Phylogenomic Analyses of Bradyrhizobium Reveal Uneven Distribution of the Lateral and Subpolar Flagellar Systems, Which Extends to Rhizobiales

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets and Phylogenomic Analysis of Bradyrhizobium
2.2. Phylogenetic Analysis of Rhizobiales Order Genomes
2.3. Phylogenetic Analysis of the Flagellar Systems
2.4. Lateral and Subpolar Flagellar Systems Identification and Synteny
3. Results and Discussion
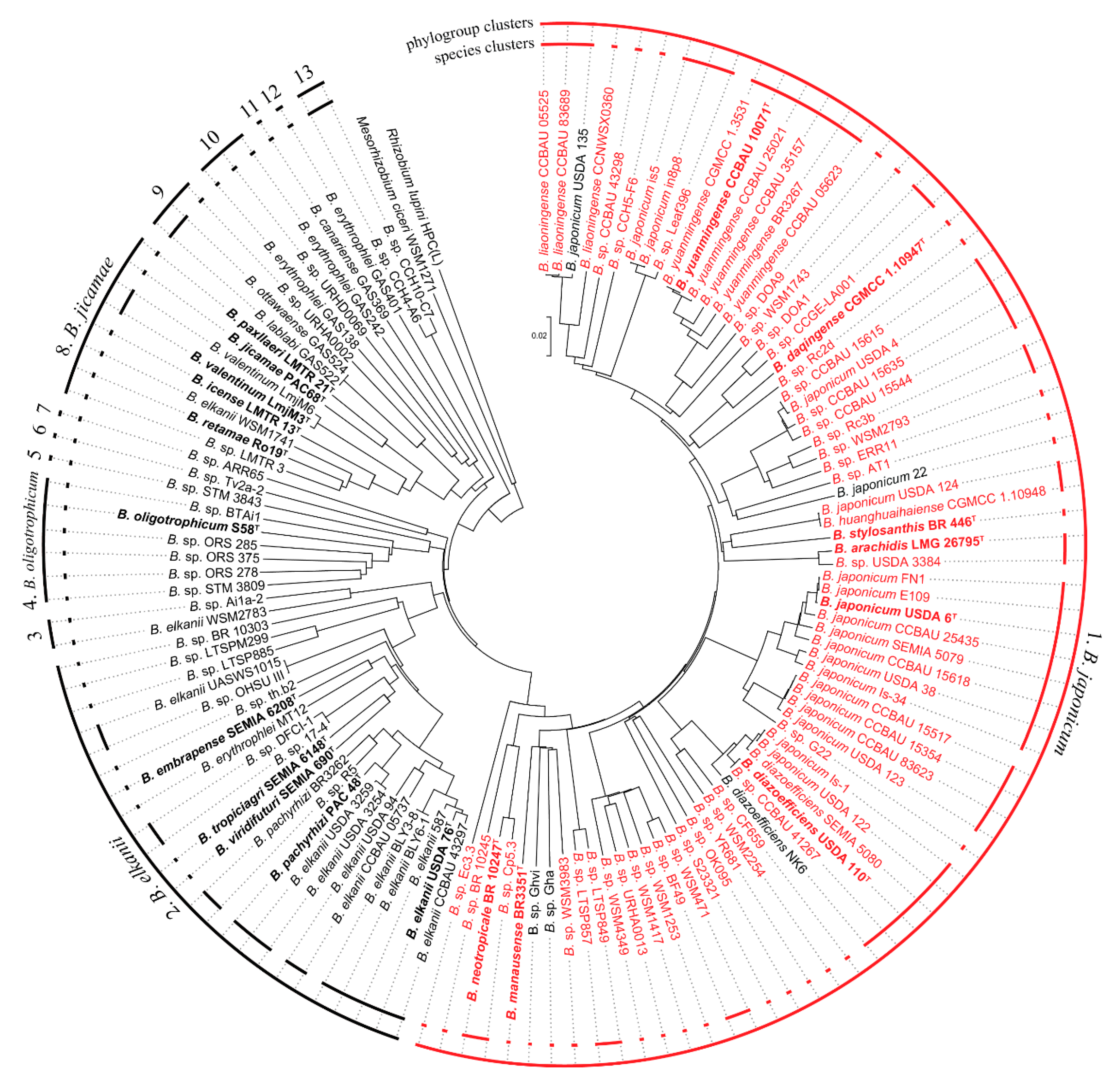
3.1. Phylogenomic Analysis of the Bradyrhizobium Genus
3.2. Lateral and Subpolar Flagellar Systems in the Bradyrhizobium Genus
3.3. Lateral Flagellar System Outside Bradyrhizobium
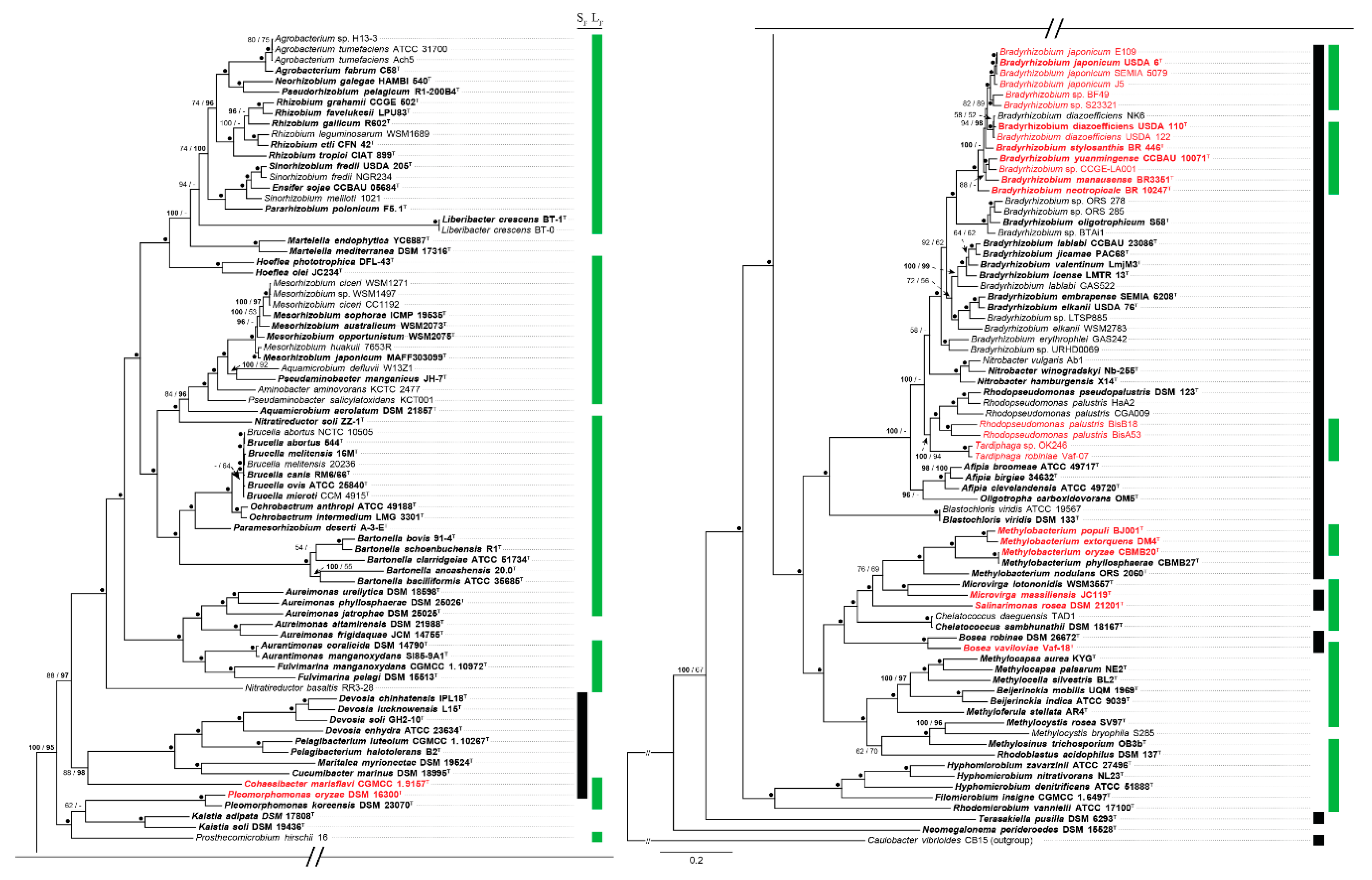
3.4. Flagellar Systems Distribution in Rhizobiales
3.5. Flagellar Systems Phylogeny in Rhizobiales
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, R.; Ochman, H. Origins of flagellar gene operons and secondary flagellar systems. J. Bacteriol. 2007, 189, 7098–7104. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Ochman, H. Stepwise formation of the bacterial flagellar system. Proc. Natl. Acad. Sci. USA 2007, 104, 7116–7121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, G.; Chevance, F.F.V.; Liou, W.; Renault, T.T.; Hughes, K.T.; Rainville, S.; Erhardt, M. Variability in bacterial flagella re-growth patterns after breakage. Sci. Rep. 2017, 7, 1282. [Google Scholar] [CrossRef] [PubMed]
- Macnab, R.M. Type III flagellar protein export and flagellar assembly. Biochim Biophys Acta 2004, 1694, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macnab, R.M. How bacteria assemble flagella. Annu Rev. Microbiol. 2003, 57, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Schuhmacher, J.S.; Thormann, K.M.; Bange, G. How bacteria maintain location and number of flagella? FEMS Microbiol. Rev. 2015, 39, 812–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarter, L.L. Dual flagellar systems enable motility under different circumstances. J. Mol. Microbiol. Biotechnol. 2004, 7, 18–29. [Google Scholar] [CrossRef]
