1. Introduction
Ever since the first methanotroph was described a century ago by Söhngen [
1], it has been known that methanotrophs are actively involved in a global cycle of methane [
2]. Aerobic methanotrophs inhabit diverse ecosystems where an anoxic/oxic interfaces exist, and oxidize anaerobically produced methane using O
2 as an electron acceptor. The enzyme responsible for the initial step of methane oxidation is methane monooxygenase (MMO), which exists in two evolutionarily unrelated forms: a soluble, cytoplasmic form (sMMO), and a particulate, membrane-bound form (pMMO). Phylogenetically, known aerobic methanotrophs belong to the proteobacterial classes
Alphaproteobacteria and
Gammaproteobacteria [
3], the phylum
Verrucomicrobia [
4,
5,
6,
7,
8,
9] and the candidate phylum NC10 [
10]. The
Alphaproteobacteria methanotrophs are further divided into two families,
Methylocystaceae and
Beijerinckiaceae [
11]. The
Beijerinckiaceae family encompasses species with diverse phenotypes, including versatile chemoorganotrophs, phototrophs, obligate methanotrophs, facultative methylotrophs and methanotrophs [
12]. The methanotrophs in this family include the only two genera known to use the sMMO enzyme exclusively to activate methane:
Methylocella and
Methyloferula [
13,
14]. All other methanotrophs possess just pMMO, or both pMMO and sMMO.
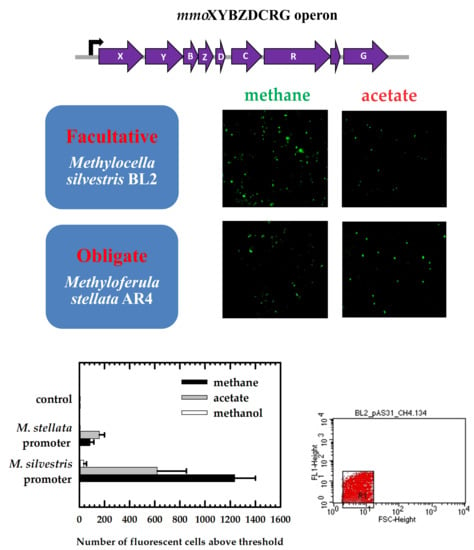
Methyloferula stellata AR4 is an obligate methanotroph, capable of growing on the C1 compounds methane or methanol as sole substrates [
14,
15]. In contrast,
Methylocella silvestris BL2 was the first documented facultative methanotroph able to grow on multicarbon compounds in addition to methane. In terms of growth substrates, it is by far the most versatile methanotroph yet discovered [
16], growing on C1 compounds (methane, methanol, formate, and methylamine) as well as organic acids (acetate, pyruvate, succinate, malate, gluconate, and propionate), alcohols (ethanol, 2-propanol, 1,2-propanediol, glycerol), short chain alkanes (ethane, propane), acetone, and methyl acetate [
17,
18,
19]. Facultative methanotrophy, the ability to grow on substrates besides methane and related C1 compounds, has now been demonstrated in a few other (pMMO-using) methanotrophs [
20,
21,
22,
23]. However, the range of growth substrates for these other facultative methanotrophs is much narrower than for
Methylocella, generally limited to acetate, ethanol, and H
2.
In
Methylocella silvestris BL2, the sMMO is repressed at the transcriptional level in the presence of alternative substrates like acetate [
17,
19]. In contrast, other facultative methanotrophs like
Methylocapsa aurea and
Methylocystis sp. H2 grow more efficiently on methane rather than acetate and/or ethanol. These other methanotrophs utilize a pMMO to convert methane to methanol and possess a well-developed intracytoplasmic membrane (ICM) in which pMMO is bound. Interestingly,
Methylocystis strain H2, which has functional genes for both sMMO and pMMO, was shown to express only pMMO regardless of tested growth conditions. Moreover, pMMO was expressed constitutively in the facultative
Methylocystis strains H2 and SB2, even in the presence of alternative substrates [
21].
Methylocella and
Methyloferula lack pMMO and extensive ICM.
The obligate methanotroph
Methyloferula stellata AR4 is closely related to
Methylocella silvestris BL2 (97.1% identity of 16S ribosomal RNA (rRNA) genes). A recently published draft genome of
Methyloferula stellata AR4 revealed little difference in functional genes involved in methane metabolism compared to
Methylocella silvestris BL2 [
14]. Both use only sMMO to convert methane to methanol, plus similar pathways for further processing of methanol. However,
Methyloferula stellata AR4 is unable to grow on any of the alternative, non-C1 substrates that
Methylocella silvestris BL2 uses. Therefore, we compared transcriptional activities of the
mmo promoters in the two organisms. Transcriptional fusions of the promoters to a promoterless reporter gene,
gfp, and their responses to growth on different substrates were tested in the genetic backgrounds of both organisms.
