Comparison of Rumen and Manure Microbiomes and Implications for the Inoculation of Anaerobic Digesters



Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
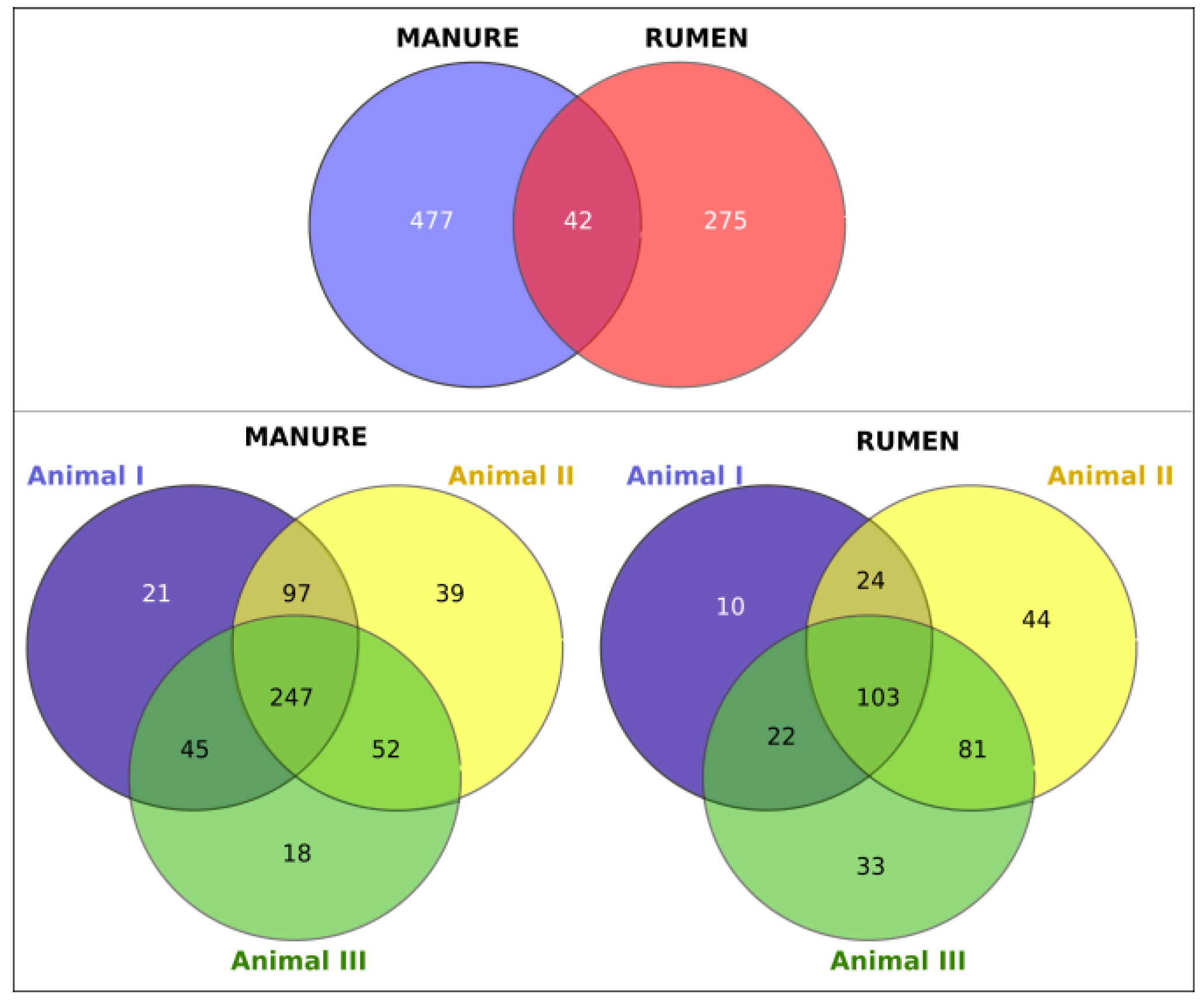
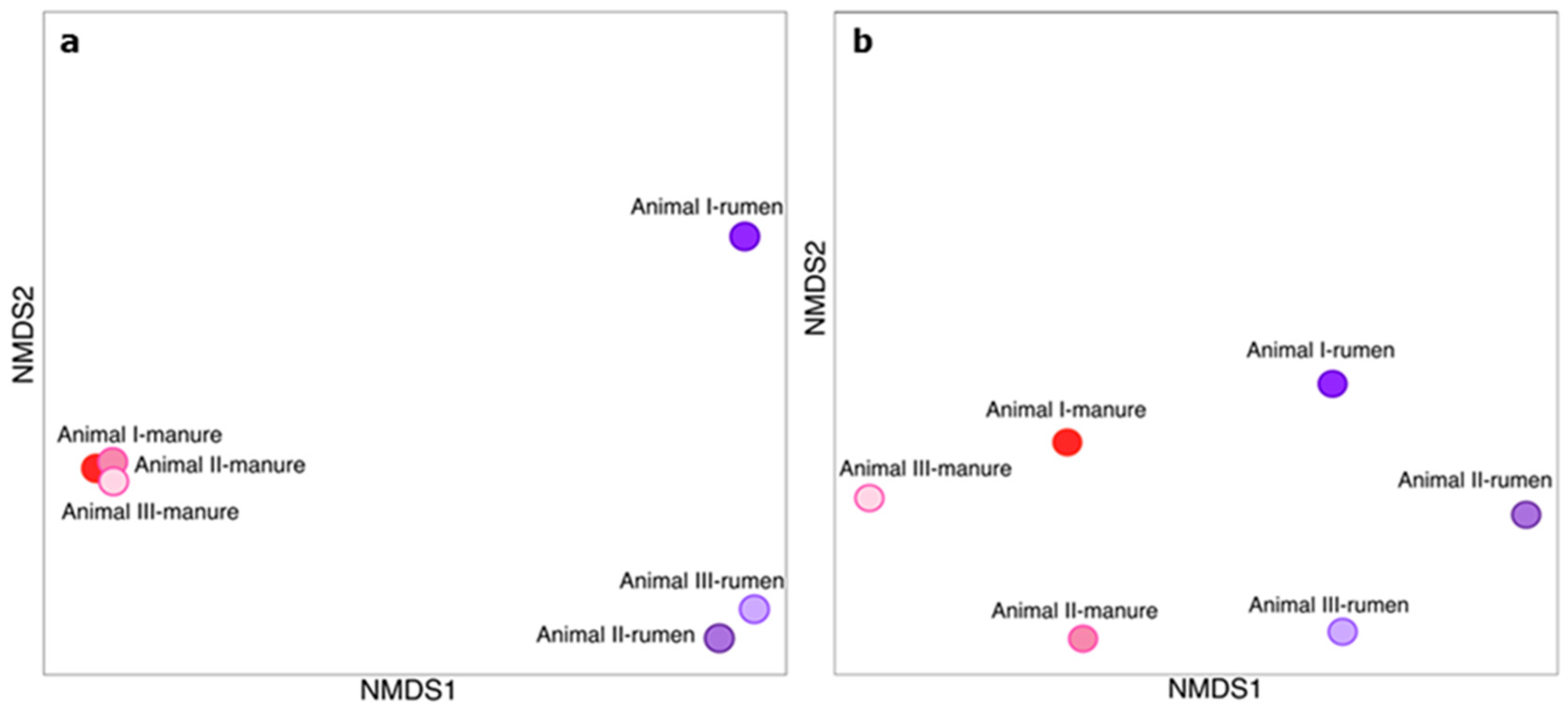
3.1. Bacterial Communities of Rumen and Manure
3.2. Methanogenic Communities of Rumen and Manure
3.3. Functional Implications of the Differences in Rumen and Manure Microbiomes
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thornton, P.K. Livestock production: Recent trends, future prospects. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2010, 365, 2853–2867. [Google Scholar] [CrossRef] [PubMed]
- Denman, S.E.; McSweeney, C.S. Development of a real-time PCR assay for monitoring anaerobic fungal and cellulolytic bacterial populations within the rumen. FEMS Microbiol. Ecol. 2006, 58, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Zened, A.; Combes, S.; Cauquil, L.; Mariette, J.; Klopp, C.; Bouchez, O.; Troegeler-Meynadier, A.; Enjalbert, F. Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets. FEMS Microbiol. Ecol. 2013, 83, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef] [PubMed]
- Khiaosa-Ard, R.; Zebeli, Q. Cattle’s variation in rumen ecology and metabolism and its contributions to feed efficiency. Livest. Sci. 2014, 162, 66–75. [Google Scholar] [CrossRef]
- Bayané, A.; Guiot, S.R. Animal digestive strategies versus anaerobic digestion bioprocesses for biogas production from lignocellulosic biomass. Rev. Environ. Sci. Biotechnol. 2011, 10, 43–62. [Google Scholar] [CrossRef]
- Hu, Z.H.; Yu, H.Q. Application of rumen microorganisms for enhanced anaerobic fermentation of corn stover. Process Biochem. 2005, 40, 2371–2377. [Google Scholar] [CrossRef]
- Alrawi, R.A.; Ahmad, A.; Ismail, N.; Kadir, M.O.A. Anaerobic co-digestion of palm oil mill effluent with rumen fluid as a co-substrate. Desalination 2011, 269, 50–57. [Google Scholar] [CrossRef]
- Jin, W.; Cheng, Y.F.; Mao, S.Y.; Zhu, W.Y. Discovery of a novel rumen methanogen in the anaerobic fungal culture and its distribution in the rumen as revealed by real-time PCR. BMC Microbiol. 2014, 14, 104. [Google Scholar] [CrossRef] [PubMed]
- Angelidaki, I.; Ellegaard, L. Codigestion of manure and organic wastes in centralized biogas plants: Status and future trends. Appl. Biochem. Biotechnol. 2003, 109, 95–105. [Google Scholar] [CrossRef]
