1. Introduction
Phytophthora capsici, an important plant-pathogenic oomycete of
Phytophthora, damages various crops including members of Solanaceae (such as pepper and tomato) and Cucurbitaceae (such as cucumber and pumpkin) [
1]. Pepper blight caused by this pathogen was first discovered in the United States in 1918, and the disease can be catastrophic in a short time, endangering the entire growth period of pepper [
2]. At present, metalaxyl is the main fungicide used for preventing and controlling plant diseases such as pepper blight. However, because this type of agent is a site-specific inhibitor and has a single site of action on pathogens,
Phytophthora can develop resistance as they are susceptible to mutations [
3,
4,
5]. Metalaxyl resistance is mainly found in
Phytophthora species such as
P. capsici,
P. infestans, and
P. parasitica. A metalaxyl-resistant strain of
P. infestans was discovered in the Netherlands in 1980 [
6]. Subsequently, European and American countries reported metalaxyl-resistant strains of
P. capsici [
7,
8,
9]. China also reported resistant
P. capsici in the 1960s [
10]. To date, metalaxyl-resistant strains of
P. capsici have been successively found in Anhui, Gansu, and Yunnan Provinces [
10,
11,
12,
13].
There are some studies on the resistance mechanism of
Phytophthora to metalaxyl. Chen et al. [
14] revealed two evolutionary pathways of resistance involving the
RPA190 gene. The results of their research indicate that changes in the activity of
Phytophthora RNA polymerase are important resistance mechanisms. Similar results were also confirmed in
P. infestans. The diversity of the RNA polymerase I large subunit sequence of
P. infestans plays a key role in its resistance to metalaxyl [
15]. The biological degradation of metalaxyl by
Phytophthora is another resistance mechanism. The RNA polymerase activity of sensitive strains was shown to be significantly inhibited by metalaxyl, while resistant strains showed only a slight inhibitory effect and had a certain degradation effect on metalaxyl [
14].
Although crop losses caused by
P. capsici have increased in recent years, we know very little about the molecular basis of its pathogenicity in peppers. Therefore, excavation of metalaxyl-resistance genes of
P. capsici at the molecular level has become an important means of developing resistance to pepper blight. With the decrease in the cost of high-throughput sequencing, transcriptome sequencing has been widely used in molecular biology research and has become one of the most commonly used high-throughput sequencing technologies [
16,
17]. It has a number of advantages, such as a wide range of applications, good repeatability, good sensitivity, and high sequencing throughput. It can be used to discover new genes, optimize structural genes, and analyze differential expression of different transcripts, making it very convenient for differentially expressed gene (DEG) screening. The genome of
P. capsici LT1534 was sequenced in 2012 and its size is 64 MB [
18]. Transcriptome information was compared against the known genome sequence of
P. capsici, and then the sequencing depth and expression of DEGs were analyzed to predict new genes and identify alternative splicing and gene fusion. This is of great significance for elucidating the resistance mechanism of
P. capsici to pesticides. Parada-Rojas et al. [
19] identified and characterized microsatellites in the
P. capsici transcriptome, and then assayed a subset of 50 microsatellites in a diverse set of
P. capsici isolates to find polymorphism. Their findings revealed that 12 microsatellites were useful to characterize the population structure of
P. capsici and were potentially transferable to closely-related
Phytophthora spp. Root rot caused by
P. capsici is the most serious disease in black pepper. Researchers performed transcriptome analysis to identify candidate genes for field tolerance to black pepper root rot [
20]. Sequence analysis revealed a series of proteins involved in black pepper tolerance to root rot, including signal proteins and defense enzymes such as premnaspirodiene oxidase, a phosphatase 2C-like domain protein, a mature protein of the nitrous oxide reductase family, disease resistance protein RGA3, asparaginase, β-glucosidase, a cytochrome P450 signal protein, serine/threonine protein kinase WAG1, and nucleoredoxin 1-1 enzyme. Chen et al. [
21] applied RNA-Seq technology to reveal a large number of genes related to pathogenicity at three stages of mycelia (MY), zoospores (ZO), and germinating cysts with germ tubes (GC) were identified, including 98 predicted effector genes. Therefore, transcriptome sequencing can be used to study the gene functions of pathogenic bacteria and analyze the expression differences between different strains to screen genes related to target traits.