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
Methylocella silvestris BL2 and
Methyloferula stellata AR4 were cultivated in DAMS (pH 5.8) and MM2 (pH 4.8–5.2) media, respectively, as described previously [
13,
15].
M. silvestris BL2 was maintained on DAMS agar plates, whereas
M. stellata AR4 was maintained on MM2 plates containing Phytagel as a solidifying agent. Plates were incubated at 25 °C in an anaerobic jar (Oxoid, Nepean, ON, Canada) containing 20% (
v/
v) methane in air. Routinely, 30 mL of liquid cultures were grown in serum bottles (120 mL) sealed with butyl rubber stoppers and supplemented with either 20% (
v/
v) methane in the headspace or 0.5% (
v/
v) methanol in a medium, and incubated at 25 °C on a shaker at 120 rpm.
M. silvestris BL2 cultures were also cultivated in 30 mL of DAMS (Diluted ammonium mineral salts) medium containing 5 mM sodium acetate as a growth substrate. For growth and methane consumption experiments, cells were cultivated in 250 mL or 1 L bottles sealed with GL45 chlorbutyl septa (Glasgerätebau Ochs, Bovenden, Niedersachsen, Germany) and open-top caps (VWR, Edmonton, AB, Canada). Growth of cells was monitored via optical density at 600 nm using an Ultrospec spectrophotometer (GE Healthcare Life Sciences, Mississauga, ON, Canada). A decrease of methane in the headspace of growth vials was quantified using an SRI 8610C Gas chromatograph equipped with a HayeSep-D column coupled to a flame ionization detector (FID) (column T 100 °C; detector T 300 °C; N
2 as carrier gas). A certified 0.5% (
v/
v) methane in air mixture was used as a standard (Praxair, Danbury, CT, USA).
E.coli DH5α strain was used to propagate plasmids for cloning in LB medium. Kanamycin (20 mg/L) and ampicillin (100 mg/L) were used to maintain plasmids in E. coli DH5α, M. silvestris BL2, and M. stellata AR4.
2.2. DNA Manipulations, Plasmid Construction
FastDNA Spin Kit (MP Biomedicals, Solon, OH, USA) and EZ-10 Spin Column Plasmid Prep kits (BioBasic Inc., Markham, ON, Canada) were used to isolate genomic and plasmid DNA, respectively, according to the manufacturer’s instructions. Polymerase chain reaction (PCR) primers were designed using the genomes of
M. silvestris BL2 and
M. stellata AR4 (Accession NC_011666 and NZ_ARWA00000000, respectively) and Vector NTI software (ThermoFischer Scientific, Waltham, MA, USA). Primers were synthesized by ThermoFischer Scientific (USA). The primers sMMO_EcoRI_f, 5′-ACTGAATTCAGCCCGTTGTCGCTTTGATA-3′, and sMMO_SacI_rev, 5-ACTGAGCTCATGTCTCCTCCTTGGTGCTC-3′, were used to amplify a 1080 bp fragment containing a promoter region of sMMO upstream of the
mmoX gene of
M. silvestris BL2. The primers Pmmo AR4_EcoRI_F, 5′-ACTGAATTCGCGAAATACGATACGCCGAC-3′ and Pmmo AR4_SacI_R, 5′-ACTGAGCTCTCATCGCTCTGGTGCTTTGA-3′ were used to amplify an 889 bp fragment containing a promoter region of sMMO upstream of the
mmoX gene of
M. stellata AR4. PCRs were performed with Phusion proofreading DNA polymerase (Bioline, Taunton, MA, USA) under conditions recommended by the manufacturer. A two-step PCR procedure was used for cycling. Initially, DNA was denaturated at 98 °C for 3 min, followed by 35 amplification cycles (denaturation at 98 °C for 20 s, annealing and extension at 72 °C for 40 s), plus a final extension at 72 °C for 7 min. PCR products were purified using an EZ-10 Spin Column PCR Products Purification Kit (BioBasic Inc., Canada), digested with
EcoRI and
SacI (ThermoFischer Scientific, USA) and ligated into pMHA200 [
19]. Promoter fusions to
gfp in the resulting plasmids, pAS31 and pAS50, were confirmed by Sanger sequencing (Eurofins MWG Operon, Louisville, KY, USA) with forward and reverse primers: seq gfp 5′-AACAATTTCACACAGGAAAC-3′, seq TF_gfpBL2 5′-ACAACAGAGCTGCCGCACTG-3′, seq TF_gfpAR4 5′-ACGACTATCGCCCCAATTTG-3′.