- Janke, L.; Leite, A.F.; Nikolausz, M.; Radetski, C.M.; Nelles, M.; Stinner, W. Comparison of start-up strategies and process performance during semi-continuous anaerobic digestion of sugarcane filter cake co-digested with bagasse. Waste Manag. 2016, 48, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Goberna, M.; Gadermaier, M.; Franke-Whittle, I.H.; García, C.; Wett, B.; Insam, H. Start-up strategies in manure-fed biogas reactors: Process parameters and methanogenic communities. Biomass Bioenergy 2015, 75, 46–56. [Google Scholar] [CrossRef]
- Kong, Y.; Teather, R.; Forster, R. Composition, spatial distribution, and diversity of the bacterial communities in the rumen of cows fed different forages. FEMS Microbiol. Ecol. 2010, 74, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Kittelmann, S.; Janssen, P.H. Characterization of rumen ciliate community composition in domestic sheep, deer, and cattle, feeding on varying diets, by means of PCR-DGGE and clone libraries. FEMS Microbiol. Ecol. 2011, 75, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Morrison, M.; Yu, Z. Status of the phylogenetic diversity census of ruminal microbiomes. FEMS Microbiol. Ecol. 2011, 76, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Dowd, S.E.; Callaway, T.R.; Wolcott, R.D.; Sun, Y.; McKeehan, T.; Hagevoort, R.G.; Edrington, T.S. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 2008, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Durso, L.M.; Harhay, G.P.; Smith, T.P.L.; Bono, J.L.; Desantis, T.Z.; Harhay, D.M.; Gary, L.; Keen, J.E.; Laegreid, W.W.; Clawson, L.; et al. Animal-to-animal variation in fecal microbial diversity among beef cattle. 2010, 76, 4858–4862. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Baldwin, R.L.; Li, W.; Li, C.; Connor, E.E.; Li, R.W. The bacterial community composition of the bovine rumen detected using pyrosequencing of 16S rRNA genes. Metagenomics 2012, 1, 1–11. [Google Scholar] [CrossRef]
- Pitta, D.W.; Kumar, S.; Veiccharelli, B.; Parmar, N.; Reddy, B.; Joshi, C.G. Bacterial diversity associated with feeding dry forage at different dietary concentrations in the rumen contents of Mehshana buffalo (Bubalus bubalis) using 16S pyrotags. Anaerobe 2014, 25, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Zhang, M.L.; Zhang, R.Y.; Zhu, W.Y.; Mao, S.Y. Comparative studies of the composition of bacterial microbiota associated with the ruminal content, ruminal epithelium and in the faeces of lactating dairy cows. Microb. Biotechnol. 2016, 9, 257–268. [Google Scholar] [CrossRef] [PubMed]