In this study, the sensitive strain SD1 and the resistant mutant strain SD1-9 were used as test materials, and their transcriptomes were sequenced and analyzed. Referring to the published P. capsici LT1534 genome, we performed data splicing, gene expression analysis, and function prediction. Our results will help to understand the molecular mechanism of metalaxyl resistance in P. capsici, provide a theoretical basis for the monitoring and treatment of metalaxyl resistance of P. capsici, and provide an important reference for studying the molecular mechanism of metalaxyl resistance in other Phytophthora species.
3. Discussion
Pathogenicity refers to the strength of the pathogenic infection of the host plant. There are differences in pathogenicity among different strains of
Phytophthora species, which is manifested by different pathogenicities of strains in the same
Phytophthora species to the same host plant and different pathogenicity of the same strain to different host plants. In traditional biology, the pathogenic inoculation methods for
Phytophthora are root inoculation, stem inoculation, and leaf spraying. These methods are relatively simple and fast, but are susceptible to variability due to factors such as host plants, inoculation methods and environmental conditions, which can make their results differ [
24]. Therefore, when measuring the pathogenicity of
Phytophthora on host plants under laboratory conditions, the experimental conditions must be consistent to make the results accurate and reliable. Tian et al. [
25] determined the virulence of
P. capsici isolates on different pumpkin varieties, and reported that
P. capsici isolates were more virulent towards jack-o-lantern pumpkins than processing pumpkins. Similarly, the pathogenicity test results of five
P. capsici strains from different regions of Guangdong on eight hot (sweet) pepper materials indicated that the pathogenicities of different strains were significantly different, with differences in pathogenicity in the same strain in different pepper materials [