- Bubendorfer, S.; Koltai, M.; Rossmann, F.; Sourjik, V.; Thormann, K.M. Secondary bacterial flagellar system improves bacterial spreading by increasing the directional persistence of swimming. Proc. Natl. Acad. Sci. USA 2014, 111, 11485–11490. [Google Scholar] [CrossRef] [Green Version]
- Poggio, S.; Abreu-Goodger, C.; Fabela, S.; Osorio, A.; Dreyfus, G.; Vinuesa, P.; Camarena, L. A complete set of flagellar genes acquired by horizontal transfer coexists with the endogenous flagellar system in Rhodobacter sphaeroides. J. Bacteriol. 2007, 189, 3208–3216. [Google Scholar] [CrossRef]
- Kanbe, M.; Yagasaki, J.; Zehner, S.; Göttfert, M.; Aizawa, S. Characterization of two sets of subpolar flagella in Bradyrhizobium japonicum. J. Bacteriol. 2007, 189, 1083–1089. [Google Scholar] [CrossRef]
- Kirov, S.M.; Tassell, B.C.; Semmler, A.B.; O’Donovan, L.A.; Rabaan, A.A.; Shaw, J.G. Lateral flagella and swarming motility in Aeromonas species. J. Bacteriol. 2002, 184, 547–555. [Google Scholar] [CrossRef] [PubMed]
- McClain, J.; Rollo, D.R.; Rushing, B.G.; Bauer, C.E. Rhodospirillum centenum utilizes separate motor and switch components to control lateral and polar flagellum rotation. J. Bacteriol. 2002, 184, 2429–2438. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.P.; Beatson, S.A.; Parkhill, J.; Pallen, M.J. The Flag-2 locus, an ancestral gene cluster, is potentially associated with a novel flagellar system from Escherichia coli. J. Bacteriol. 2005, 187, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Croess, C.; Moens, S.; Van Bastelaere, E.; Vanderleyden, J.; Michiels, K. The polar flagellum mediates Azospirillum brasilense adsorption to wheat roots. J. Gen. Microbiol. 1993, 139, 2261–2269. [Google Scholar] [CrossRef]