- De Menezes, A.B.; Lewis, E.; O’Donovan, M.; O’Neill, B.F.; Clipson, N.; Doyle, E.M. Microbiome analysis of dairy cows fed pasture or total mixed ration diets. FEMS Microbiol. Ecol. 2011, 78, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Ziganshin, A.M.; Liebetrau, J.; Pröter, J.; Kleinsteuber, S. Microbial community structure and dynamics during anaerobic digestion of various agricultural waste materials. Appl. Microbiol. Biotechnol. 2013, 97, 5161–5174. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, L.M.; Regan, J.M. Phylogenetic comparison of the methanogenic communities from an acidic, oligotrophic fen and an anaerobic digester treating municipal wastewater sludge. Appl. Environ. Microbiol. 2008, 74, 6663–6671. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Kuchenbuch, A.; Fetzer, I.; Harms, H.; Kleinsteuber, S. Long-term monitoring reveals stable and remarkably similar microbial communities in parallel full-scale biogas reactors digesting energy crops. FEMS Microbiol. Ecol. 2015, 91, fiv004. [Google Scholar] [CrossRef] [PubMed]
- Popp, D.; Plugge, C.M.; Kleinsteuber, S.; Harms, H.; Sträuber, H. Inhibitory effect of coumarin on syntrophic fatty acid-oxidizing and methanogenic cultures and biogas reactor microbiomes. Appl. Environ. Microbiol. 2017, 83, e00438-17. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. 2007–2015. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 9 January 2018).
- Shanks, O.C.; Kelty, C.A.; Archibeque, S.; Jenkins, M.; Newton, R.J.; McLellan, S.L.; Huse, S.M.; Sogin, M.L. Community structures of fecal bacteria in cattle from different animal feeding operations. Appl. Environ. Microbiol. 2011, 77, 2992–3001. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Zhang, R.; Wang, D.; Zhu, W. The diversity of the fecal bacterial community and its relationship with the concentration of volatile fatty acids in the feces during subacute rumen acidosis in dairy cows. BMC Vet. Res. 2012, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Pope, P.B.; Eijsink, V.G.H.; Schnürer, A. Characterization of microbial community structure during continuous anaerobic digestion of straw and cow manure. Microb. Biotechnol. 