26]. In this study, the results of the pathogenicity test showed that each strain had different pathogenicity on different host materials. The phenotypes of strains SD1 and SD1-9 on green peppers and pepper leaves showed that their pathogenicity gradually weakened as the metalaxyl concentration increased.
Previous genetic studies have shown that insensitivity to metalaxyl is regulated by one or two major MEX loci, while other genes are less affected [
27,
28,
29,
30,
31]. Mionor sites that cause chemical insensitivity may include non-specific efflux pumps and detoxification. These functions can be performed by proteins such as the ATP-binding cassette (ABC) transporter and the cytochrome P450 protein, respectively [
32,
33]. However,
P. infestans isolates that have essentially non-specific insensitivity to chemicals have not shown a corresponding increase in transcriptional abundance from genes encoding ABC transporters [
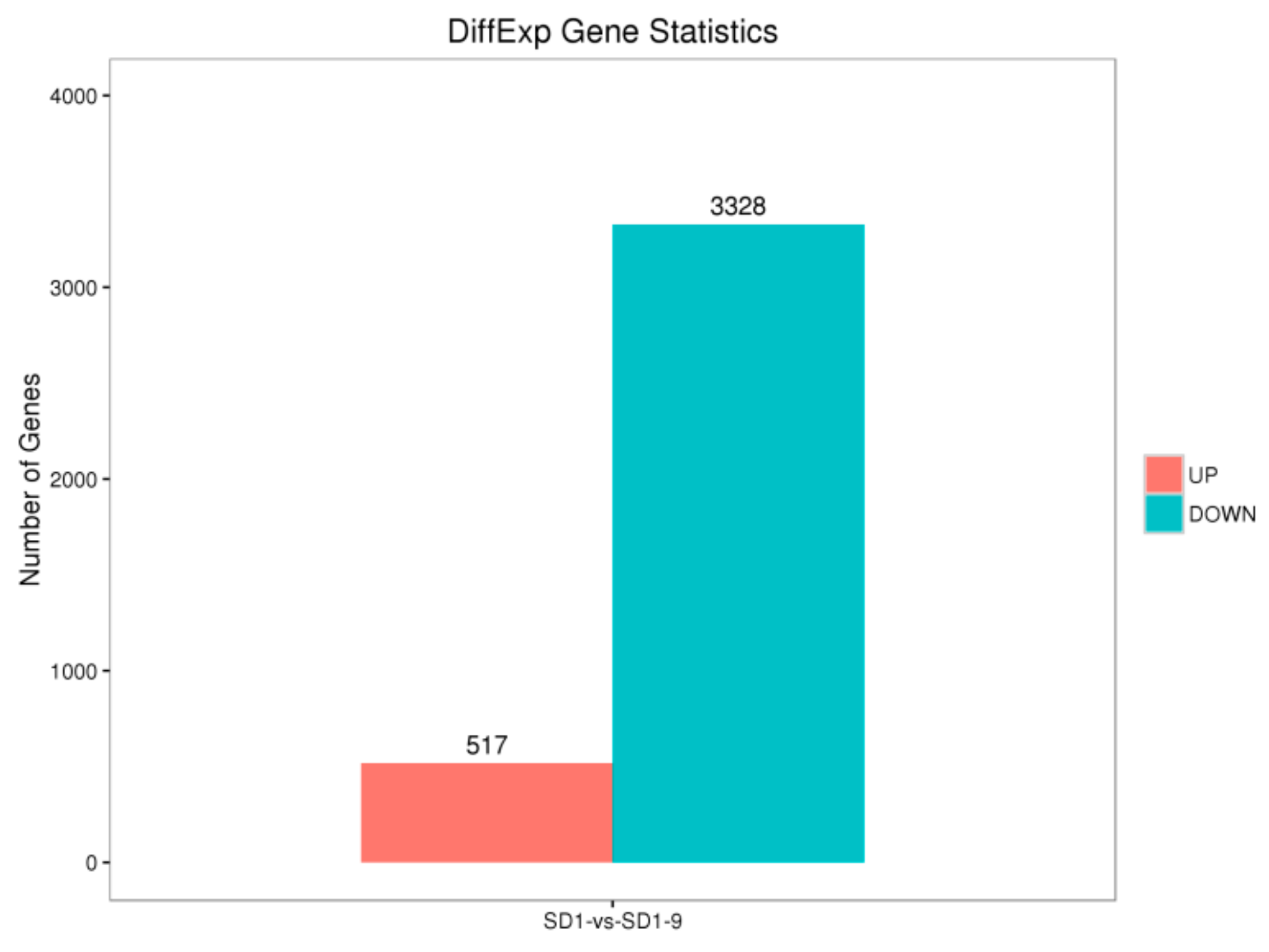
29]. Transcriptome sequencing was used to analyze the gene functions and expression of the sensitive strain SD1 and metalaxyl-resistant mutant strain SD1-9. The results showed that compared with the sensitive strain SD1, the mutant strain SD1-9 treated with metalaxyl had 517 significantly upregulated genes and 3328 significantly downregulated genes. The functions of these DEGs varied. Researchers [
34,
35,
36] have used
3H uridine incorporation biochemical analysis to show that metalaxyl has specific effects on the synthesis of RNA, especially ribosomal RNA (rRNA), while messenger (mRNA) and transport RNA (tRNA) synthesis is less affected. These effects are related to RNA polymerase I (RNApolI) because it transcribes rRNA. In addition, another study showed that metalaxyl exerts its activity when the RNA polymerase complex binds to DNA [
34]. RNA polymerases that rely on eukaryotic DNA are multi-subunit complexes, and up to seven subunits can be shared among the three major RNA polymerases [
37,
38]. Other proteins such as topoisomerases and transcription factors may also affect RNA polymerase activity [
39]. These factors complicate the identification of metalaxyl-targeted RNA polymerase subunits and sequence variations that lead to insensitivity.
