Diversity of Bacterial Biosynthetic Genes in Maritime Antarctica

 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
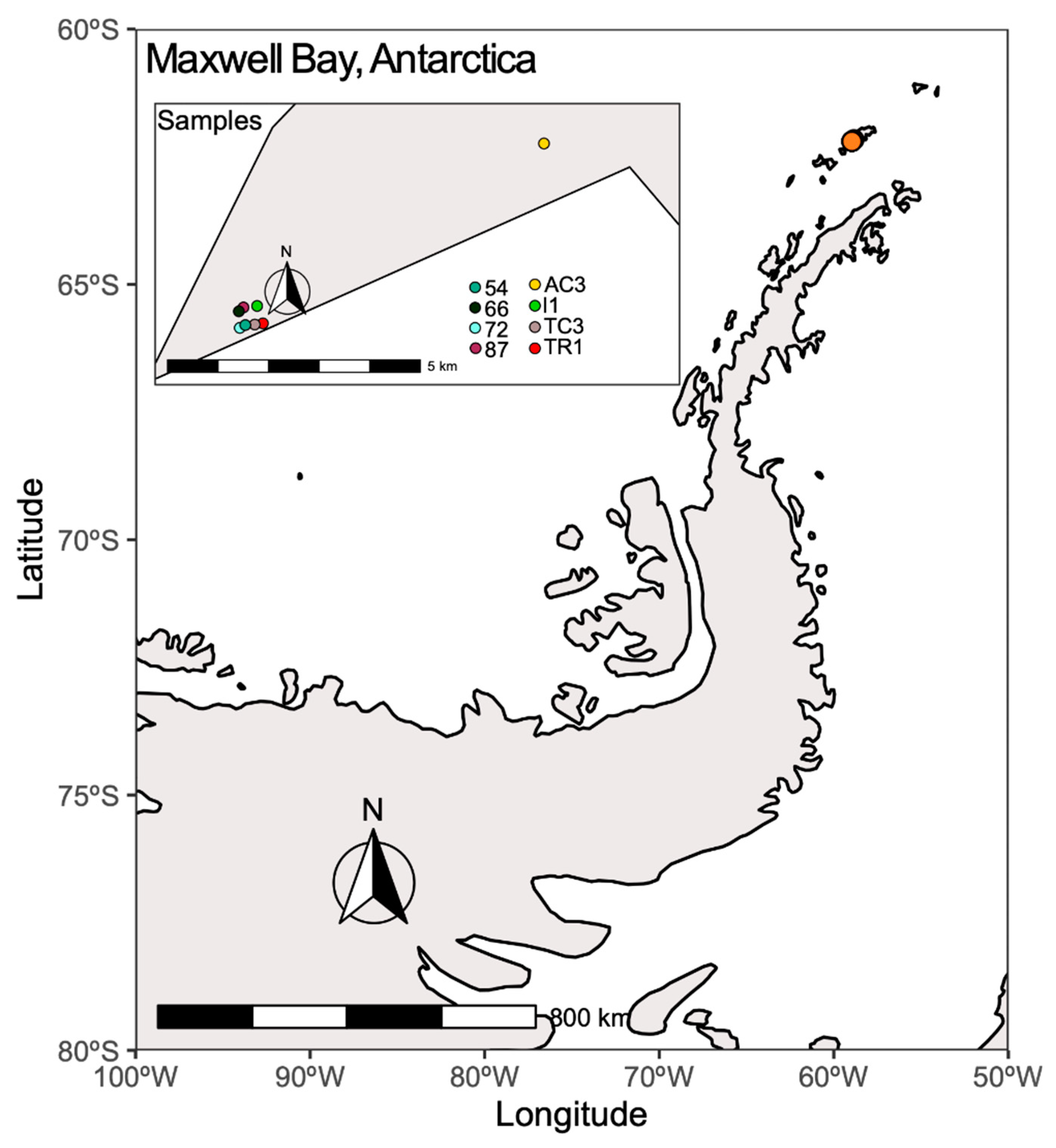
2.1. Sampling Area and Environmental Variables
2.2. DNA Extraction and 16S rRNA Gene Amplification and Sequencing