2015, 8, 815–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyonyo, T.; Shinkai, T.; Mitsumori, M. Improved culturability of cellulolytic rumen bacteria and phylogenetic diversity of culturable cellulolytic and xylanolytic bacteria newly isolated from the bovine rumen. FEMS Microbiol. Ecol. 2014, 88, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Naas, A.E.; Mackenzie, A.K.; Mravec, J.; Schückel, J.; Willats, W.G.T.; Eijsink, V.G.H.; Pope, P.B. Do rumen Bacteroidetes utilize an alternative mechanism for cellulose degradation? MBio 2014, 5, e01401-14. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hatem, A.; Catalyurek, U.V.; Morrison, M.; Yu, Z. Metagenomic insights into the carbohydrate-active enzymes carried by the microorganisms adhering to solid digesta in the rumen of cows. PLoS ONE 2013, 8, e78507. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.W.; Saddler, J.N.; Patel, G.B.; Colvin, J.R.; Martin, S.M. Degradation of cellulose by a newly isolated mesophilic anaerobe, Bacteroidaceae family. FEMS Microbiol. Lett. 1980, 7, 47–50. [Google Scholar] [CrossRef]
- Ransom-Jones, E.; Jones, D.L.; McCarthy, A.J.; McDonald, J.E. The Fibrobacteres: An important phylum of cellulose-degrading bacteria. Microb. Ecol. 2012, 63, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Suen, G.; Weimer, P.J.; Stevenson, D.M.; Aylward, F.O.; Boyum, J.; Deneke, J.; Drinkwater, C.; Ivanova, N.N.; Mikhailova, N.; Chertkov, O.; et al. The complete genome sequence of Fibrobacter succinogenes s85 reveals a cellulolytic and metabolic specialist. PLoS ONE 2011, 6, e18814. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Groeneveld, J.; Harms, H.; Johst, K.; Frank, K.; Kleinsteuber, S. A critical evaluation of ecological indices for the comparative analysis of microbial communities based on molecular datasets. FEMS Microbiol. Ecol. 2017, 93, fiw209. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, C.; Mackie, R. Micro-Organisms and Ruminant Digestion: State of knowledge, Trends and Future Prospects. Commission on Genetic Resources for Food and Agriculture. Background Study Paper No. 61. 2012. Available online: http://www.fao.org/docrep/016/me992e/me992e.pdf (accessed on 9 January 2018).
