Microorganisms, 12(1), 144. https://doi.org/10.3390/microorganisms12010144