- Barahona, E.; Navazo, A.; Garrido-Sanz, D.; Muriel, C.; Martínez-Granero, F.; Redondo-Nieto, M.; Martín, M.; Rivilla, R. Pseudomonas fluorescens F113 Can Produce a Second Flagellar Apparatus, Which Is Important for Plant Root Colonization. Front. Microbiol. 2016, 7, 1471. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.; Wang, H.; Zeng, X.; Xiong, L.; Wang, F.; Xiao, X. Characterization of the relationship between polar and lateral flagellar structural genes in the deep-sea bacterium Shewanella piezotolerans WP3. Sci. Rep. 2016, 6, 39758. [Google Scholar] [CrossRef] [PubMed]
- Paulick, A.; Koerdt, A.; Lassak, J.; Huntley, S.; Wilms, I.; Narberhaus, F.; Thormann, K.M. Two different stator systems drive a single polar flagellum in Shewanella oneidensis MR-1. Mol. Microbiol. 2009, 71, 836–850. [Google Scholar] [CrossRef] [PubMed]
- Canals, R.; Altarriba, M.; Vilches, S.; Horsburgh, G.; Shaw, J.G.; Tomás, J.M.; Merino, S. Analysis of the lateral flagellar gene system of Aeromonas hydrophila AH-3. J. Bacteriol. 2006, 188, 852–862. [Google Scholar] [CrossRef]
- Althabegoiti, M.J.; Covelli, J.M.; Pérez-Giménez, J.; Quelas, J.I.; Mongiardini, E.J.; López, M.F.; López-García, S.L.; Lodeiro, A.R. Analysis of the role of the two flagella of Bradyrhizobium japonicum in competition for nodulation of soybean. FEMS Microbiol. Lett. 2011, 319, 133–139. [Google Scholar] [CrossRef]
- Althabegoiti, M.J.; López-García, S.L.; Piccinetti, C.; Mongiardini, E.J.; Perez-Gimenez, J.; Quelas, J.I.; Perticari, A.; Lodeiro, A.R. Strain selection for improvement of Bradyrhizobium japonicum competitiveness for nodulation of soybean. FEMS Microbiol. Lett. 2008, 282, 115–123. [Google Scholar] [CrossRef]
- Mongiardini, E.J.; Quelas, J.I.; Dardis, C.; Althabegoiti, M.J.; Lodeiro, A.R. Transcriptional Control of the Lateral-Flagellar Genes of Bradyrhizobium diazoefficiens. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef] [PubMed]
- Quelas, J.I.; Althabegoiti, M.J.; Jimenez-Sanchez, C.; Melgarejo, A.A.; Marconi, V.I.; Mongiardini, E.J.; Trejo, S.A.; Mengucci, F.; Ortega-Calvo, J.J.; Lodeiro, A.R. Swimming performance of Bradyrhizobium diazoefficiens is an emergent property of its two flagellar systems. Sci. Rep. 2016, 6, 23841. [Google Scholar] [CrossRef] [PubMed]