2.3. Preparation of Competent Cells and Electroporation into M. silvestris BL2 and M. stellata AR4
Cells were grown in batch culture. A stock culture was added to either 500 mL DAMS medium for M. silvestris or 500 mL of MM2 medium for M. stellata in 1 L bottles. Methane (20% v/v) was added to the headspaces. Altogether, six 1 L bottles containing bacterial inoculum were incubated at 25 °C, shaken at 120 rpm, for either 9 days (M. silvestris BL2) or 21 days (M. stellata AR4). Exponentially growing cells were cooled on ice for 15 min, harvested by centrifugation (6000× g for 15 min at 4 °C), washed twice in ice-cold water (4 °C), and finally resuspended in 10 mL of 10% glycerol (m/v). Finally, 300–400 µL of suspended cells were aliquoted in 1.5 mL centrifuge tubes, frozen in liquid nitrogen, and stored at −80 °C until used. For electroporation, 200 µL of competent cell suspension was gently mixed with 400–500 ng of plasmid DNA and transferred to a 2-mm cooled electroporation cuvette (Eppendorf Canada, Mississauga, ON, Canada). Electroporation was performed at 2.2 kV for 4–5 ms, using an Eppendorf Eporator (Eppendorf Canada, Mississauga, ON, Canada). Cells were washed from the cuvette with either 1 mL of DAMS containing 0.5% ethanol for M. silvestris or 1 mL of MM2 containing 0.5% methanol for M. stellata. Cells were allowed to recover overnight at 25 °C with shaking at 120 rpm. Cells were pelleted by centrifugation (1500× g, 8 min) at room temperature and finally resuspended in 250 µL of DAMS or MM2 medium before plating out either on DAMS-agar plates for M. silvestris or on MM2-phytagel plates for M. stellata, with kanamycin as an antibiotic for positive selection. After incubation for three weeks in an atmosphere containing methane, kanamycin-resistant colonies appeared. A few colonies were transferred twice onto new plates with kanamycin and cells were then were transferred to liquid medium containing kanamycin, supplied with 20% (v/v) methane in the headspace. If transformants