Geographical Isolation, Buried Depth, and Physicochemical Traits Drive the Variation of Species Diversity and Prokaryotic Community in Three Typical Hypersaline Environments


Abstract
:1. Introduction
2. Materials and Methods
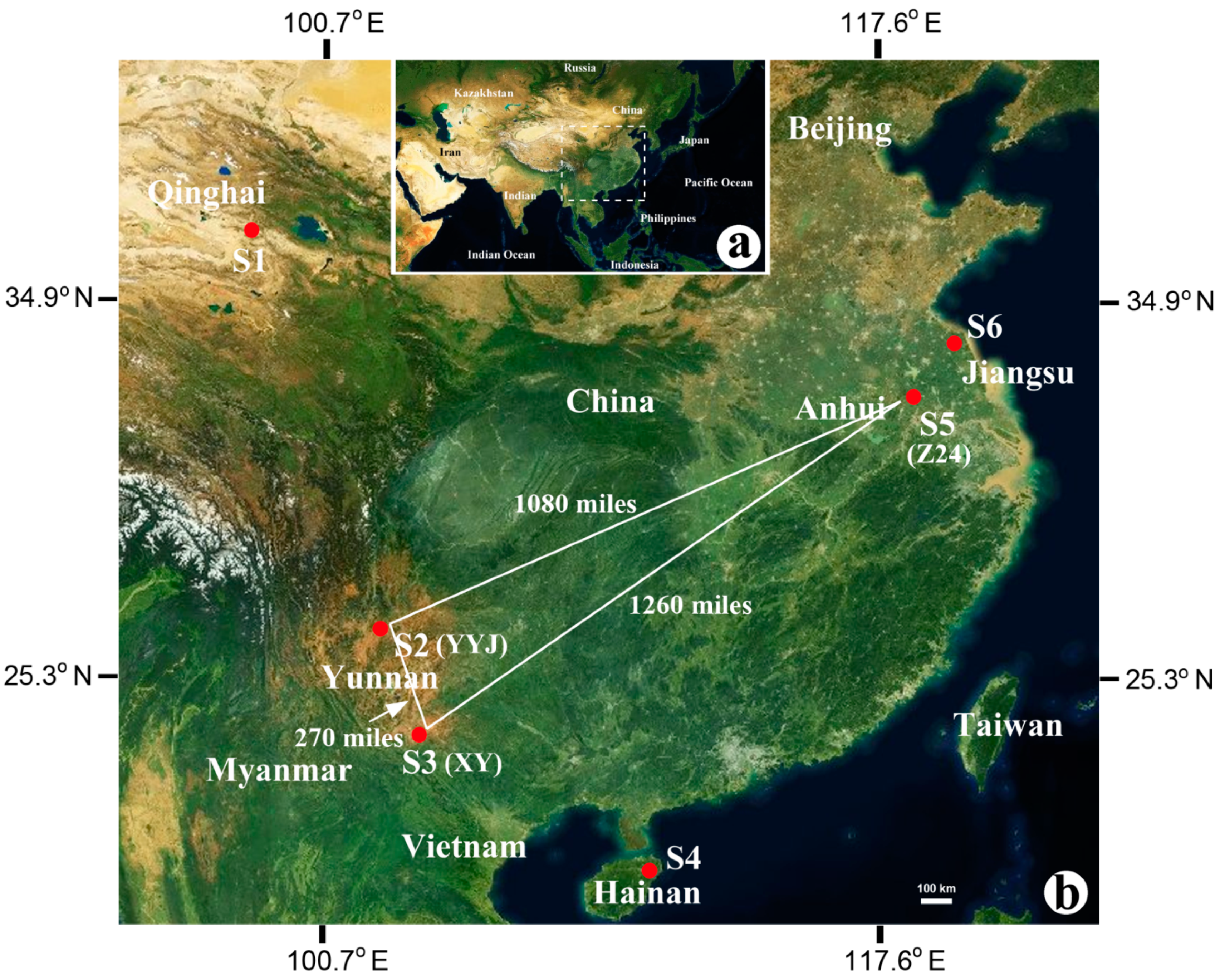
2.1. Sample Collection
2.2. Chemical Analyses of Salt Mines
2.3. Total Environmental DNA Extraction
2.4. Clone Library Construction and DNA Sequencing
2.5. High-Throughput Sequencing
2.6. Operational Taxonomic Unit (OTU) Assignment
2.7. Species Diversity Evaluation
3. Results
3.1. Determination of the Physicochemical Parameters
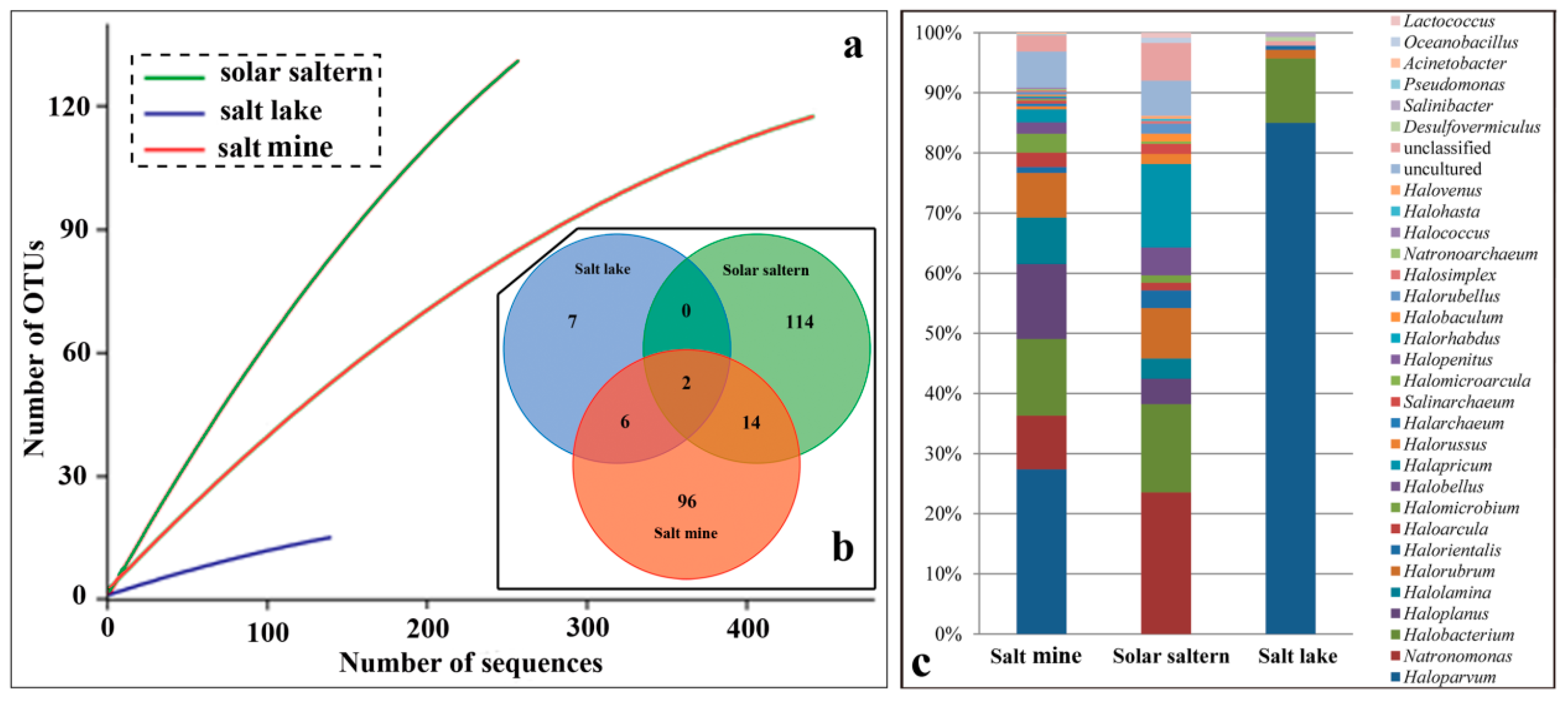
3.2. Type of Hypersaline Environments and Microbial Communities
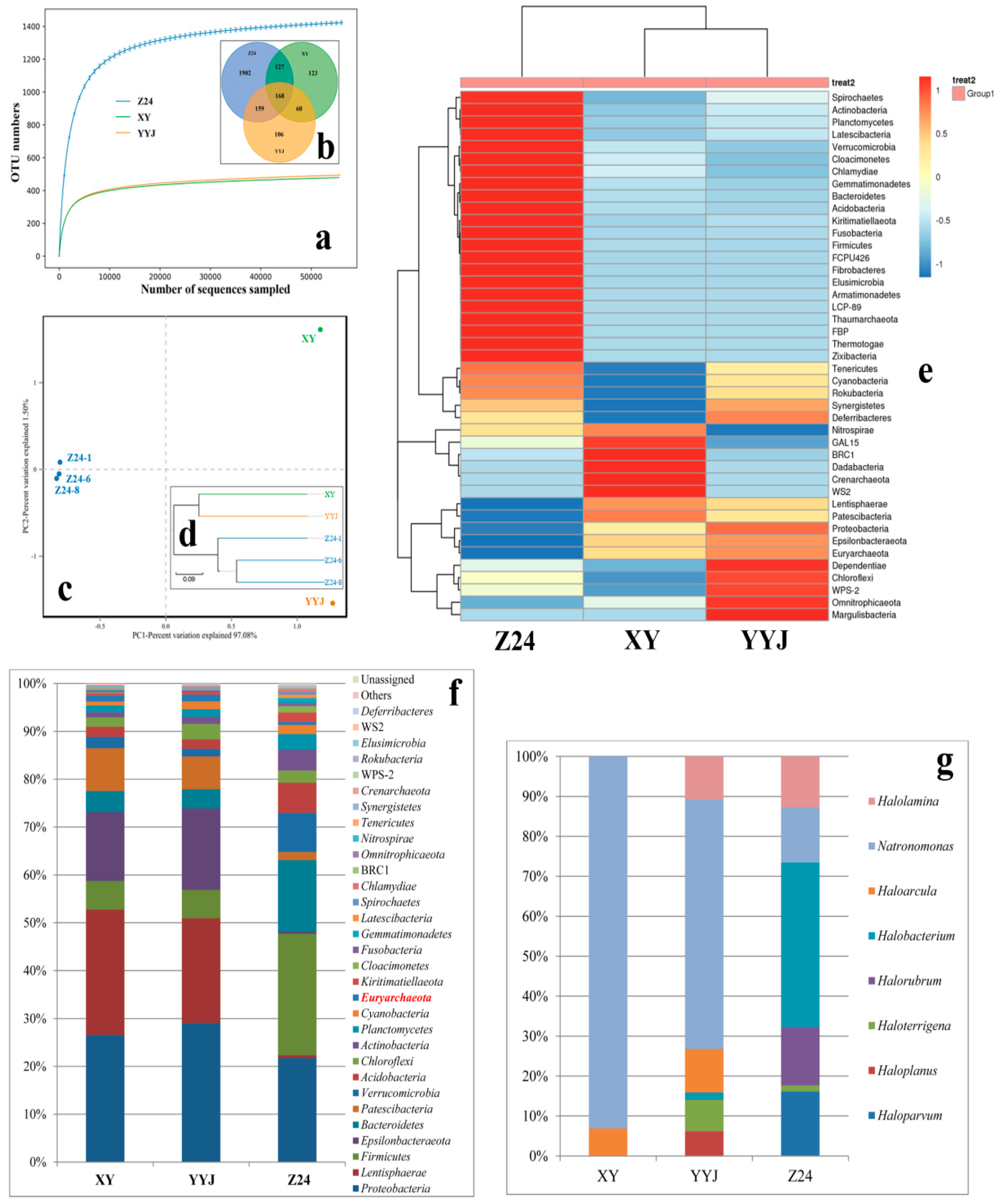
3.3. Geographical Isolation and Microbial Communities
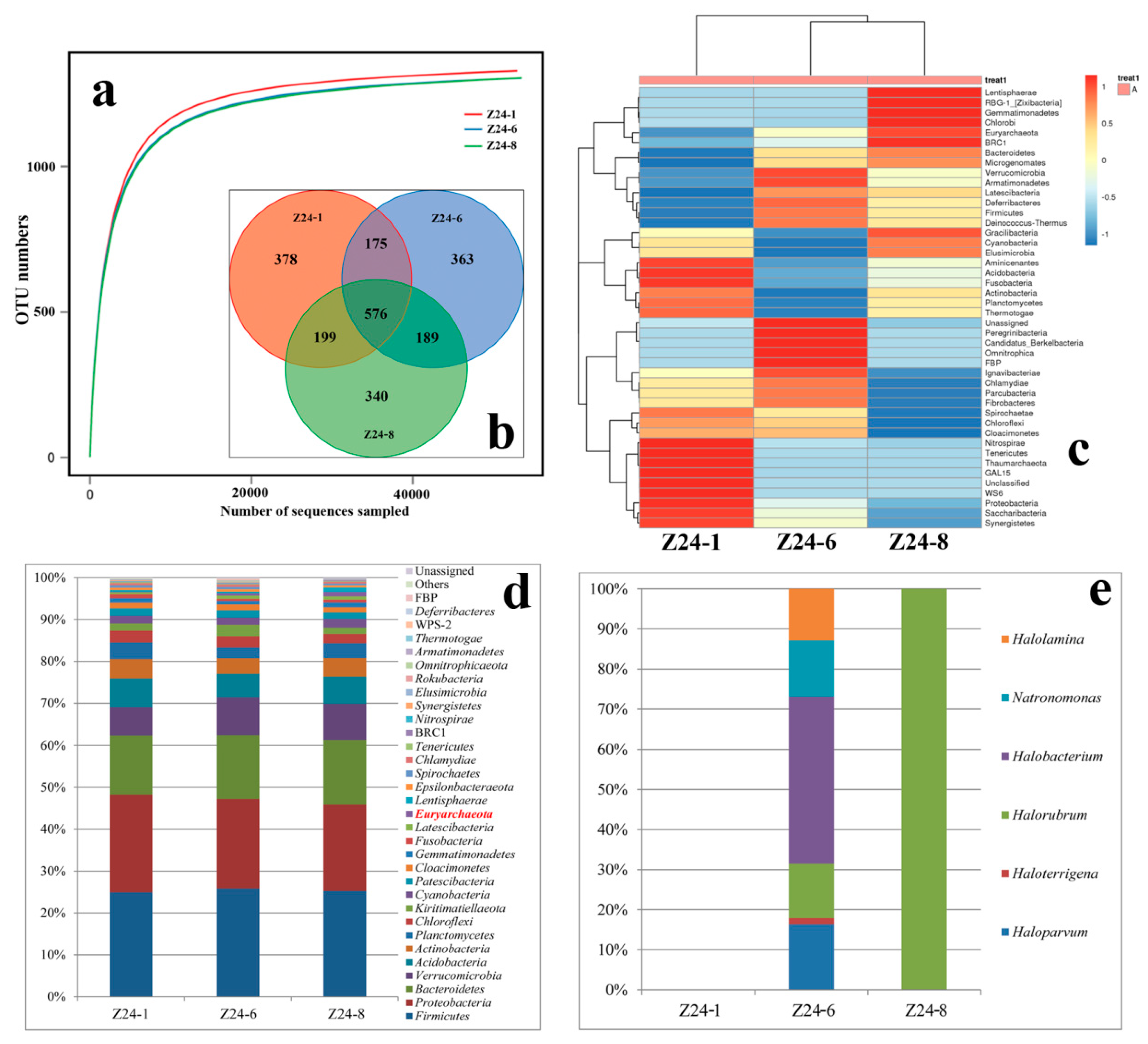
3.4. The Buried Depth and Microbial Communities
4. Discussion
4.1. Species Diversity Varies from Different Types of Hypersaline Environment
4.2. Geographical Isolation Influences the Microbial Community
4.3. Species Diversity Along with Different Buried Depths
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oren, A. Halophilic microbial communities and their environments. Curr. Opin. Biotechnol. 2015, 33, 119–124. [Google Scholar] [CrossRef]
- Di Meglio, L.; Santos, F.; Gomariz, M.; Almansa, C.; Lopez, C.; Anton, J.; Nercessian, D. Seasonal dynamics of extremely halophilic microbial communities in three Argentinian salterns. