Inflammatory Bowel Disease: A Potential Result from the Collusion between Gut Microbiota and Mucosal Immune System


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Microbiota Dysbiosis as a Potential Trigger for IBD
2.1. Specific Pathogenic Microbes in IBD
2.2. Profiles of the Intestinal Bacteria and IBD
2.3. Fungal Microbiota and IBD
2.4. Enteric Virome and IBD
2.5. Protozoans and IBD
2.6. Helminths and IBD
3. Mucosal Immune System and Intestinal Homeostasis
3.1. Composition of the Mucosal Immune System
3.2. Mucus Layer
3.3. Single Layer of IECs
3.4. Intestinal Immune Cells
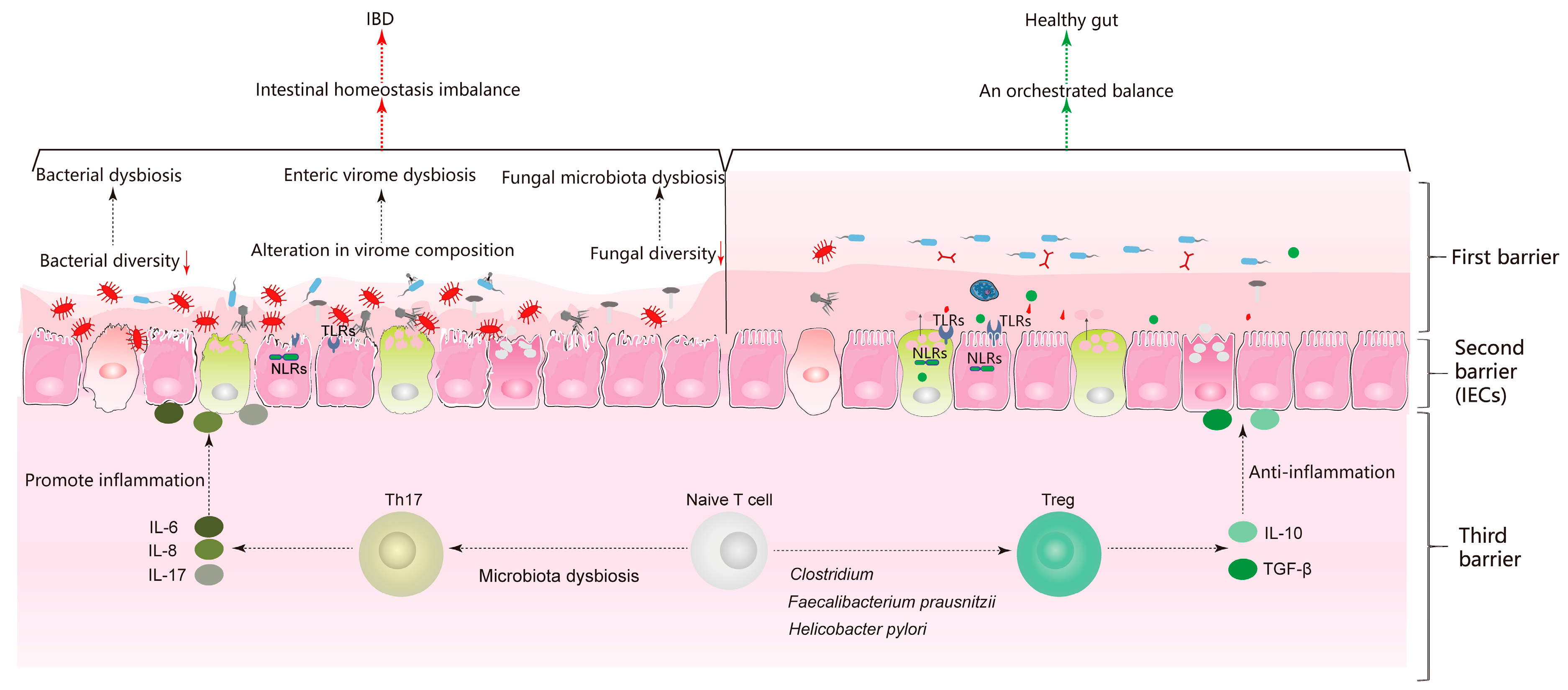
4. Orchestrated Balance between Mucosal Immune System and Gut Microbiota
4.1. Interaction between Treg/Th17 Axis and Gut Microbiota
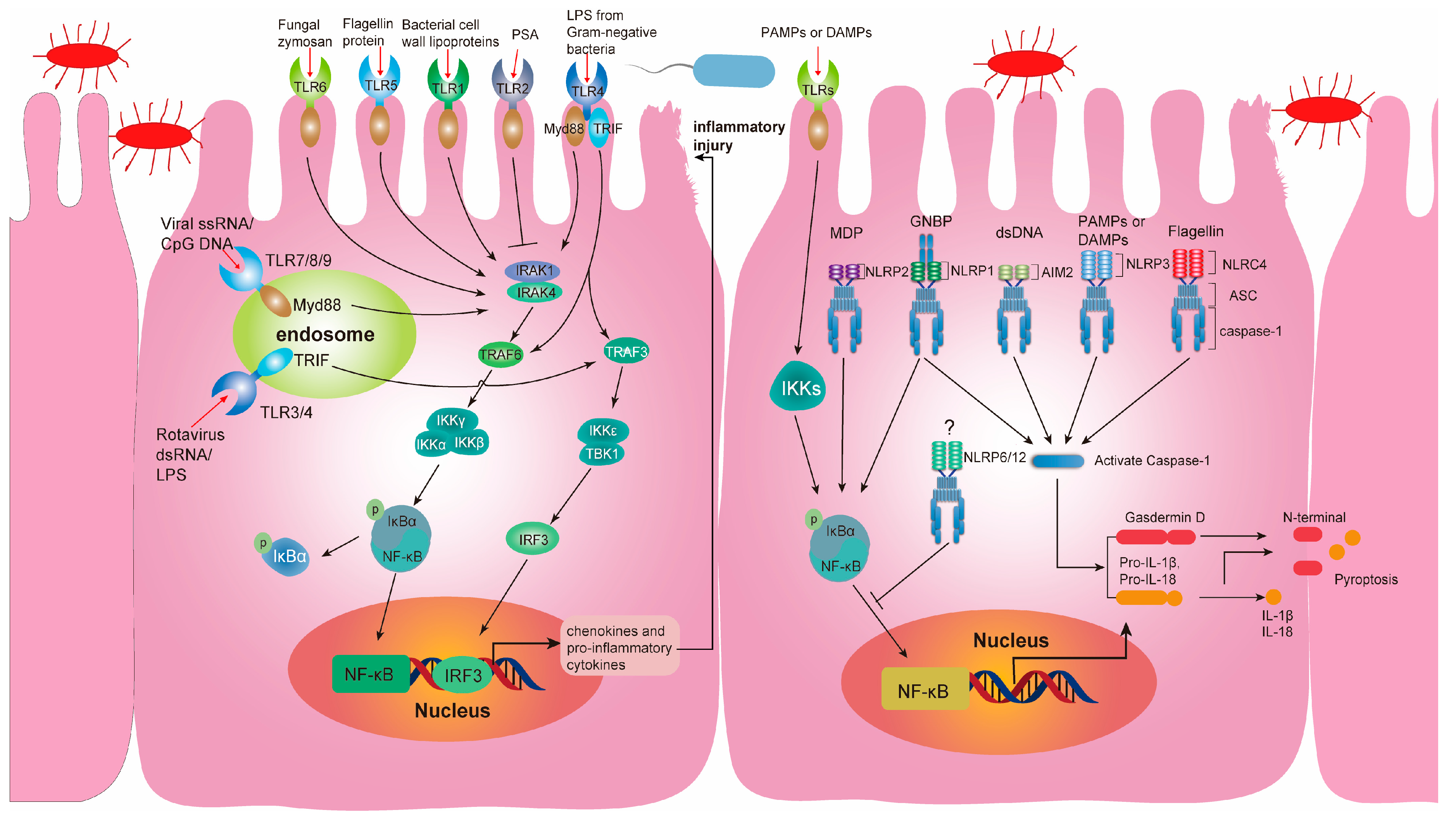
4.2. Communication between Pattern Recognition Receptors (PRRs) and Gut Microbiota
4.3. TLRs and the Gut Microbiota
4.4. NLRs and the Gut Microbiota
5. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AIEC | adherent-invasive Escherichia coli |
| AMPs | antimicrobial peptides |
| ASCA | anti-Saccharomyces cerevisiae antibodies |
| ASC | apoptosis-associated speck-like protein containing a CARD |
| CD | Crohn’s disease |
| DAMPs | damage-associated molecular patterns |
| DCs | dendritic cells |
| DSS | dextran sulfate sodium |
| IBDs | inflammatory bowel diseases |
| ILCs | innate lymphoid cells |
| IECs | intestinal epithelial cells |
| LGP2 | laboratory of genetics and physiology gene 2 |
| LPS | lipopolysaccharides |
| MDA5 | melanoma differentiation-associated gene 5 |
| MUC2 | mucin 2 |
| MDP | muramyl dipeptide |
| MNV | murine norovirus |
| MyD88 | myeloid differentiation primary response gene 88 |
| NLRs | NOD domain-like receptors |
| NF-κB | nuclear factor kappa-B |
| PAMPs | pathogen-associated molecular patterns |
| PRRs | pattern recognition receptors |
| PSA | polysaccharide A |
| Treg | regulatory T |
| RIG-I | retinoid acid-inducible gene-I |
| SIgA | secretory immunoglobulin A |
| SCFA | short-chain fatty acids |
| TLRs | toll-like receptors |
| ILC2s | type 2 innate lymphoid cells |
| UC | ulcerative colitis |
References
- Yue, B.; Ren, Y.J.; Zhang, J.J.; Luo, X.P.; Yu, Z.L.; Ren, G.Y.; Sun, A.N.; Deng, C.; Wang, Z.T.; Dou, W. Anti-Inflammatory Effects of Fargesin on Chemically Induced Inflammatory Bowel Disease in Mice. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.G.; Chang, E.B. The intestinal microbiota in the pathogenesis of inflammatory bowel diseases: New insights into complex disease. Clin. Sci. (Lond.) 2018, 132, 2013–2028. [Google Scholar] [CrossRef] [PubMed]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Winglee, K.; Howard, A.G.; Sha, W.; Gharaibeh, R.Z.; Liu, J.; Jin, D.; Fodor, A.A.; Gordon-Larsen, P. Recent urbanization in China is correlated with a Westernized microbiome encoding increased virulence and antibiotic resistance genes. Microbiome 2017, 5, 121. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Kamm, M.A.; Colombel, J.F.; Ng, S.C. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Curciarello, R.; Canziani, K.E.; Docena, G.H.; Muglia, C.I. Contribution of Non-immune Cells to Activation and Modulation of the Intestinal Inflammation. Front Immunol. 2019, 10, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, L.J.; Cho, J.H.; Gevers, D.; Chu, H. Genetic Factors and the Intestinal Microbiome Guide Development of Microbe-Based Therapies for Inflammatory Bowel Diseases. Gastroenterology 2019, 156, 2174–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef]
- Gao, X.; Cao, Q.; Cheng, Y.; Zhao, D.; Wang, Z.; Yang, H.; Wu, Q.; You, L.; Wang, Y.; Lin, Y.; et al. Chronic stress promotes colitis by disturbing the gut microbiota and triggering immune system response. Proc. Natl. Acad. Sci. USA 2018, 115, E2960–E2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond D, T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Ihara, S.; Koike, K. Targeting the complex interactions between microbiota: Host epithelial and immune cells in inflammatory bowel disease. Pharmacol. Res. 2016, 113, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, R.J.; Van Kruiningen, H.J.; Thayer, W.R.; Merkal, R.S.; Coutu, J.A. Possible role of mycobacteria in inflammatory bowel disease. I. An unclassified Mycobacterium species isolated from patients with Crohn’s disease. Dig. Dis. Sci. 1984, 29, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.N.; Shanahan, F. Disorders of a modern lifestyle: Reconciling the epidemiology of inflammatory bowel diseases. Gut 2008, 57, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Feller, M.; Huwiler, K.; Stephan, R.; Altpeter, E.; Shang, A.; Furrer, H.; Pfyffer, G.E.; Jemmi, T.; Baumgartner, A.; Egger, M. Mycobacterium avium subspecies paratuberculosis and Crohn’s disease: A systematic review and meta-analysis. Lancet. Infect. Dis. 2007, 7, 607–613. [Google Scholar] [CrossRef]