- Covelli, J.M.; Althabegoiti, M.J.; López, M.F.; Lodeiro, A.R. Swarming motility in Bradyrhizobium japonicum. Res. Microbiol 2013, 164, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Cogo, C.; Pérez-Giménez, J.; Rajeswari, C.B.; Luna, M.F.; Lodeiro, A.R. Induction by Bradyrhizobium diazoefficiens of Different Pathways for Growth in D-mannitol or L-arabinose Leading to Pronounced Differences in CO2 Fixation, O2 Consumption, and Lateral-Flagellum Production. Front. Microbiol. 2018, 9, 1189. [Google Scholar] [CrossRef]
- Siqueira, A.F.; Ormeño-Orrillo, E.; Souza, R.C.; Rodrigues, E.P.; Almeida, L.G.; Barcellos, F.G.; Batista, J.S.; Nakatani, A.S.; Martínez-Romero, E.; Vasconcelos, A.T.; et al. Comparative genomics of Bradyrhizobium japonicum CPAC 15 and Bradyrhizobium diazoefficiens CPAC 7: Elite model strains for understanding symbiotic performance with soybean. BMC genomics 2014, 15, 420. [Google Scholar] [CrossRef]
- VanInsberghe, D.; Maas, K.R.; Cardenas, E.; Strachan, C.R.; Hallam, S.J.; Mohn, W.W. Non-symbiotic Bradyrhizobium ecotypes dominate North American forest soils. ISME J. 2015, 9, 2435–2441. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-González, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef]
- NCBI ftp Server. Available online: https://ftp.ncbi.nlm.nih.gov/ (accessed on 1 October 2017).
- JGI IMG Database. Available online: http://img.jgi.doe.gov (accessed on 1 October 2017).
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 2013, 14, 60. [Google Scholar] [CrossRef]
- Genome-to-Genome Distance Calculator (GGDC) 2.1. Available online: http://ggdc.dsmz.de/ggdc.php (accessed on 1 October 2017).
- Richter, M.; Rosselló-Morá, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Calculate_ani.py. Available online: https://github.com/widdowquinn/scripts/blob/master/bioinformatics/calculate_ani.py (accessed on 1 October 2017).