could not grow on liquid medium with kanamycin, they were discarded. After a few passages in liquid medium with kanamycin, transformants were subjected to microscopy studies to analyze their fluorescence. Genomic DNA was isolated from one transformant for each transcriptional fusion in the respective host. A 1.2-kb fragment for the M. silvestris promoter and a 1-kb fragment for the M. stellata promoter were amplified from genomic DNAs using the sequencing primers seq gfp, seq TF_gfpBL2, seq TF_gfpAR4 (see above), to prove the presence of transcriptional fusions in the respective host.
2.4. Fluorescence Microscopic Detection of GFP-Labelled Cells
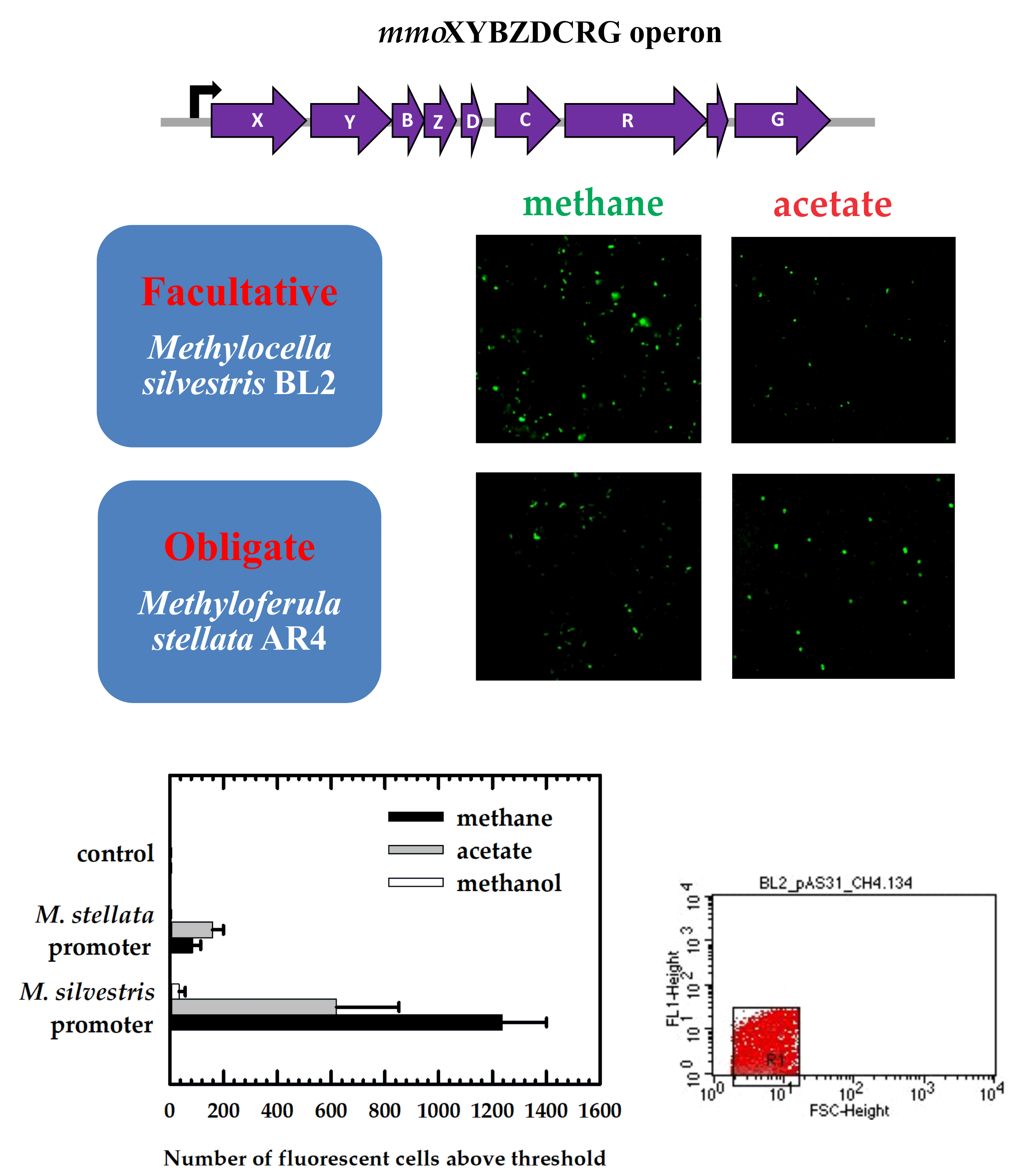
To assess promoter activities, we transferred colonies of the positive transformants into liquid media with methane (20% v/v), methanol (0.5% v/v), or acetate (5 mM) as the sole energy source. Six ml of bacterial cultures after 3–4 weeks of growth were harvested by centrifugation, resuspended in 25 µL of PBS buffer, and 10 µL of the cell suspension was added to a cover slip. A drop of Prolong Diamond Antifade Mountant (ThermoFischer Scientific, USA) was added to a glass slide. Subsequently, the cover slip was placed sample-side down onto the mountant on the glass slide. To cure, the glass slide was incubated at room temperature for 24 hours. Fluorescence of single cells was detected using an Olympus BX51 fluorescence microscope (Olympus Canada Inc., Richmond Hill, ON, Canada) with an excitation filter at 460–490 nm and an emission filter at wavelengths >520 nm, using a 100× oil immersion objective. Image-Pro Express (version 6.0) software was used to view images.
2.5. Flow Cytometry Analysis of Promoter Fusion to gfp
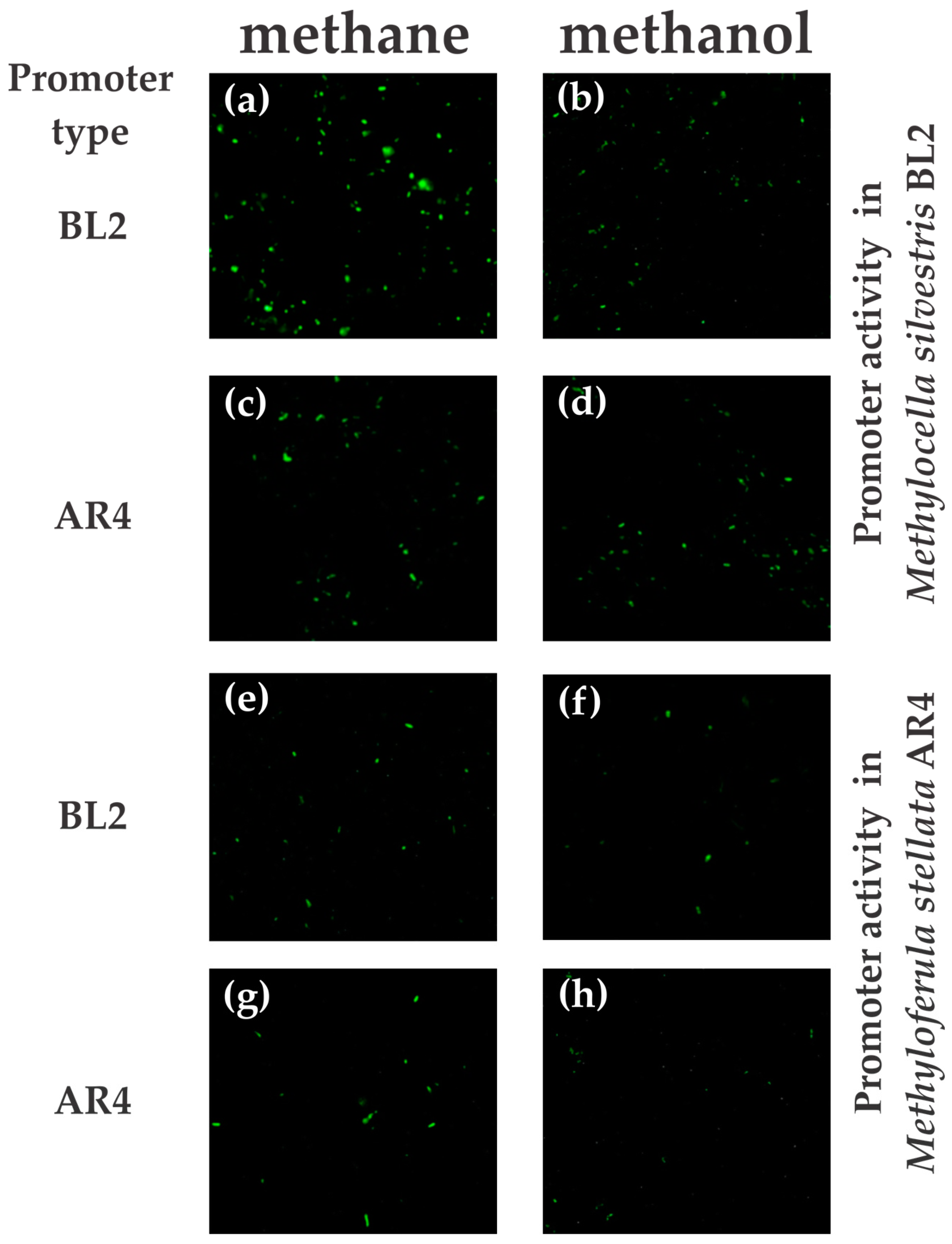