4. Materials and Methods
4.1. Tested Strain
P. capsici strain SD1 was isolated from diseased plants with the typical symptoms of pepper blight. The plants were collected from a metalaxyl-free pepper field in Taian, Shandong Province. According to the metalaxyl sensitivity test, the EC50 value was 0.4 µg·mL−1, indicating that the strain was sensitive to metalaxyl. The strain has been deposited to the Fungal Laboratory of Anhui Agricultural University.
Generation of the metalaxyl-resistant mutant strains: The metalaxyl-sensitive
P. capsici strain SD1 was used as the wild type strain. After the strain was cultured on 10% V
8 (V
8 juice 10 mL, H
2O 90 mL, CaCO
3 0.02 g, agar 3 g) plates for 5–7 d, the mycelium discs with a diameter of 6–10 mm were transferred onto 10% V
8 solid medium containing 10 µg·mL
−1 metalaxyl [
40], and incubated in a 25 °C incubator. After 7 d of incubation, the growth of the colonies was observed. After 10–14 d of incubation, if a rapidly growing sector (mycelial growth rate > 6 mm·d
−1) appeared, the colonies in that sector were transferred onto 10% V
8 solid medium containing 10 µg·mL
−1 metalaxyl. A normal mycelial growth rate (3–6 mm·d
−1) was considered to be a sign of acquisition of metalaxyl-resistance and such strains were chosen for further study.
4.2. Pathogenicity Determination
Pathogenicity on different vegetables: Green bell peppers, squash, red peppers, purple eggplants, and cucumbers that were fresh, healthy, and basically the same size were purchased from a local market in Hefei. Strains SD1 and SD1-9 were transferred to 10% V8 medium and cultured in the dark at 25 °C for 5–7 days. Mycelium discs with a diameter of 6–10 mm were cut out. Three small insect needles were used to puncture the surface of each vegetable, and the discs were applied with the mycelium side facing the wound. Absorbent cotton was dipped in water and placed over the wound for 24 h, and each treatment was repeated four times, with strain SD1 as a control. The samples were then cultured in a 25 °C light incubator, and lesion size was measured after 72 h.
Pathogenicity under different metalaxyl concentrations: Pepper leaves (variety Xinsujiao 5) and green peppers that were fresh, healthy and consistent in size were selected. Strains SD1 and SD1-9 were transferred to 10% V8 medium and cultured in the dark at 25 °C for 5–7 days. Mycelium discs with a diameter of 6–10 mm were cut out. Three small insect needles were used to puncture the surfaces of the pepper leaves and green peppers, and the discs were applied with the mycelium side facing the wound. Each wound was sprayed with 1 mL of metalaxyl medicinal solution, at concentrations of 0, 5, and 100 µg·mL−1. After this treatment, absorbent cotton was put on the wound and moistened for 24 h. Each treatment was repeated four times. A blank 10% V8 medium block was inoculated and sprayed with sterile water as a control. After inoculation, the samples were cultured in a 25 °C light incubator. Lesion size was measured after 72 h and the test was repeated twice.