- Okura, H.; Toft, N.; Nielsen, S.S. Occurrence of Mycobacterium avium subsp. paratuberculosis in milk at dairy cattle farms: A systematic review and meta-analysis. Vet. Microbiol. 2012, 157, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef]
- Carrillo-Larco, R.M.; Bernabe-Ortiz, A.; Pillay, T.D.; Gilman, R.H.; Sanchez, J.F.; Proterico, J.A.; Quispe, R.; Smeeth, L.; Miranda, J.J. Obesity risk in rural, urban and rural-to-urban migrants: Prospective results of the PERU MIGRANT study. Int. J. Obes. (Lond.) 2016, 40, 181–185. [Google Scholar] [CrossRef] [PubMed]
- McPhee, J.B.; Small, C.L.; Reid-Yu, S.A.; Brannon, J.R.; Le Moual, H.; Coombes, B.K. Host defense peptide resistance contributes to colonization and maximal intestinal pathology by Crohn’s disease-associated adherent-invasive Escherichia coli. Infect. Immun. 2014, 82, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Jarry, A.; Cremet, L.; Caroff, N.; Bou-Hanna, C.; Mussini, J.M.; Reynaud, A.; Servin, A.L.; Mosnier, J.F.; Lievin-Le Moal, V.; Laboisse, C.L. Subversion of human intestinal mucosa innate immunity by a Crohn’s disease-associated E. coli. Mucosal. Immunol. 2015, 8, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Viladomiu, M.; Kivolowitz, C.; Abdulhamid, A.; Dogan, B.; Victorio, D.; Castellanos, J.G.; Woo, V.; Teng, F.; Tran, N.L.; Sczesnak, A.; et al. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Tremlett, H.; Bauer, K.C.; Appel-Cresswell, S.; Finlay, B.B.; Waubant, E. The gut microbiome in human neurological disease: A review. Ann. Neurol. 2017, 81, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Huttenhower, C.; Kostic, A.D.; Xavier, R.J. Inflammatory bowel disease as a model for translating the microbiome. Immunity 2014, 40, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Nava, G.M.; Stappenbeck, T.S. Host genetic susceptibility, dysbiosis and viral triggers in inflammatory bowel disease. Curr. Opin. Gastroenterol. 2011, 27, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The microbiome in infectious disease and inflammation. Annu. Rev. Immunol. 2012, 30, 759–795. [Google Scholar] [CrossRef] [PubMed]
- Coleman, O.I.; Haller, D. Bacterial Signaling at the Intestinal Epithelial Interface in Inflammation and Cancer. Front. Immunol. 2017, 8, 1927. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wu, W.; Liu, Z.; Cong, Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J. Gastroenterol. 2017, 52, 1–8. [Google Scholar] [CrossRef]
- Lepage, P.; Häsler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Doré, J.; Raedler, A.; et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, H.; Kataoka, K.; Ishikawa, H.; Ikata, K.; Arimochi, H.; Iwasaki, T.; Ohnishi, Y.; Kuwahara, T.; Yasutomo, K. Reduced diversity and imbalance of fecal microbiota in patients with ulcerative colitis. Dig. Dis. Sci. 2012, 57, 2955–2964. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, H.E.; Abrahamsson, T.R.; Jenmalm, M.C.; Harris, K.; Quince, C.; Jernberg, C.; Björkstén, B.; Engstrand, L.; Andersson, A.F. Decreased gut microbiota diversity, delayed Bacteroidetes colonization and reduced Th1 responses in infants delivered by caesarean section. Gut 2014, 63, 559–566. [Google Scholar] [CrossRef]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.A.; Frank, D.N.; Pace, N.R.; Gordon, J.I. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe 2008, 3, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.; Moran, C.; Shanahan, F. The microbiota in inflammatory bowel disease. J. Gastroenterol. 2015, 50, 495–507. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- De-Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, F.; Adam, N.; Johansson, M.E.; Xia, L.; Hansson, G.C.; Bäckhed, F. Altered mucus glycosylation in core 1 O-glycan-deficient mice affects microbiota composition and intestinal architecture. PLoS ONE 2014, 9, e85254. [Google Scholar] [CrossRef] [PubMed]
- Kleessen, B.; Kroesen, A.J.; Buhr, H.J.; Blaut, M. Mucosal and invading bacteria in patients with inflammatory bowel disease compared with controls. Scand. J. Gastroenterol. 2002, 37, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Ocvirk, S.; Sava, I.G.; Lengfelder, I.; Lagkouvardos, I.; Steck, N.; Roh, J.H.; Tchaptchet, S.; Bao, Y.; Hansen, J.J.; Huebner, J.; et al. Surface-Associated Lipoproteins Link Enterococcus faecalis Virulence to Colitogenic Activity in IL-10-Deficient Mice Independent of Their Expression Levels. PLoS Pathog. 2015, 11, e1004911. [Google Scholar] [CrossRef] [PubMed]