To quantify green fluorescent protein (GFP)-fluorescent cells, the cell suspension in PBS buffer that was used for fluorescence microscopy was diluted to 2–3 mL using sheath fluid (BD Biosciences, Mississauga, ON, Canada) to adjust the concentration of cells to 108 cells/mL. A proper dilution to 108 cells/mL was achieved by measuring optical density at 600 nm. A sample was injected into a FACScalibur (BD Biosciences, USA) flow cytometer equipped with a 15-mW air-cooled argon-ion laser as the excitation light source (488 nm). Fluorescence in the range of 515–545 nm (FL1) was detected via a fluorescence detector set at a photomultiplier tube range of either 525 V (for M. silvestris) or 580 V (for M. stellata) with logarithmic gain. Forward scatter (FSC) was collected by a diode with an amplification factor of E00 and processed in logarithmic gain. Side scatter (SSC) was detected in logarithmic gain by a photomultiplier tube set at 356 V. Each sample was collected for 60 s at high flow rate. A total of 10,000 events were collected for each measurement. The BD CellQuest Pro software was used to generate histograms, plots and perform analysis (BD Biosciences, Mississauga, ON, Canada).
2.6. Quantitative Measurements of Promoter Activities
To quantify promoter activities, replicate samples of harvested bacterial cultures were resuspended in 200 µL of PBS buffer. Cell suspensions were placed into wells of a black microtiter plate. Subsequently, the emission of GFP-labeled cells was recorded on an EnSpire Multimode Plate Reader (PerkinElmer, Woodbridge, ON, Canada). All fluorescence values were normalized by total cell protein concentrations. First, cells suspensions were transferred into 1.5 mL tubes and processed through 5 times freeze-thaw cycles (from −80 °C to room temperature). Second, proteins were precipitated by adding an equal volume of cold (4 °C) 10% trichloroacetic acid (TCA) and denatured by boiling for 5–10 min. After centrifugation for 5 min, the protein pellet was dissolved in 100 µL of 1 M NaOH and diluted to 1 mL with 900 µL ddH2O. Finally, the protein concentration was measured using a Qubit protein assay kit and Qubit fluorimeter (ThermoFischer Scientific, Waltham, MA, USA).
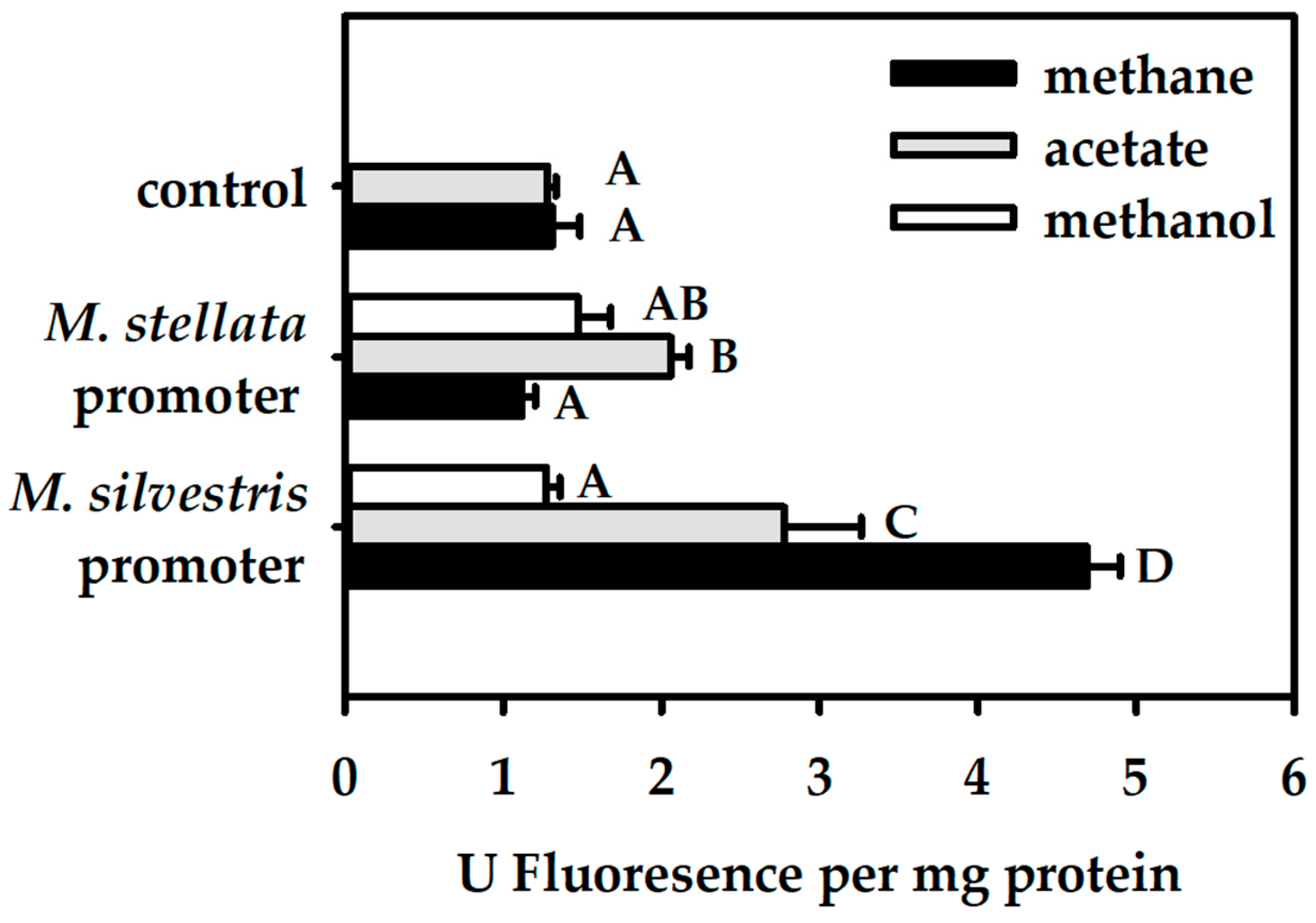