- Garrido-Sanz, D.; Meier-Kolthoff, J.P.; Göker, M.; Martín, M.; Rivilla, R.; Redondo-Nieto, M. Genomic and Genetic Diversity within the Pseudomonas fluorescens Complex. PloS ONE 2016, 11, e0150183. [Google Scholar] [CrossRef]
- Garrido-Sanz, D.; Arrebola, E.; Martínez-Granero, F.; García-Méndez, S.; Muriel, C.; Blanco-Romero, E.; Martín, M.; Rivilla, R.; Redondo-Nieto, M. Classification of Isolates from the Pseudomonas fluorescens Complex into Phylogenomic Groups Based in Group-Specific Markers. Front. Microbiol. 2017, 8, 413. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Göker, M.; García-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PloS ONE 2009, 4, e6319. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083(T), the type strain (U5/41(T)) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genomic Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Method. Mol. Biol 2014, 1079, 105–116. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef]
- Pattengale, N.D.; Alipour, M.; Bininda-Emonds, O.R.; Moret, B.M.; Stamatakis, A. How many bootstrap replicates are necessary? J. Comput. Biol. 2010, 17, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. Phylogenetic Analysis using Parsimony; Illinois Natural History Survey: Champaign, IL, USA, 1985. [Google Scholar]
- Tavaré, S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences; Lectures on Mathematics in the Life Sciences; The American Mathematical Society: Providence, RI, USA, 1986; Volume 17, pp. 57–86. [Google Scholar]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC genomics 2008, 9, 75. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Menna, P.; Hungria, M.; Barcellos, F.G.; Bangel, E.V.; Hess, P.N.; Martínez-Romero, E. Molecular phylogeny based on the 16S rRNA gene of elite rhizobial strains used in Brazilian commercial inoculants. Syst. Appl. Microbiol. 2006, 29, 315–332. [Google Scholar] [CrossRef]
- Vinuesa, P.; Rademaker, J.L.; de Bruijn, F.J.; Werner, D. Genotypic characterization of Bradyrhizobium strains nodulating endemic woody legumes of the Canary Islands by PCR-restriction fragment length polymorphism analysis of genes encoding 16S rRNA (16S rDNA) and 16S-23S rDNA intergenic spacers, repetitive extragenic palindromic PCR genomic fingerprinting, and partial 16S rDNA sequencing. Appl. Environ. Microbiol. 1998, 64, 2096–2104. [Google Scholar] [PubMed]
- Menna, P.; Barcellos, F.G.; Hungria, M. Phylogeny and taxonomy of a diverse collection of Bradyrhizobium strains based on multilocus sequence analysis of the 16S rRNA gene, ITS region and glnII, recA, atpD and dnaK genes. Int. J. Syst. Evol. Microbiol. 2009, 59, 2934–2950. [Google Scholar] [CrossRef]
- Vinuesa, P.; Rojas-Jiménez, K.; Contreras-Moreira, B.; Mahna, S.K.; Prasad, B.N.; Moe, H.; Selvaraju, S.B.; Thierfelder, H.; Werner, D. Multilocus sequence analysis for assessment of the biogeography and evolutionary genetics of four Bradyrhizobium species that nodulate soybeans on the asiatic continent. Appl. Environ. Microbiol. 2008, 74, 6987–6996. [Google Scholar] [CrossRef]
- Delamuta, J.R.; Ribeiro, R.A.; Menna, P.; Bangel, E.V.; Hungria, M. Multilocus sequence analysis (MLSA) of Bradyrhizobium strains: Revealing high diversity of tropical diazotrophic symbiotic bacteria. Braz. J. Microbiol. 2012, 43, 698–710. [Google Scholar] [CrossRef]