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, R.; Zhang, X.; Liu, J.; Long, Q.F.; Chen, L.S.; Liu, D.L.; Zhu, D.R. Microbial community structure and diversity within hypersaline Keke Salt Lake environments. Can. J. Microbiol. 2017, 63, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.H.; Hussein, E.I.; Shakhatreh, M.A.K.; Cornelison, C.T. Microbial community analysis of the hypersaline water of the Dead Sea using high-throughput amplicon sequencing. MicrobiologyOpen 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Barbero, M.D.; Martinez, J.M.; Almansa, C.; Rodriguez, N.; Villamor, J.; Gomariz, M.; Escudero, C.; Rubin, S.D.C.; Anton, J.; Martinez-Garcia, M.; et al. Prokaryotic and viral community structure in the singular chaotropic salt lake Salar de Uyuni. Environ. Microbiol. 2019, 21, 2029–2042. [Google Scholar] [CrossRef] [Green Version]
- Dillon, J.G.; Carlin, M.; Gutierrez, A.; Nguyen, V.; McLain, N. Patterns of microbial diversity along a salinity gradient in the Guerrero Negro solar saltern, Baja CA Sur, Mexico. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Cinar, S.; Mutlu, M.B. Comparative analysis of prokaryotic diversity in solar salterns in eastern Anatolia (Turkey). Extremophiles 2016, 20, 589–601. [Google Scholar] [CrossRef]
- Vreeland, R.H.; Piselli, A.F.; McDonnough, S.; Meyers, S.S. Distribution and diversity of halophilic bacteria in a subsurface salt formation. Extremophiles 1998, 2, 321–331. [Google Scholar] [CrossRef]