- Sobieszczańska, B.A.; Duda-Madej, A.B.; Turniak, M.B.; Franiczek, R.; Kasprzykowska, U.; Duda, A.K.; Rzeszutko, M.; Iwańczak, B. Invasive properties: Adhesion patterns and phylogroup profiles among Escherichia coli strains isolated from children with inflammatory bowel disease. Adv. Clin. Exp. Med. 2012, 21, 591–599. [Google Scholar] [PubMed]
- Boudeau, J.; Glasser, A.L.; Julien, S.; Colombel, J.F.; Darfeuille-Michaud, A. Inhibitory effect of probiotic Escherichia coli strain Nissle 1917 on adhesion to and invasion of intestinal epithelial cells by adherent-invasive E. coli strains isolated from patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2003, 18, 45–56. [Google Scholar] [CrossRef]
- Sartor, R.B.; Wu, G.D. Roles for Intestinal Bacteria, Viruses and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 2017, 152, 327–339. [Google Scholar] [CrossRef]
- Russell, R.K.; Ip, B.; Aldhous, M.C.; MacDougall, M.; Drummond, H.E.; Arnott, I.D.; Gillett, P.M.; McGrogan, P.; Weaver, L.T.; Bisset, W.M.; et al. Anti-Saccharomyces cerevisiae antibodies status is associated with oral involvement and disease severity in Crohn disease. J. Pediatr. Gastroenterol. Nutr. 2009, 48, 161–167. [Google Scholar] [CrossRef]
- Colombel, J.F.; Sendid, B.; Jouault, T.; Poulain, D. Secukinumab failure in Crohn’s disease: The yeast connection? Gut 2013, 62, 800–801. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.L.; Lamas, B.; Liguori, G.; Hoffmann, T.W.; Sokol, H. Gut fungal microbiota: The Yin and Yang of inflammatory bowel disease. Inflamm. Bowel. Dis. 2015, 21, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Jawhara, S.; Poulain, D. Saccharomyces boulardii decreases inflammation and intestinal colonization by Candida albicans in a mouse model of chemically-induced colitis. Med. Mycol. 2007, 45, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Jawhara, S.; Thuru, X.; Standaert-Vitse, A.; Jouault, T.; Mordon, S.; Sendid, B.; Desreumaux, P.; Poulain, D. Colonization of mice by Candida albicans is promoted by chemically induced colitis and augments inflammatory responses through galectin-3. J. Infect. Dis. 2008, 197, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.L.; Limon, J.J.; Bar, A.S.; Leal, C.A.; Gargus, M.; Tang, J.; Brown, J.; Funari, V.A.; Wang, H.L.; Crother, T.R.; et al. Immunological Consequences of Intestinal Fungal Dysbiosis. Cell Host Microbe 2016, 19, 865–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, J.K.; Virgin, H.W. Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Science 2016, 351. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W. The virome in mammalian physiology and disease. Cell 2014, 157, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Karst, S.M. Viral, Safeguard: The Enteric Virome Protects against Gut Inflammation. Immunity 2016, 44, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Basic, M.; Keubler, L.M.; Buettner, M.; Achard, M.; Breves, G.; Schröder, B.; Smoczek, A.; Jörns, A.; Wedekind, D.; Zschemisch, N.H.; et al. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflamm. Bowel. Dis. 2014, 20, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Patel, K.K.; Maloney, N.S.; Liu, T.C.; Ng, A.C.; Storer, C.E.; Head, R.D.; Xavier, R.; Stappenbeck, T.S.; Virgin, H.W. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 2010, 141, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J.; Auro, R.; Furlan, M.; Whiteson, K.L.; Erb, M.L.; Pogliano, J.; Stotland, A.; Wolkowicz, R.; Cutting, A.S.; Doran, K.S.l.; et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 10771–10776. [Google Scholar] [CrossRef] [PubMed]
- Kernbauer, E.; Ding, Y.; Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014, 516, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome. Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Waller, A.S.; Yamada, T.; Kristensen, D.M.; Kultima, J.R.; Sunagawa, S.; Koonin, E.V.; Bork, P. Classification and quantification of bacteriophage taxa in human gut metagenomes. ISME J. 2014, 8, 1391–1402. [Google Scholar] [CrossRef]
- Escalante, N.K.; Lemire, P.; Cruz-Tleugabulova, M.; Prescott, D.; Mortha, A.; Streutker, C.J.; Girardin, S.E.; Philpott, D.J.; Mallevaey, T. The common mouse protozoa Tritrichomonas muris alters mucosal T cell homeostasis and colitis susceptibility. J. Exp. Med. 2016, 213, 2841–2850. [Google Scholar] [CrossRef]