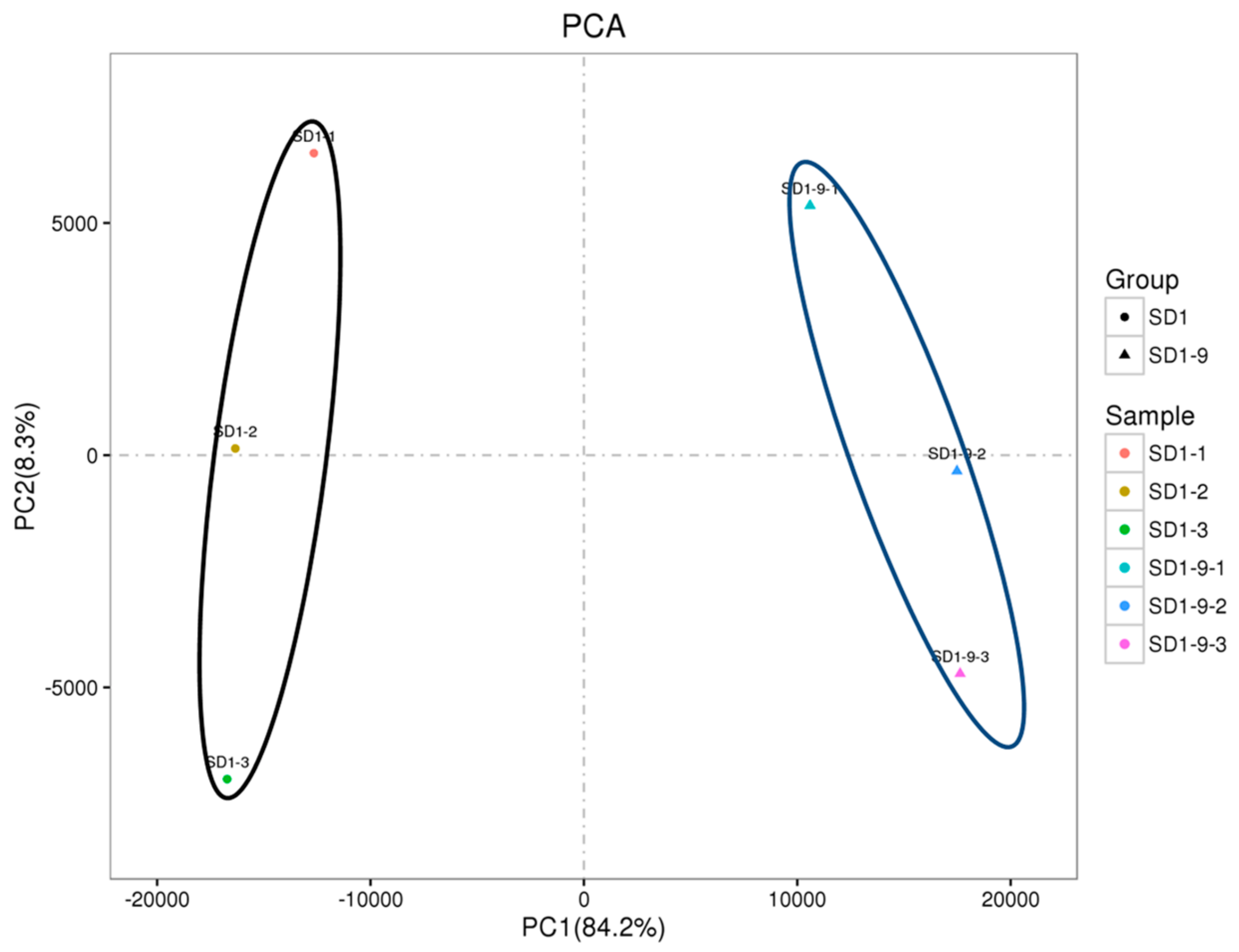
4.3. RNA Extraction, cDNA Library Preparation and Transcriptome Sequencing
The P. capsici strains SD1 and SD1-9 were transferred to 10% V8 medium and cultured in the dark at 25 °C for 5–7 days. Mycelium discs with a diameter of 5 mm were transferred to 10% V8 liquid medium (V8 juice 10 mL, H2O 90 mL, CaCO3 0.02 g). Ten mycelium discs per dish and 10 dishes per strain were placed in the dark at 25 °C for 3 days. After 3 days, the mycelium was filtered with gauze, rinsed with 25% ethanol, and repeatedly washed with deionized water three to four times. The excess water was squeezed out to collect mycelia. The collected mycelia were quickly frozen and ground into powder with liquid nitrogen for RNA extraction. Each strain was repeated three times. RNA extraction was performed using the Takara RNA extraction kit, following the method provided. The concentration and purity of the RNA samples were quantified using a spectrophotometer (NanoDrop ND-1000; Thermo Fisher Scientific, Waltham, MA, USA), and the RNA degradation of the six samples was assessed in 1% agarose gels. The RNA integrity was assessed using the RNA Nano 6000 Assay Kit with the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Sequencing libraries were generated using a NEBNext Ultra™ RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA), following the manufacturer’s recommendations. The effective concentration of each library was accurately quantified using qPCR to ensure library quality, and then cDNA library sequencing was conducted with an Illumina high-throughput sequencing platform (HiSeq™ 2500) by Genedenovo Biotechnology Co., Ltd. (Guangzhou, China).