- Yildiz, E.; Ozcan, B.; Caliskan, M. Isolation, Characterization and Phylogenetic Analysis of Halophilic Archaea from a Salt Mine in Central Anatolia (Turkey). Pol. J. Microbiol. 2012, 61, 111–117. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, Z.G.; Wang, Y.X.; Schneegurt, M.A.; Li, Z.Y.; Lai, Y.H.; Zhang, S.Y.; Wen, M.L.; Cui, X.L. Comparative molecular analysis of the prokaryotic diversity of two salt mine soils in southwest China. J. Basic Microb. 2013, 53, 942–952. [Google Scholar] [CrossRef]
- Almeida-Dalmet, S.; Litchfield, C.D.; Gillevet, P.; Baxter, B.K. Differential Gene Expression in Response to Salinity and Temperature in a Haloarcula Strain from Great Salt Lake, Utah. Genes 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.P.; Liu, Y.; Miao, L.L.; Wang, F.; Chu, L.M.; Wang, J.L.; Liu, Z.P. Prokaryotic Community Structure Driven by Salinity and Ionic Concentrations in Plateau Lakes of the Tibetan Plateau. Appl. Environ. Microb. 2016, 82, 1846–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, B.A.; Lowenstein, T.K.; Timofeeff, M.N.; Parker, M.A. Halophilic Archaea cultured from ancient halite, Death Valley, California. Environ. Microbiol. 2010, 12, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, M.B.; Martiez-Garcia, M.; Santos, F.; Pena, A.; Guven, K.; Anton, J. Prokaryotic diversity in Tuz Lake, a hypersaline environment in Inland Turkey. FEMS Microbiol. Ecol. 2008, 65, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Vafina, G.; Zainutdinova, E.; Bulatov, E.; Filimonova, M.N. Endonuclease from Gram-negative bacteria Serratia marcescens is as effective as pulmozyme in the hydrolysis of DNA in sputum. Front. Pharmacol. 2018, 9, 114. [Google Scholar] [CrossRef]