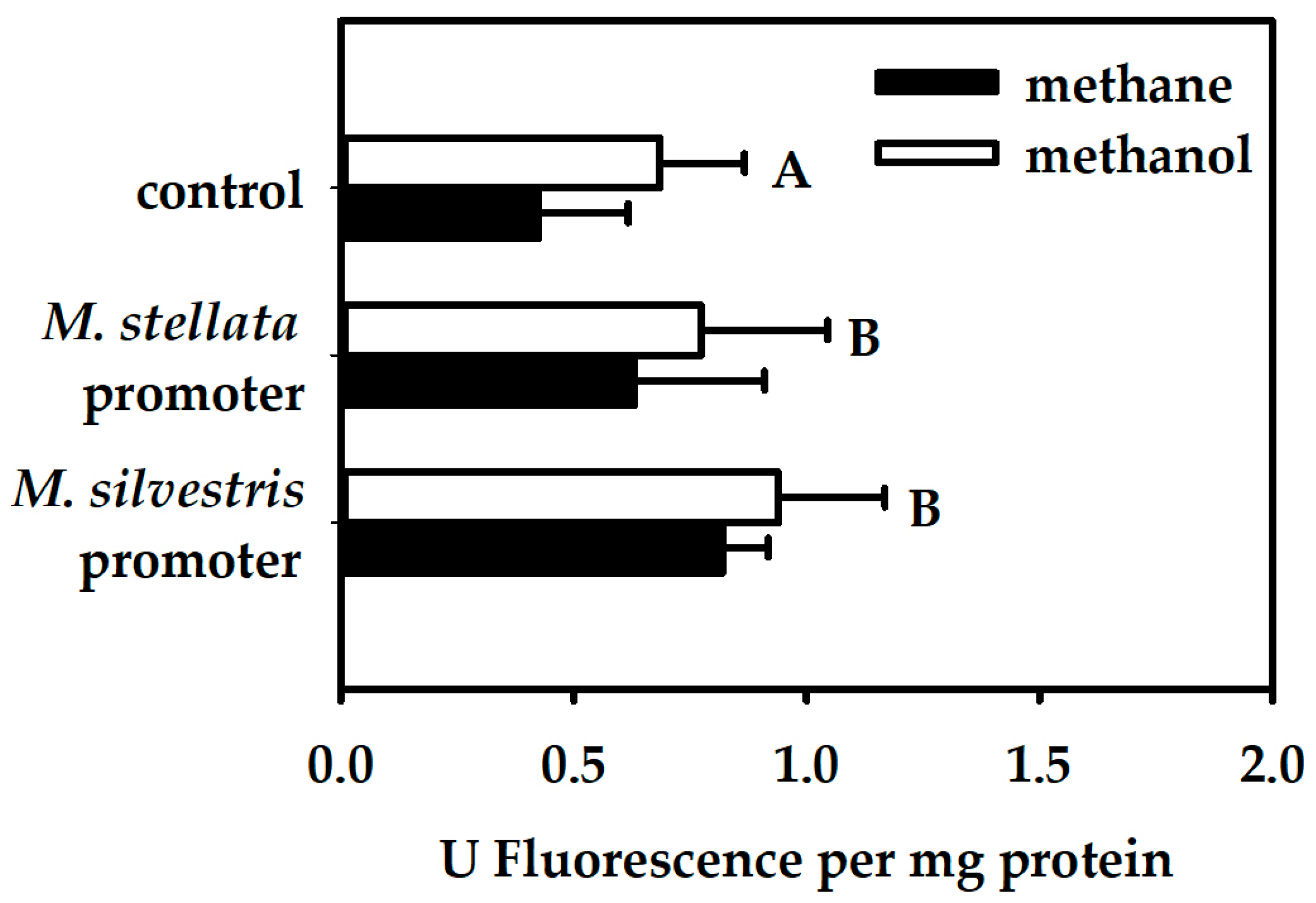
For M. silvestris cells, fluorescence was tested in a single trial with 3–5 replicate cultures. For M. stellata, because the differences in fluorescence were noticeably smaller, the fluorescence was tested in 3 separate trials with 2–4 replicate cultures each. Fluorescence of control M. stellata cultures (i.e., cultures with gfp fused to no promoter) varied between the different trials. Therefore, for statistical testing, data were blocked by the trial, although only the overall averages are shown in the relevant figures. A General Linear Model (analysis of variance; ANOVA) was performed using Systat v.13 (Systat Software Inc., San Jose, CA, USA) with: Blocks (four trials), Host organisms (two levels), Promoters (three levels; two promoters + one control with no promoter); and Growth substrates (three levels). The design was incomplete because some treatments were impossible (e.g., growth on acetate in M. stellata). Each treatment combination tested contained at least three replicates.
4. Discussion
In this study, we demonstrated that transcriptional activation of the mmo promoter in the facultative methanotroph Methylocella silvestris BL2 was dependent on the presence of methane as the growth substrate, whereas transcriptional activation of the mmo promoter in the obligate methanotroph Methyloferula stellata AR4, was constitutive regardless of the growth substrate. Methane dramatically enhances transcriptional activation of the mmo promoter in M. silvestris. Other growth substrates either downregulate or repress transcriptional activation of the mmo promoter. Interestingly, the mmo promoter of M. stellata was constitutively expressed either in the M. silvestris or M. stellata cells and the promoter activity was low regardless growth substrate. Even though M. silvestris and M. stellata are closely related organisms (97.1% identity of 16S rRNA genes), and their proteins involved in methane oxidation share high degrees of similarity, their mode of regulation of the mmo promoter is very different.