4.4. RNA-Seq Data Analysis
The constructed libraries were sequenced with an Illumina HiSeq™ 2500. After filtering the sequencing data to obtain clean data, the reads were compared to the P. capsici LT1534 genome and Cufflinks was used to splice the reads to obtain transcript data. Then, the obtained genes were statistically analyzed, and differential expression and functional enrichment analyses were performed. The edgeR software was used to analyze the differences in gene expression between groups. FDR (corrected p-value, indicating significance) and log2FC (FC is the fold change multiple) were used to screen for DEGs. The screening criteria were FDR < 0.05 and |log2FC| > 1. The library corresponding to the SD1 sample was used as a control.
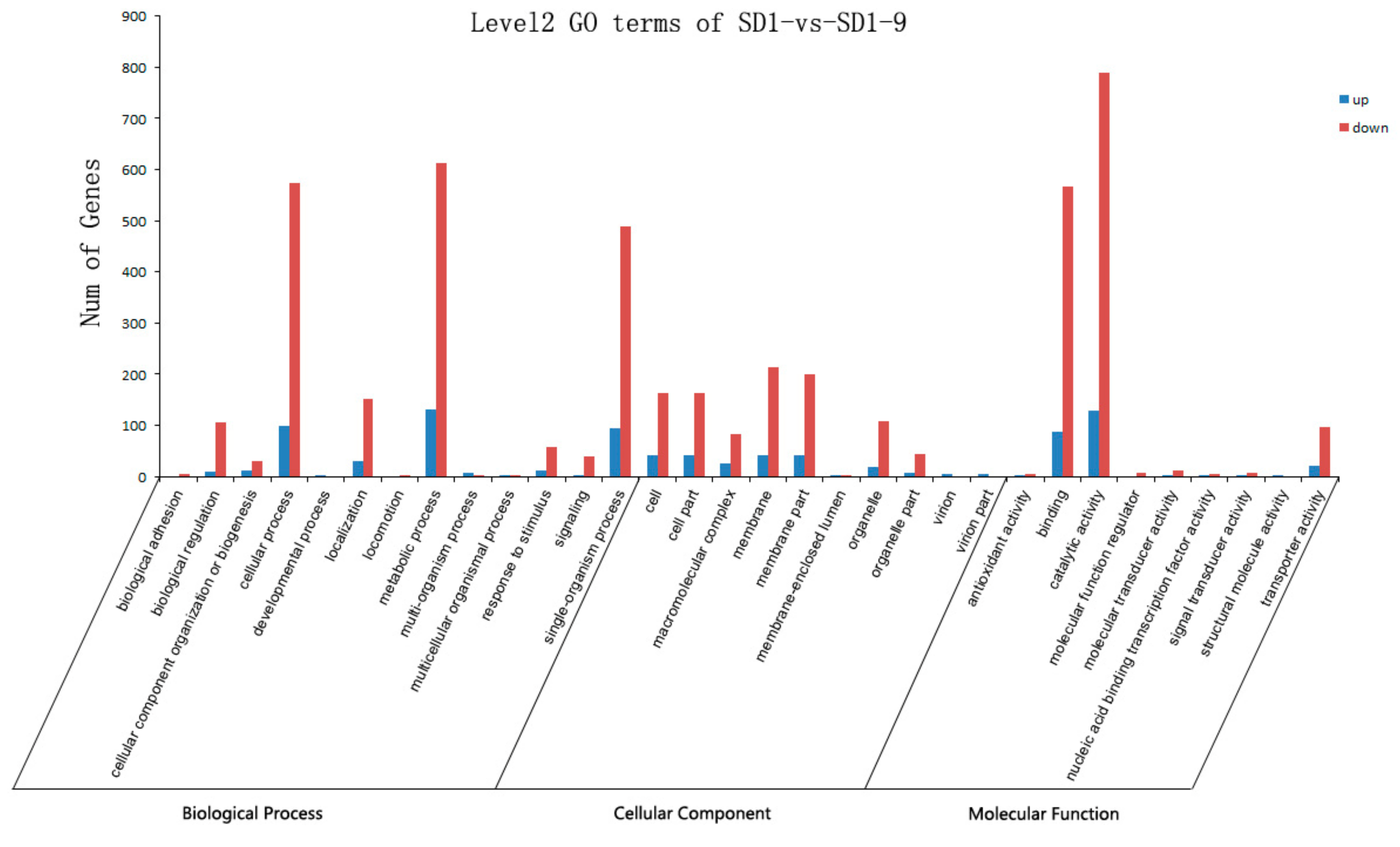
4.5. Gene Annotation
GO annotation is based on the significant enrichment of GO functions to analyze DEGs and related gene modules for bioinformatics analysis. Referring to the annotation information in the NCBI non-redundant (Nr) database, the Blast2 GO software (version 3.0,
https://www.blast2go.com/, BioBam Bioinformatics S.L., Valencia, Spain) was used to perform GO annotation on the core DEGs and the co-expressed gene modules, and the WEGO software was used to annotate and statistically analyze the GO functional classifications of all genes. GO covers three aspects of biology: cellular components, molecular functions, and biological processes. The KEGG database (
https://www.kegg.jp/) can systematically classify and annotate the metabolic pathways of genes and can be used to study genes and their expression information at a general level. The KO-BAS software (version 2.0, KOBAS, Surrey, UK) was used to detect the enrichment of DEGs in KEGG pathways, and the biological functions of specific genes of