- Stentiford, G.D.; Becnel, J.; Weiss, L.M.; Keeling, P.J.; Didier, E.S.; Williams, B.A.P.; Bjornson, S.; Kent, M.L.; Freeman, M.A.; Brown, M.J.F.; et al. Microsporidia—Emergent Pathogens in the Global Food Chain. Trends Parasitol. 2016, 32, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Moonah, S.N.; Jiang, N.M.; Petri, W.A., Jr. Host immune response to intestinal amebiasis. PLoS Pathog. 2013, 9, e1003489. [Google Scholar] [CrossRef]
- Molloy, M.J.; Grainger, J.R.; Bouladoux, N.; Hand, T.W.; Koo, L.Y.; Naik, S.; Quinones, M.; Dzutsev, A.K.; Gao, J.L.; Trinchieri, G.; et al. Intraluminal containment of commensal outgrowth in the gut during infection-induced dysbiosis. Cell Host Microbe. 2013, 14, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Kotloff, K.L.; Nataro, J.P.; Blackwelder, W.C.; Nasrin, D.; Farag, T.H.; Panchalingam, S.; Wu, Y.; Sow, S.O.; Sur, D.; Breiman, R.F.; et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): A prospective, case-control study. Lancet 2013, 382, 209–222. [Google Scholar] [CrossRef]
- Lewthwaite, P.; Gill, G.V.; Hart, C.A.; Beeching, N.J. Gastrointestinal parasites in the immunocompromised. Curr. Opin. Infect. Dis. 2005, 18, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Lukeš, J.; Stensvold, C.R.; Jirků-Pomajbiková, K.; Wegener-Parfrey, L. Are Human Intestinal Eukaryotes Beneficial or Commensals? PLoS Pathog. 2015, 11, e1005039. [Google Scholar] [CrossRef] [PubMed]
- Parfrey, L.W.; Walters, W.A.; Lauber, C.L.; Clemente, J.C.; Berg-Lyons, D.; Teiling, C.; Kodira, C.; Mohiuddin, M.; Brunelle, J.; Driscoll, M.; et al. Communities of microbial eukaryotes in the mammalian gut within the context of environmental eukaryotic diversity. Front. Microbiol. 2014, 5, 298. [Google Scholar] [CrossRef] [PubMed]
- Parfrey, L.W.; Walters, W.A.; Knight, R. Microbial eukaryotes in the human microbiome, ecology, evolution, and future directions. Front. Microbiol. 2011, 2, 153. [Google Scholar] [CrossRef] [PubMed]
- Chudnovskiy, A.; Mortha, A.; Kana, V.; Kennard, A.; Ramirez, J.D.; Rahman, A.; Remark, R.; Mogno, I.; Ng, R.; Gnjatic, S.; et al. Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell 2016, 167, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Howitt, M.R.; Lavoie, S.; Michaud, M.; Blum, A.M.; Tran, S.V.; Weinstock, J.V.; Gallini, C.A.; Redding, K.; Margolskee, R.F.; Osborne, L.C.; et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016, 351, 1329–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitetta, L.; Saltzman, E.T.; Nikov, T.; Ibrahim, I.; Hall, S. Modulating the Gut Micro-Environment in the Treatment of Intestinal Parasites. J. Clin. Med. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, J.V.; Elliott, D.E. Helminths and the IBD hygiene hypothesis. Inflamm. Bowel. Dis. 2009, 15, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, H.; Xu, L.; Liao, Q.; Wan, S.; Yu, Z.; Lin, D.; Zhang, B.; Lv, Z.; Wu, Z.; et al. rSj16 Protects against DSS-Induced Colitis by Inhibiting the PPAR-alpha Signaling Pathway. Theranostics 2017, 7, 3446–3460. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, J.P.; Grainger, J.R.; Maizels, R.M. Helminth immunoregulation: The role of parasite secreted proteins in modulating host immunity. Mol. Biochem. Parasitol. 2009, 167, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, D.; Bowcutt, R.; Lee, S.C.; Tang, M.S.; Kurtz, Z.D.; Ding, Y.; Honda, K.; Gause, W.C.; Blaser, M.J.; Bonneau, R.A.; et al. Helminth infection promotes colonization resistance via type 2 immunity. Science 2016, 352, 608–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomin, P.; Agha, Z.; Loukas, A. Helminths and Intestinal Flora Team Up to Improve Gut Health. Trends Parasitol. 2016, 32, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lopez, A.; Behnsen, J.; Nuccio, S.P.; Raffatellu, M. Mucosal immunity to pathogenic intestinal bacteria. Nat. Rev. Immunol. 2016, 16, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Atuma, C.; Strugala, V.; Allen, A.; Holm, L. The adherent gastrointestinal mucus gel layer: Thickness and physical state in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G922–G929. [Google Scholar] [CrossRef]
- Barrett, J.C.; Lee, J.C.; Lees, C.W.; Prescott, N.J.; Anderson, C.A.; Phillips, A.; Wesley, E.; Parnell, K.; Zhang, H.; Drummond, H.; et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat. Genet. 2009, 41, 1330–1334. [Google Scholar]
- Van-der-Sluis, M.; De-Koning, B.A.; De-Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van-Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van-Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef]
- Martens, E.C.; Neumann, M.; Desai, M.S. Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nat. Rev. Microbiol. 2018. [Google Scholar] [CrossRef]