- Lizama, C.; Monteoliva-Sanchez, M.; Prado, B.; Ramos-Cormenzana, A.; Weckesser, J.; Campos, V. Taxonomic study of extreme halophilic archaea isolated from the Salar de Atacama, Chile. Syst. Appl. Microbiol. 2001, 24, 464–474. [Google Scholar] [CrossRef] [Green Version]
- Derakhshani, H.; Tun, H.M.; Khafipour, E. An extended single-index multiplexed 16S rRNA sequencing for microbial community analysis on MiSeq illumina platforms. J. Basic Microb. 2016, 56, 321–326. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Dubois, P.C.A.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.R.; Adany, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.C.; Dong, H.L.; Zhang, G.X.; Yu, B.S.; Chapman, L.R.; Fields, M.W. Microbial diversity in water and sediment of Lake Chaka, an athalassohaline lake in northwestern China. Appl. Environ. Microb. 2006, 72, 3832–3845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botero, C.A.; Dor, R.; McCain, C.M.; Safran, R.J. Environmental harshness is positively correlated with intraspecific divergence in mammals and birds. Mol. Ecol. 2014, 23, 259–268. [Google Scholar] [CrossRef]
- Ochsenreiter, T.; Pfeifer, F.; Schleper, C. Diversity of Archaea in hypersaline environments characterized by molecular-phylogenetic and cultivation studies. Extremophiles 2002, 6, 267–274. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Sample | Habitat | Location | Altitude (m) | Buried Depth (m) |
|---|---|---|---|---|---|
| S1 | CK | Salt lake | N 36°39′37.53″ E 99°08′49.95″ | 3059 | 0 |
| S2 | YYJ | Salt mine | N 25°16′49.61″ E 101°54′02.75″ | 2059 | unknown |
| S3 | XY | Salt mine | N 23°24′30.54″ E 100°39′56.51″ | 967 | unknown |