P. capsici were considered and evaluated at a macro level.
4.6. qRT-PCR
The remaining
P. capsici SD1 and SD1-9 samples were taken for RNA extraction and purification treatment experiments and then reverse transcribed into cDNA. Ten differentially expressed genes (including genes that were upregulated and downregulated) were randomly selected, and their relative expression levels were verified using a quantitative real-time PCR method. Primer 3.0 was used to design qRT-PCR primers online. The gene names and quantitative primers are shown in
Table S1. Quantification was performed following the operation method of the CFX96 Real-time PCR Detection System and the TB Green™ Premix Ex Taq™ II kit. Each analysis consisted of three biological replicates and three technical replicates per biological replicate. Quantitative fluorescence analysis was performed using the
β-actin gene of
P. capsici as an internal reference. The mRNA levels were normalized with the relative mRNA level of the
P. capsici β-actin gene using the 2
−ΔΔCt method [
41].
4.7. RNA Extraction at Various Growth Stages of P. capsici
Total RNA was extracted from
P. capsici SD1 and SD1-9 at 10 stages, including the mycelia (MY), zoosporangia (SP), zoospores (ZO), cysts (CYST), germinating cysts (GC), and infected pepper leaves at 1.5, 3, 6, 12, and 24 h. The RNA extraction method in each period followed Zhang [
42]. The cDNA was synthesized using the TaKaRa PrimeScript™ RT reagent Kit with gDNA Eraser and used as the template for qRT-PCR. The qRT-PCR primers are listed in
Table S1.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}