From previous studies [
19,
31,
32] we know that the transcription of
mmo genes in
Methylosinus trichosporium OB3b,
Methylococcus capsulatus Bath, and
Methylocella silvestris BL2 is RpoN-dependent. Inactivation of
rpoN, which encodes σ
54 factor, resulted in abolition of
mmo gene expression in
M. trichosporium OB3b [
32]. A-24-12 recognition motif (YTGGCACGrNNNTTGCW) characteristic of σ
54-type promoters [
29] can be identified upstream of every sequenced
mmoX gene [
19], including sequences in
M. silvestris BL2 and
M. stellata AR4. Unlike the major σ
70-factor, the σ
54-factor relies on a specialized class of transcriptional activators (enhancers), which uses ATP binding and hydrolysis to catalyze the formation of open promoter complexes for transcriptional initiation. In numerous cases such activators, for instance NtrC, are members of two-component signal transduction pathways, and require phosphorylation of an N-terminal receiver domain to ‘translate’ environmental cues into a coherent transcriptional activation of genes [
29]. A gene encoding a σ
54-dependent transcriptional activator,
mmoR, is located in
mmo operons of both
M. silvestris and
M. stellata. Homologues of
mmoR are also located in the vicinity of the
mmo operons in other methanotrophs [
33]. Notably, a gene encoding a cognate histidine protein kinase, another member of two-component regulatory system, which is usually located adjacent to a response regulator, is not found in the sMMO-coding regions of the genomes of
M. silvestris and
M.
stellata. The GasS/GacA two-component regulatory system provides an example where the
gacS gene coding for a histidine kinase is physically unlinked on bacterial chromosome to the
gacA gene encoding a response regulator [
34]. However, a receiver domain, which contains a site being phosphorylated by a cognate histidine kinase, was not identified in MmoR proteins by a search of conserved domains in our study.