| S4 | HND | Solar saltern | N 19°09′23.06″ E 108°40′45.37″ | 35 | 0 |
| S5 | Z24-1 | Salt mine | N 32°30′23.88″ E 117°29′44.16″ | 73 | 410 |
| S5 | Z24-6 | Salt mine | N 32°30′23.88″ E 117°29′44.16″ | 73 | 370 |
| S5 | Z24-8 | Salt mine | N 32°30′23.88″ E 117°29′44.16″ | 73 | 310 |
| S6 | LYG | Solar saltern | N 34°40′43.31″ E 119°13′47.07″ | 30 | 0 |
| Name | Sequence (5′–3′) |
|---|---|
| F8 | TTGATCCTGCCGGAGGCCATTG |
| R1462 | ATCCAGCCGCAGATTCCCCTAC |
| F338 | ACTCCTACGGGAGGCAGCA |
| R806 | GGACTACHVGGGTWTCTAAT |
| Item | Z24-1 | Z24-6 | Z24-8 | YYJ | XY |
|---|---|---|---|---|---|
| Fe (mg/kg) | 151.7 | 180.8 | 278.9 | 32.79 | 32.17 |
| Mg (mg/kg) | 243.9 | 92.16 | 609.0 | 24.53 | 28.76 |
| Ca (mg/kg) | 1417 | 1259 | 1259 | 6775 | 4460 |
| K (mg/kg) | 198.0 | 304.2 | 332.1 | 163.9 | 311.3 |
| Na (%) | 73.32 | 75.07 | 70.22 | 76.40 | 74.13 |
| Mn (μg/kg) | 2874.24 | 2531.11 | 6970.51 | 775.70 | 891.31 |
| Cl− (mg/kg) | 733,610 | 716,109 | 730,420 | 719,048 | 709,146 |
| SO42− (mg/kg) | 483.84 | 7075.31 | 1893.51 | 28,982.68 | 10,735.84 |
| Soluble component (w/w) | 72.13 | 72.13 | 72.13 | 17.8 | 27.2 |
| pH | 7.02 | 7.02 | 7.02 | 7.08 | 6.81 |
| C (%) | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 |
| N (%) | 0.23 | 0.21 | 0.32 | 0.36 | 0.40 |
| Source | ACE | Chao1 | Simpson | Shannon | Good’s Coverage |
|---|---|---|---|---|---|
| Salt mine | 427.82 | 301.88 | 0.08 | 3.57 | 0.83 |
| Salt lake | 111.54 | 42.50 | 0.62 | 0.98 | 0.92 |
| Solar saltern | 1085.00 | 382.16 | 0.02 | 4.36 | 0.62 |
| Sample | ACE | Chao1 | Simpson | Shannon |