- Johansson, M.E.; Hansson, G.C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 2016, 16, 639–649. [Google Scholar] [CrossRef]
- Javitt, G.; Calvo, M.L.G.; Albert, L.; Reznik, N.; Ilani, T.; Diskin, R.; Fass, D. Intestinal gel-forming mucins polymerize by disulfide-mediated dimerization of d3 domains. J. Mol. Biol. 2019, 431, 3740–3752. [Google Scholar] [CrossRef] [PubMed]
- Petta, I.; Fraussen, J.; Somers, V.; Kleinewietfeld, M. Interrelation of Diet, Gut Microbiome, and Autoantibody Production. Front. Immunol. 2018, 9, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ijssennagger, N.; van-der-Meer, R.; van-Mil, S.W.C. Sulfide as a Mucus Barrier-Breaker in Inflammatory Bowel Disease? Trends Mol. Med. 2016, 22, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Gustafsson, J.K.; Holmen-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjövall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Ivanov, I.I. Intestinal epithelial cells as mediators of the commensal-host immune crosstalk. Immunol. Cell Biol. 2013, 91, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Kurashima, Y.; Kiyono, H. Mucosal Ecological Network of Epithelium and Immune Cells for Gut Homeostasis and Tissue Healing. Annu. Rev. Immunol. 2017, 35, 119–147. [Google Scholar] [CrossRef] [PubMed]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Gribblem, F.M.; Reimannm, F. Enteroendocrine Cells: Chemosensors in the Intestinal Epithelium. Annu. Rev. Physiol. 2016, 78, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Gerbe, F.; van-Es, J.H.; Makrini, L.; Brulin, B.; Mellitzer, G.; Robine, S.; Romagnolo, B.; Shroyer, N.F.; Bourgaux, J.F.; Pignodel, C.; et al. Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J. Cell Biol. 2011, 192, 767–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henson, C.C.; Burden, S.; Davidson, S.E.; Lal, S. Nutritional interventions for reducing gastrointestinal toxicity in adults undergoing radical pelvic radiotherapy. Cochrane. Database. Syst. Rev. 2013, D9896. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, D.; Chaparro, M.; Gisbert, J.P. Human Intestinal Dendritic Cells in Inflammatory Bowel Diseases. Mol. Nutr. Food Res. 2018, 62, e1700931. [Google Scholar] [CrossRef] [PubMed]
- Meroni, E.; Stakenborg, N.; Viola, M.F.; Boeckxstaens, G.E. Intestinal macrophages and their interaction with the enteric nervous system in health and inflammatory bowel disease. Acta. Physiol. (Oxf.) 2019, 225, e13163. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di-Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.; Prescott, N.; Lord, G.M.; MacDonald, T.T.; Powell, N. The unusual suspects--innate lymphoid cells as novel therapeutic targets in IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Cader, M.Z.; Kaser, A. Recent advances in inflammatory bowel disease: Mucosal immune cells in intestinal inflammation. Gut 2013, 62, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Rossi, O.; van-Baarlen, P.; Wells, J.M. Host-recognition of pathogens and commensals in the mammalian intestine. Curr. Top Microbiol. Immunol. 2013, 358, 291–321. [Google Scholar] [PubMed]
- Sarra, M.; Pallone, F.; Macdonald, T.T.; Monteleone, G. IL-23/IL-17 axis in IBD. Inflamm. Bowel. Dis. 2010, 16, 1808–1813. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, C.; Xu, H.; Jia, J.; Wei, Z.; Guo, R.; Jia, Y.; Wu, Y.; Li, Y.; Qi, X.; et al. The Immunoregulation of Th17 in Host against Intracellular Bacterial Infection. Mediators. Inflamm. 2018, 2018, 6587296. [Google Scholar] [CrossRef]
- Pandiyan, P.; Bhaskaran, N.; Zou, M.; Schneider, E.; Jayaraman, S.; Huehn, J. Microbiome Dependent Regulation of Tregs and Th17 Cells in Mucosa. Front. Immunol. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; He, C.; Cong, Y.; Liu, Z. Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal. Immunol. 2015, 8, 969–978. [Google Scholar] [CrossRef]
- Okumura, R.; Takeda, K. Maintenance of gut homeostasis by the mucosal immune system. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2016, 92, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostman, S.; Rask, C.; Wold, A.E.; Hultkrantz, S.; Telemo, E. Impaired regulatory T cell function in germ-free mice. Eur. J. Immunol. 2006, 36, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Geva-Zatorsky, N.; Sefik, E.; Kua, L.; Pasman, L.; Tan, T.G.; Ortiz-Lopez, A.; Yanortsang, T.B.; Yang, L.; Jupp, R.; Mathis, D.; et al. Mining the Human Gut Microbiota for Immunomodulatory Organisms. Cell 2017, 168, 928–943. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, M.; Wang, Y.; Dorfman, R.G.; Liu, H.; Yu, T.; Chen, X.; Tang, D.; Xu, L.; Yin, Y.; et al. Faecalibacterium prausnitzii Produces Butyrate to Maintain Th17/Treg Balance and to Ameliorate Colorectal Colitis by Inhibiting Histone Deacetylase 1. Inflamm. Bowel. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dai, Y.; Liu, Y.; Wu, T.; Li, J.; Wang, X.; Wang, W. Helicobacter pylori Colonization Protects Against Chronic Experimental Colitis by Regulating Th17/Treg Balance. Inflamm. Bowel. Dis. 2018, 24, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; et al. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORgammat (+) Regulatory T Cells and Exacerbate Colitis in Mice. Immunity 2019, 50, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.; Blume, J.; Roy, U.; Teh, P.P.; Vasanthakumar, A.; Beller, A.; Liao, Y.; Heinrich, F.; Arenzana, T.L.; Hackney, J.A.; et al. c-Maf-dependent Treg cell control of intestinal TH17 cells and IgA establishes host-microbiota homeostasis. Nat. Immunol. 2019, 20, 471–481. [Google Scholar] [CrossRef]
- Kumar, P.; Monin, L.; Castillo, P.; Elsegeiny, W.; Horne, W.; Eddens, T.; Vikram, A.; Good, M.; Schoenborn, A.A.; Bibby, K.; et al. Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity 2016, 44, 659–671. [Google Scholar] [CrossRef] [Green Version]
- Campbell, C.; Dikiy, S.; Bhattarai, S.K.; Chinen, T.; Matheis, F.; Calafiore, M.; Hoyos, B.; Hanash, A.; Mucida, D.; Bucci, V.; et al. Extrathymically Generated Regulatory T Cells Establish a Niche for Intestinal Border-Dwelling Bacteria and Affect Physiologic Metabolite Balance. Immunity 2018, 48, 1245–1257. [Google Scholar] [CrossRef] [PubMed]
- Spiljar, M.; Merkler, D.; Trajkovski, M. The Immune System Bridges the Gut Microbiota with Systemic Energy Homeostasis: Focus on TLRs, Mucosal Barrier and SCFAs. Front. Immunol. 2017, 8, 1353. [Google Scholar] [CrossRef] [PubMed]
- Salonen, A.; de-Vos, W.M. Impact of diet on human intestinal microbiota and health. Annu. Rev. Food Sci. Technol. 2014, 5, 239–262. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Yang, Y.; Zhu, W. Crosstalk Between the Immune Receptors and Gut Microbiota. Curr. Protein Pept. Sci. 2015, 16, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Fawkner-Corbett, D.; Simmons, A.; Parikh, K. Microbiome: Pattern recognition receptor function in health and inflammation. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Mazmanian, S.K. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat. Immunol. 2013, 14, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Liston, A.; Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208–214. [Google Scholar] [CrossRef]
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016, 16, 35–50. [Google Scholar] [CrossRef]
- de-Kivit, S.; Tobin, M.C.; Forsyth, C.B.; Keshavarzian, A.; Landay, A.L. Regulation of Intestinal Immune Responses through TLR Activation: Implications for Pro- and Prebiotics. Front. Immunol. 2014, 5, 60. [Google Scholar] [CrossRef] [Green Version]
- Otte, J.M.; Cario, E.; Podolsky, D.K. Mechanisms of cross hyporesponsiveness to Toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology 2004, 126, 1054–1070. [Google Scholar] [CrossRef]
- Buwitt-Beckmann, U.; Heine, H.; Wiesmüller, K.H.; Jung, G.; Brock, R.; Akira, S.; Ulmer, A.J. TLR1- and TLR6-independent recognition of bacterial lipopeptides. J. Biol. Chem. 2006, 281, 9049–9057. [Google Scholar] [CrossRef] [PubMed]
- Nigar, S.; Yamamoto, Y.; Okajima, T.; Sato, T.; Ogita, T.; Shimosato, T. Immune synergistic oligodeoxynucleotide from Lactobacillus rhamnosus GG enhances the immune response upon co-stimulation by bacterial and fungal cell wall components. Anim. Sci. J. 2018, 89, 1504–1511. [Google Scholar] [CrossRef] [PubMed]
- Roeder, A.; Kirschning, C.J.; Rupec, R.A.; Schaller, M.; Weindl, G.; Korting, H.C. Toll-like receptors as key mediators in innate antifungal immunity. Med. Mycol. 2004, 42, 485–498. [Google Scholar] [CrossRef]
- Balka, K.R.; De-Nardo, D. Understanding early TLR signaling through the Myddosome. J. Leukoc. Biol. 2019, 105, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Cash, H.L.; Whitham, C.V.; Behrendt, C.L.; Hooper, L.V. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 2006, 313, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Lee, S.M.; Li, J.; Tran, G.; Jabri, B.; Chatila, T.A.; Mazmanian, S.K. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 2011, 332, 974–977. [Google Scholar] [CrossRef]
- Kayama, H.; Takeda, K. Polysaccharide A of Bacteroides fragilis: Actions on dendritic cells and T cells. Mol. Cell 2014, 54, 206–207. [Google Scholar] [CrossRef]