2.3. Amplification and Sequencing of KS and AD Domain Sequences
2.4. Sequence Analysis and Taxonomic Assignment of 16S rRNA Gene Amplicons
2.5. Sequence Analysis and Taxonomic Assignment of KS and AD Domains
2.6. Phylogenetic Analysis
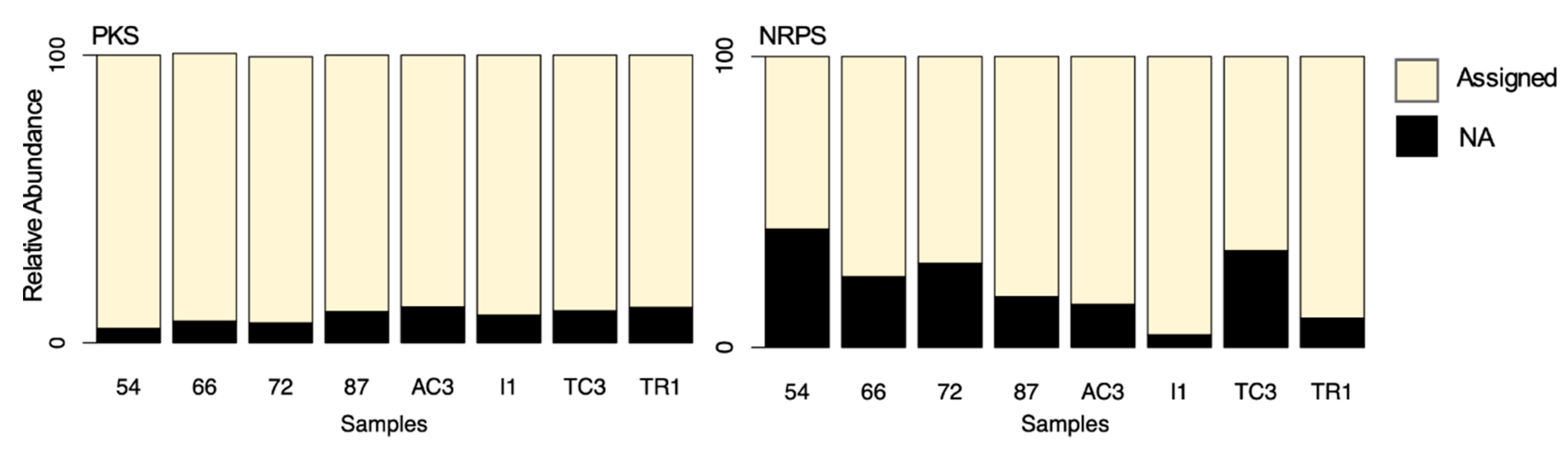