|---|---|---|---|---|
| XY | 511.75 | 534.89 | 0.05 | 4.08 |
| YYJ | 527.81 | 569.56 | 0.06 | 4.08 |
| Z24 | 2364.64 | 2366.11 | 0.01 | 6.65 |
| Sample | ACE | Chao1 | Simpson | Shannon |
|---|---|---|---|---|
| Z24-1 | 1347.94 | 1357.32 | 0.0090 | 6.14 |
| Z24-6 | 1322.35 | 1322.60 | 0.0088 | 6.15 |
| Z24-8 | 1332.73 | 1340.65 | 0.0089 | 6.09 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Xu, Y.; Helfant, L. Geographical Isolation, Buried Depth, and Physicochemical Traits Drive the Variation of Species Diversity and Prokaryotic Community in Three Typical Hypersaline Environments. Microorganisms 2020, 8, 120. https://doi.org/10.3390/microorganisms8010120
Chen S, Xu Y, Helfant L. Geographical Isolation, Buried Depth, and Physicochemical Traits Drive the Variation of Species Diversity and Prokaryotic Community in Three Typical Hypersaline Environments. Microorganisms. 2020; 8(1):120. https://doi.org/10.3390/microorganisms8010120
Chicago/Turabian StyleChen, Shaoxing, Yao Xu, and Libby Helfant. 2020. "Geographical Isolation, Buried Depth, and Physicochemical Traits Drive the Variation of Species Diversity and Prokaryotic Community in Three Typical Hypersaline Environments" Microorganisms 8, no. 1: 120. https://doi.org/10.3390/microorganisms8010120
APA StyleChen, S., Xu, Y., & Helfant, L. (2020). Geographical Isolation, Buried Depth, and Physicochemical Traits Drive the Variation of Species Diversity and Prokaryotic Community in Three Typical Hypersaline Environments. Microorganisms, 8(1), 120. https://doi.org/10.3390/microorganisms8010120