- Vinuesa, P.; León-Barrios, M.; Silva, C.; Willems, A.; Jarabo-Lorenzo, A.; Pérez-Galdona, R.; Werner, D.; Martínez-Romero, E. Bradyrhizobium canariense sp. nov., an acid-tolerant endosymbiont that nodulates endemic genistoid legumes (Papilionoideae: Genisteae) from the Canary Islands, along with Bradyrhizobium japonicum bv. genistearum, Bradyrhizobium genospecies alpha and Bradyrhizobium genospecies beta. Int. J. Syst. Evol. Microbiol. 2005, 55, 569–575. [Google Scholar] [CrossRef]
- Delamuta, J.R.M.; Menna, P.; Ribeiro, R.A.; Hungria, M. Phylogenies of symbiotic genes of Bradyrhizobium symbionts of legumes of economic and environmental importance in Brazil support the definition of the new symbiovars pachyrhizi and sojae. Syst. Appl. Microbiol. 2017, 40, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Benzine, J.; Shelobolina, E.; Xiong, M.Y.; Kennedy, D.W.; McKinley, J.P.; Lin, X.; Roden, E.E. Fe-phyllosilicate redox cycling organisms from a redox transition zone in Hanford 300 Area sediments. Front. Microbiol. 2013, 4, 388. [Google Scholar] [CrossRef] [PubMed]
- Van Berkum, P.; Elia, P.; Song, Q.; Eardly, B.D. Development and application of a multilocus sequence analysis method for the identification of genotypes within genus Bradyrhizobium and for establishing nodule occupancy of soybean (Glycine max L. Merr). Mol. Plant Microbe Interact. 2012, 25, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Minamisawa, K.; Uchiumi, T.; Sasamoto, S.; Watanabe, A.; Idesawa, K.; Iriguchi, M.; Kawashima, K.; et al. Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110 (supplement). DNA Res. 2002, 9, 225–256. [Google Scholar] [CrossRef] [PubMed]
- Scharf, B.; Schuster-Wolff-Bühring, H.; Rachel, R.; Schmitt, R. Mutational analysis of the Rhizobium lupini H13-3 and Sinorhizobium meliloti flagellin genes: Importance of flagellin A for flagellar filament structure and transcriptional regulation. J. Bacteriol. 2001, 183, 5334–5342. [Google Scholar] [CrossRef] [PubMed]
- Tambalo, D.D.; Bustard, D.E.; Del Bel, K.L.; Koval, S.F.; Khan, M.F.; Hynes, M.F. Characterization and functional analysis of seven flagellin genes in Rhizobium leguminosarum bv. viciae. BMC Microbiol. 2010, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.P.; Sobral, B.W.; Dickerman, A.W. A robust species tree for the alphaproteobacteria. J. Bacteriol. 2007, 189, 4578–4586. [Google Scholar] [CrossRef]
- Gupta, R.S.; Mok, A. Phylogenomics and signature proteins for the alpha proteobacteria and its main groups. BMC Microbiol. 2007, 7, 106. [Google Scholar] [CrossRef]
- Simon, M.; Scheuner, C.; Meier-Kolthoff, J.P.; Brinkhoff, T.; Wagner-Döbler, I.; Ulbrich, M.; Klenk, H.P.; Schomburg, D.; Petersen, J.; Göker, M. Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non-marine habitats. ISME J. 2017, 11, 1483–1499. [Google Scholar] [CrossRef]
- Braeken, K.; Daniels, R.; Vos, K.; Fauvart, M.; Bachaspatimayum, D.; Vanderleyden, J.; Michiels, J. Genetic determinants of swarming in Rhizobium etli. Microb. Ecol. 2008, 55, 54–64. [Google Scholar] [CrossRef]
- Soto, M.J.; Fernández-Pascual, M.; Sanjuan, J.; Olivares, J. A fadD mutant of Sinorhizobium meliloti shows multicellular swarming migration and is impaired in nodulation efficiency on alfalfa roots. Mol. Microbiol. 2002, 43, 371–382. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Genus (Sequenced Species No.) | Subpolar Flagellum | Lateral Flagellum |
|---|---|---|---|
| Aurantimonadaceae | Aurantimonas (2) | - | + |
| Aureimonas (5) | - | + | |
| Fulvimarina (2) | - | + | |
| Martelella (2) | n.m./n.d. | ||
| Bartonellaceae | Bartonella (27) | - | + |
| Beijerinckiaceae | Beijerinckia (2) | - | + |
| Chelatococcus (2) | - | + | |
| Methylocapsa (3) | - | + | |
| Methylocella (1) | - | + | |