- Cario, E.; Gerken, G.; Podolsky, D.K. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology 2007, 132, 1359–1374. [Google Scholar] [CrossRef]
- Heimesaat, M.M.; Fischer, A.; Siegmund, B.; Kupz, A.; Niebergall, J.; Fuchs, D.; Jahn, H.K.; Freudenberg, M.; Loddenkemper, C.; Batra, A.; et al. Shift towards pro-inflammatory intestinal bacteria aggravates acute murine colitis via Toll-like receptors 2 and 4. PLoS ONE 2007, 2, e662. [Google Scholar] [CrossRef]
- Chassaing, B.; Koren, O.; Carvalho, F.A.; Ley, R.E.; Gewirtz, A.T. AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut 2014, 63, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R.; Shelling, A.N.; Browning, B.L.; Huebner, C.; Petermann, I. Genes, diet and inflammatory bowel disease. Mutat. Res. 2007, 622, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Dheer, R.; Santaolalla, R.; Davies, J.M.; Lang, J.K.; Phillips, M.C.; Pastorini, C.; Vazquez-Pertejo, M.T.; Abreu, M.T. Intestinal Epithelial Toll-Like Receptor 4 Signaling Affects Epithelial Function and Colonic Microbiota and Promotes a Risk for Transmissible Colitis. Infect. Immun. 2016, 84, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, K.; Marteyn, B.; Sansonetti, P.J.; Tang, C.M. Life on the inside: The intracellular lifestyle of cytosolic bacteria. Nat. Rev. Microbiol. 2009, 7, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Kagan, J.C. How Inflammasomes Inform Adaptive Immunity. J. Mol. Biol. 2018, 430, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 2010, 10, 688–698. [Google Scholar] [CrossRef] [Green Version]
- Rubino, S.J.; Selvanantham, T.; Girardin, S.E.; Philpott, D.J. Nod-like receptors in the control of intestinal inflammation. Curr. Opin. Immunol. 2012, 24, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017, 214, 1737–1752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.X.; Wang, Z.T.; Lu, X.X.; Wang, Y.G.; Zhong, J.; Liu, J. NLRP3 gene is associated with ulcerative colitis (UC), but not Crohn’s disease (CD) in Chinese Han population. Inflamm. Res. 2014, 63, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Claes, A.K.; Zhou, J.Y.; Philpott, D.J. NOD-Like Receptors: Guardians of Intestinal Mucosal Barriers. Physiology (Bethesda) 2015, 30, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corridoni, D.; Arseneau, K.O.; Cifone, M.G.; Cominelli, F. The dual role of nod-like receptors in mucosal innate immunity and chronic intestinal inflammation. Front. Immunol. 2014, 5, 317. [Google Scholar] [CrossRef]
- Girardin, S.E.; Boneca, I.G.; Carneiro, L.A.; Antignac, A.; Jéhanno, M.; Viala, J.; Tedin, K.; Taha, M.K.; Labigne, A.; Zähringer, U.; et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 2003, 300, 1584–1587. [Google Scholar] [CrossRef] [PubMed]
- Girardin, S.E.; Boneca, I.G.; Viala, J.; Chamaillard, M.; Labigne, A.; Thomas, G.; Philpott, D.J.; Sansonetti, P.J. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 2003, 278, 8869–8872. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Inohara, N.; Benito, A.; Chen, F.F.; Yamaoka, S.; Nunez, G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J. Biol. Chem. 2001, 276, 4812–4818. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Munoz-Planillo, R.; Nunez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010, 59, 1192–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.M.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Amer, A.; Body-Malapel, M.; Kanneganti, T.D.; Ozoren, N.; Jagirdar, R.; Inohara, N.; Vandenabeele, P.; Bertin, J.; Coyle, A.; et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 2006, 7, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Kofoed, E.M.; Vance, R.E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 2011, 477, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Mao, D.P.; Yudkovsky, N.; Bonneau, R.; Lorang, C.G.; Warren, S.E.; Leaf, I.A.; Aderem, A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2010, 107, 3076–3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchi, L.; Kamada, N.; Nakamura, Y.; Burberry, A.; Kuffa, P.; Suzuki, S.; Shaw, M.H.; Kim, Y.G.; Núñez, G. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol. 2012, 13, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Normand, S.; Delanoye-Crespin, A.; Bressenot, A.; Huot, L.; Grandjean, T.; Peyrin-Biroulet, L.; Lemoine, Y.; Hot, D.; Chamaillard, M. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl. Acad. Sci. USA 2011, 108, 9601–9606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wilson, J.E.; Koenigsknecht, M.J.; Chou, W.C.; Montgomery, S.A.; Truax, A.D.; Brickey, W.J.; Packey, C.D.; Maharshak, N.; Matsushima, G.K.; et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat. Immunol. 2017, 18, 541–551. [Google Scholar] [CrossRef]