- King, E.E.; Smith, R.P.; St-Pierre, B.; Wright, A.D.G. Differences in the rumen methanogen populations of lactating Jersey and holstein dairy cows under the same diet regimen. Appl. Environ. Microbiol. 2011, 77, 5682–5687. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Schuldes, J.; Klingl, A.; Poehlein, A.; Daniel, R.; Brune, A. New mode of energy metabolism in the seventh order of methanogens as revealed by comparative genome analysis of “Candidatus Methanoplasma termitum”. Appl. Environ. Microbiol. 2015, 81, 1338–1352. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.G.; Smithard, R.R. The fate of carbohydrates in the small and large intestines of the ruminant. Proc. Nutr. Soc. 1979, 38, 283–294. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.N.V.; Jewell, K.A.; Freitas, F.S.; Benjamin, L.A.; Tótola, M.R.; Borges, A.C.; Moraes, C.A.; Suen, G. Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer. Vet. Microbiol. 2013, 164, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Myer, P.R.; Wells, J.E.; Smith, T.P.L.; Kuehn, L.A.; Freetly, H.C. Microbial community profiles of the colon from steers differing in feed efficiency. SpringerPlus 2015, 4, 454. [Google Scholar] [CrossRef] [PubMed]
- Hook, S.E.; Wright, A.D.G.; McBride, B.W. Methanogens: Methane producers of the rumen and mitigation strategies. Archaea 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhang, G.; Li, J.; Zhao, Z.; Kang, X. Effect of endogenous hydrolytic enzymes pretreatment on the anaerobic digestion of sludge. Bioresour. Technol. 2013, 146, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.B.; Li, W.W.; Yu, H.Q. Application of rumen microorganisms for anaerobic bioconversion of lignocellulosic biomass. Bioresour. Technol. 2013, 128, 738–744. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | No. of Reads | No. of OTUs | Chao1 | Shannon | Simpson | Pielou’s Evenness |
|---|---|---|---|---|---|---|
| Rumen bacteria | 6998 | 217 | 293 | 5.26 | 0.99 | 0.92 |
| Manure bacteria | 12,232 | 402 | 513 | 5.89 | 0.99 | 0.94 |
| Rumen methanogens | 9906 | 190 | 292 | 4.09 | 0.97 | 0.76 |
| Manure methanogens | 7915 | 91 | 161 | 2.39 | 0.78 | 0.51 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozbayram, E.G.; Ince, O.; Ince, B.; Harms, H.; Kleinsteuber, S. Comparison of Rumen and Manure Microbiomes and Implications for the Inoculation of Anaerobic Digesters. Microorganisms 2018, 6, 15. https://doi.org/10.3390/microorganisms6010015
Ozbayram EG, Ince O, Ince B, Harms H, Kleinsteuber S. Comparison of Rumen and Manure Microbiomes and Implications for the Inoculation of Anaerobic Digesters. Microorganisms. 2018; 6(1):15. https://doi.org/10.3390/microorganisms6010015
Chicago/Turabian StyleOzbayram, Emine Gozde, Orhan Ince, Bahar Ince, Hauke Harms, and Sabine Kleinsteuber. 2018. "Comparison of Rumen and Manure Microbiomes and Implications for the Inoculation of Anaerobic Digesters" Microorganisms 6, no. 1: 15. https://doi.org/10.3390/microorganisms6010015
APA StyleOzbayram, E. G., Ince, O., Ince, B., Harms, H., & Kleinsteuber, S. (2018). Comparison of Rumen and Manure Microbiomes and Implications for the Inoculation of Anaerobic Digesters. Microorganisms, 6(1), 15. https://doi.org/10.3390/microorganisms6010015