Although Theisen et al. [
19] claimed that the conserved Asp residue (phosphorylation site) was found in an alignment of the MmoR sequences of
M. trichosporium,
M. capsulatus,
M. silvestris and selected other σ
54—transcriptional regulators, it is not certain that MmoR is activated via phosphorylation by a cognate histidine kinase, since if this were so then potential cognate histidine kinases should also be highly homologous and the mode of regulation of
mmo genes should therefore been similar in all methanotrophs. Our result contradicts this assumption. Not all σ
54—transcriptional regulators require phosphorylation to regulate transcription in response to an environmental cue. Either a ligand binding or protein-protein interactions can also play a role in regulation. Different sensory domains are present in an N-terminus of σ
54—transcriptional regulators depending on an environmental signal to be detected [
29]. In fact, MmoR proteins of
M. silvestris BL2 and
M. stellata AR4 are closely related to a group of σ
54—transcriptional regulators that contain a GAF sensory domain. This group includes NifA, a regulator of nitrogen fixation, FhlA, a regulator of formate metabolism, and NorR regulator for NO detoxification [
29]. The binding of 2-oxoglutarate to the GAF domain of NifA prevents the NifL-NifA protein interaction to ensure that NifA is not inhibited by NifL under nitrogen-fixing conditions [
35]. FhlA contains two GAF domains that bind formate to activate the transcription of the formate hydrogen lyase system [
36]. NorR contains a single GAF domain, which binds NO to activate transcription of
norVW genes encoding flavorubredoxin and an associated flavoprotein, which reduce NO to nitrous oxide [
37]. Our analysis of domain organization for MmoR proteins of
M. silvestris and
M. stellata revealed that the N-termini of both proteins contain a GAF domain not found in
M. trichosporium OB3b or
M. capsulatus Bath. Therefore, we cannot exclude the possibility that GAF domains of MmoR of either protein bind an unknown signal molecule to activate transcription of the
mmo operon. However, even this doesn’t account for a different mode of regulation for the
mmo genes. It is possible that MmoR is involved in protein-protein interactions with another protein to repress or activate transcriptional activation of
mmo genes.
Using gel-shifts assays, Scanlan et al. [
33] demonstrated that in order to bind upstream of the
mmo promoter in
M. trichosporium OB3b, MmoR requires a putative GroEL-like chaperon, MmoG. MmoG proteins are present and highly homologous in
M. silvestris and
M. stellata, and their presence or absence alone doesn’t explain a different mode of regulation. MmoG may be required to interact with another transcriptional regulator to adjust expression of the
mmo operon in
M. silvestris with respect to growth on different substrates.
Growth on methane is slower than on acetate or other multicarbon substrates for
M. silvestris [
17]. Methane is the least preferred substrate for
M. silvestris. Because all methanotrophs demonstrate slow growth rates and the catalytic efficiencies (kcat/K
m) of methane-oxidizing enzymes are lower than the median value of enzymes involved in central metabolism [
38], it has been speculated that high levels of MMO expression could help methanotrophs to overcome limitations in carbon uptake. Therefore, even though
M. silvestris cells grow more slowly on methane than on multicarbon substrates [
17], high levels of sMMO expression partially compensate for that.
M. stellata cells grow on methane more slowly than
M. silvestris cells do, and indeed show lower levels of sMMO gene expression. An environmental cue that might enhance transcriptional activation of the
mmo operon in
M. stellata hasn’t been identified yet. We tested an influence of carbon dioxide (CO
2) and cerium (III) on transcriptional activation of the
mmo promoter in
M. stellata. Neither CO
2 nor cerium (III) could enhance transcriptional activation of the
mmo promoter in
M. stellata. It has been shown in
Methylosinus trichosporium [
26] and
Methylomicrobium buryatense [
28] that the addition of light REE (La, Ce, Pr, and Nd) upregulates the expression of a gene encoding XoxF methanol dehydrogenase and downregulates the expression of a gene coding for another type of methanol dehydrogenase, MxaF. Moreover, REE were essential as cofactor in the XoxF enzyme of the verrucomicrobial methanotroph,
Methylacidiphilum fumariolicum SolV [
27]. Farhan et al. [
26] also suggested that in
Methylosinus trichosporium, XoxF can only serve as the methanol dehydrogenase under conditions in which sMMO is expressed. In our experiments, the addition of cerium (III) did not affect the growth, methane consumption and
mmo promoter activity in
M. stellata. Therefore, a question about an environmental cue to enhance transcriptional activation of the
mmo operon in
M. stellata remains to be elucidated.
The most important issue, however, is still the question of what kind of transcriptional regulator(s) governs transcription of mmo genes in either M. stellata or M. silvestris, and why these differ so dramatically. A more detailed study is required to identify a potential regulator in each organism. In this study, we provide solid proof that a different mode of transcriptional regulation of sMMO contributes to the facultative lifestyle of Methylocella silvetris BL2 as compared to its sibling obligate methanotroph Methyloferula stellata AR4.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