- Abraham, C.; Medzhitov, R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 2011, 140, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Michel, M.L.; Waldschmitt, N. Card9 mediates susceptibility to intestinal pathogens through microbiota modulation and control of bacterial virulence. Gastroenterology 2018, 10, 1836–1844. [Google Scholar] [CrossRef]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353. [Google Scholar] [CrossRef]
- Didelot, X.; Walker, A.S.; Peto, T.E.; Crook, D.W.; Wilson, D.J. Within-host evolution of bacterial pathogens. Nat. Rev. Microbiol. 2016, 14, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Jelsbak, L.; Marvig, R.L.; Damkiaer, S.; Workman, C.T.; Rau, M.H.; Hansen, S.K.; Folkesson, A.; Johansen, H.K.; Ciofu, O.; et al. Evolutionary dynamics of bacteria in a human host environment. Proc. Natl. Acad. Sci. USA 2011, 108, 7481–7486. [Google Scholar] [CrossRef] [Green Version]
- De-Filippo, C.; Di-Paola, M.; Ramazzotti, M.; Albanese, D.; Pieraccini, G.; Banci, E.; Miglietta, F.; Cavalieri, D.; Lionetti, P. Diet, Environments, and Gut Microbiota. A Preliminary Investigation in Children Living in Rural and Urban Burkina Faso and Italy. Front. Microbiol. 2017, 8, 1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakoff-Nahoum, S.; Medzhitov, R. Innate immune recognition of the indigenous microbial flora. Mucosal. Immunol. 2008, 1 (Suppl. 1), S10–S14. [Google Scholar] [CrossRef]
- Kim, Y.G.; Sakamoto, K.; Seo, S.U.; Pickard, J.M.; Gillilland, M.G., III; Pudlo, N.A.; Hoostal, M.; Li, X.; Wang, T.D.; Feehley, T.; et al. Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science 2017, 356, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.A.M.; Ganal-Vonarburg, S.C.; Fuhrer, T.; Rupp, S.; Uchimura, Y.; Li, H.; Steinert, A.; Heikenwalder, M.; Hapfelmeier, S.; Sauer, U.; et al. The maternal microbiota drives early postnatal innate immune development. Science 2016, 351, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- El, A.S.; Hooiveld, G.; Tremaroli, V.; Backhed, F.; Kleerebezem, M. The gut microbiota and mucosal homeostasis: Colonized at birth or at adulthood, does it matter? Gut Microbes. 2013, 4, 118–124. [Google Scholar]
- Llewellyn, S.R.; Britton, G.J.; Contijoch, E.J.; Vennaro, O.H.; Mortha, A.; Colombel, J.F.; Grinspan, A.; Clemente, J.C.; Merad, M.; Faith, J.J. Interactions Between Diet and the Intestinal Microbiota Alter Intestinal Permeability and Colitis Severity in Mice. Gastroenterology 2018, 154, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; McCoy, K.D.; Johansen, F.E.; Brandtzaeg, P. The immune geography of IgA induction and function. Mucosal. Immunol. 2008, 1, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Itav, S.; Rothschild, D.; Meijer, M.T.; Levy, M.; Moresi, C.; Dohnalova, L.; Braverman, S.; Rozin, S.; Malitsky, S.; et al. Persistent microbiome alterations modulate the rate of post-dieting weight regain. Nature 2016, 540, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Khosravi, A.; Kusumawardhani, I.P.; Kwon, A.H.; Vasconcelos, A.C.; Cunha, L.D.; Mayer, A.E.; Shen, Y.; Wu, W.L.; Kambal, A.; et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 2016, 352, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.N.; Robertson, C.E.; Hamm, C.M.; Kpadeh, Z.; Zhang, T.; Chen, H.; Zhu, W.; Sartor, R.B.; Boedeker, E.C.; Harpaz, N.; et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm. Bowel. Dis. 2011, 17, 179–184. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, B.; Luo, X.; Yu, Z.; Mani, S.; Wang, Z.; Dou, W. Inflammatory Bowel Disease: A Potential Result from the Collusion between Gut Microbiota and Mucosal Immune System. Microorganisms 2019, 7, 440. https://doi.org/10.3390/microorganisms7100440
Yue B, Luo X, Yu Z, Mani S, Wang Z, Dou W. Inflammatory Bowel Disease: A Potential Result from the Collusion between Gut Microbiota and Mucosal Immune System. Microorganisms. 2019; 7(10):440. https://doi.org/10.3390/microorganisms7100440
Chicago/Turabian StyleYue, Bei, Xiaoping Luo, Zhilun Yu, Sridhar Mani, Zhengtao Wang, and Wei Dou. 2019. "Inflammatory Bowel Disease: A Potential Result from the Collusion between Gut Microbiota and Mucosal Immune System" Microorganisms 7, no. 10: 440. https://doi.org/10.3390/microorganisms7100440
APA StyleYue, B., Luo, X., Yu, Z., Mani, S., Wang, Z., & Dou, W. (2019). Inflammatory Bowel Disease: A Potential Result from the Collusion between Gut Microbiota and Mucosal Immune System. Microorganisms, 7(10), 440. https://doi.org/10.3390/microorganisms7100440