| Methyloferula (1) | - | + | |
| Bradyrhizobiaceae | Afipia (5) | + | - |
| Bosea (4) | + | +/- | |
| Bradyrhizobium (25) | + | +/- | |
| Nitrobacter (3) | + | - | |
| Oligotropha (1) | + | - | |
| Rhodoblastus (1) | - | + | |
| Rhodopseudomonas (2) | + | +/- | |
| Salinarimonas (1) | + | + | |
| Tardiphaga (1) | + | + | |
| Brucellaceae | Brucella (11) | - | + |
| Ochrobactrum (9) | - | + | |
| Cohaesibacteraceae | Cohaesibacter (1) | + | + |
| Hyphomicrobiaceae | Blastochloris (1) | + | - |
| Cucumibacter (1) | + | - | |
| Devosia (11) | + | - | |
| Filomicrobium (1) | - | + | |
| Hyphomicrobium (5) | - | + | |
| Maritalea (1) | + | - | |
| Pelagibacterium (2) | + | - | |
| Prosthecomicrobium (1) | + | - | |
| Rhodomicrobium (2) | - | + | |
| Meganema (1) | n.m./n.d. | ||
| Methylobacteriaceae | Methylobacterium (20) | + | +/- |
| Microvirga (6) | +/- | + | |
| Neomegalonema (1) | n.d. | ||
| Methylocystaceae | Methylocystis (3) | - | + |
| Methylosinus (1) | - | + | |
| Pleomorphomonas (2) | + | +/- | |
| Terasakiella (1) | + | - | |
| Phyllobacteriaceae | Aminobacter (1) | - | + |
| Aquamicrobium (2) | - | + | |
| Hoeflea (2) | - | + | |
| Mesorhizobium (17) | - | + | |
| Nitratireductor (5) | - | + | |
| Paramesorhizobium (1) | - | + | |
| Pseudaminobacter (2) | - | + | |
| Rhizobiaceae | Kaistia (3) | n.m./n.d. | |
| Agrobacterium (9) | - | + | |
| Neorhizobium (1) | - | + | |
| Pararhizobium (2) | - | + | |
| Pseudorhizobium (1) | - | + | |
| Rhizobium (42) | - | + | |
| Ensifer (5) | - | + | |
| Sinorhizobium (6) | - | + | |
| Xanthobacteraceae | Ancylobacter (1) | - | + |
| Azorhizobium (2) | - | + | |
| Pseudoxanthobacter (1) | - | + | |
| Starkeya (1) | n.m./n.d. | ||
| Xanthobacter (1) | n.d. | ||
| unclassified Rhizobiales | Bauldia (1) | n.d. | |
| Methylobrevis (1) | + | - | |
| Methyloceanibacter (5) | n.d. | ||
| Methyloligella (1) | n.m./n.d. | ||
| Pseudorhodoplanes (1) | + | - | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrido-Sanz, D.; Redondo-Nieto, M.; Mongiardini, E.; Blanco-Romero, E.; Durán, D.; Quelas, J.I.; Martin, M.; Rivilla, R.; Lodeiro, A.R.; Althabegoiti, M.J. Phylogenomic Analyses of Bradyrhizobium Reveal Uneven Distribution of the Lateral and Subpolar Flagellar Systems, Which Extends to Rhizobiales. Microorganisms 2019, 7, 50. https://doi.org/10.3390/microorganisms7020050
Garrido-Sanz D, Redondo-Nieto M, Mongiardini E, Blanco-Romero E, Durán D, Quelas JI, Martin M, Rivilla R, Lodeiro AR, Althabegoiti MJ. Phylogenomic Analyses of Bradyrhizobium Reveal Uneven Distribution of the Lateral and Subpolar Flagellar Systems, Which Extends to Rhizobiales. Microorganisms. 2019; 7(2):50. https://doi.org/10.3390/microorganisms7020050
Chicago/Turabian StyleGarrido-Sanz, Daniel, Miguel Redondo-Nieto, Elías Mongiardini, Esther Blanco-Romero, David Durán, Juan I. Quelas, Marta Martin, Rafael Rivilla, Aníbal R. Lodeiro, and M. Julia Althabegoiti. 2019. "Phylogenomic Analyses of Bradyrhizobium Reveal Uneven Distribution of the Lateral and Subpolar Flagellar Systems, Which Extends to Rhizobiales" Microorganisms 7, no. 2: 50. https://doi.org/10.3390/microorganisms7020050
APA StyleGarrido-Sanz, D., Redondo-Nieto, M., Mongiardini, E., Blanco-Romero, E., Durán, D., Quelas, J. I., Martin, M., Rivilla, R., Lodeiro, A. R., & Althabegoiti, M. J. (2019). Phylogenomic Analyses of Bradyrhizobium Reveal Uneven Distribution of the Lateral and Subpolar Flagellar Systems, Which Extends to Rhizobiales. Microorganisms, 7(2), 50. https://doi.org/10.3390/microorganisms7020050