2.7. Functional Annotation of KS and AD Domain Sequences to the MiBIG Database
3. Results and Discussion
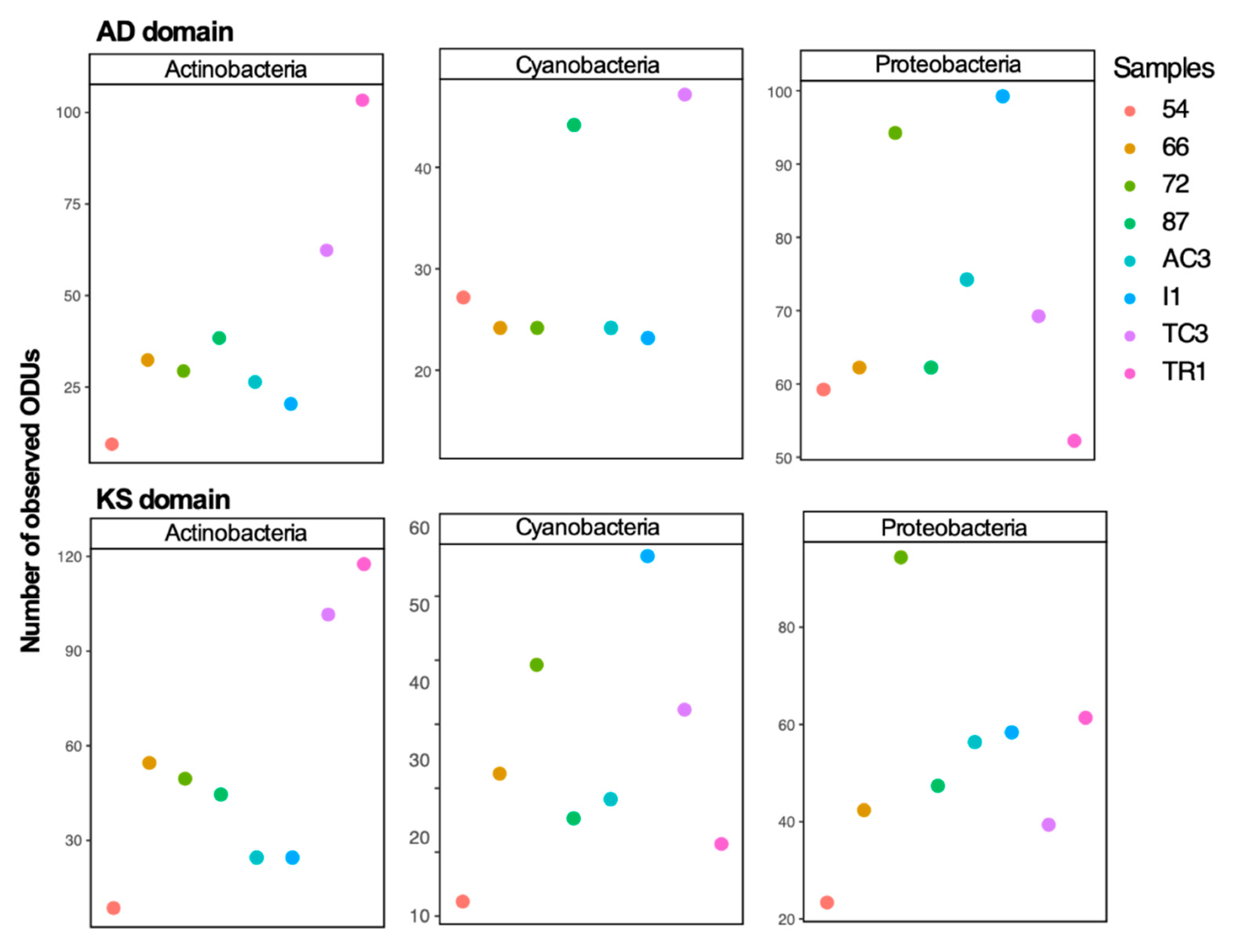
3.1. Diversity of 16S rRNA Gene, KS and AD Domains in the Maxwell Bay Peninsula, Antarctica
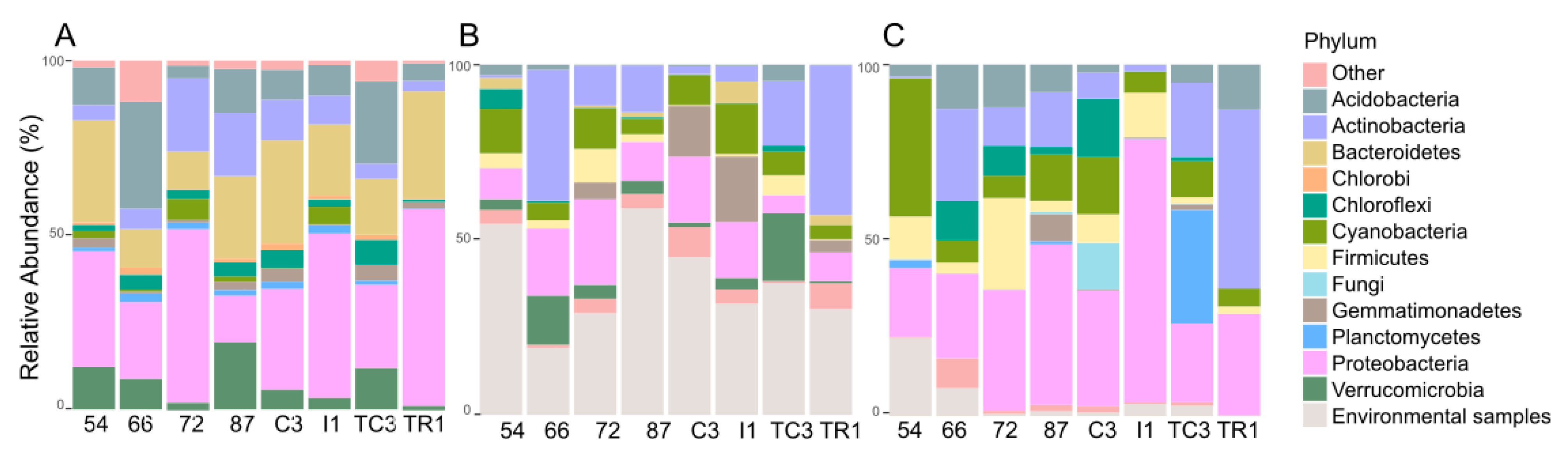
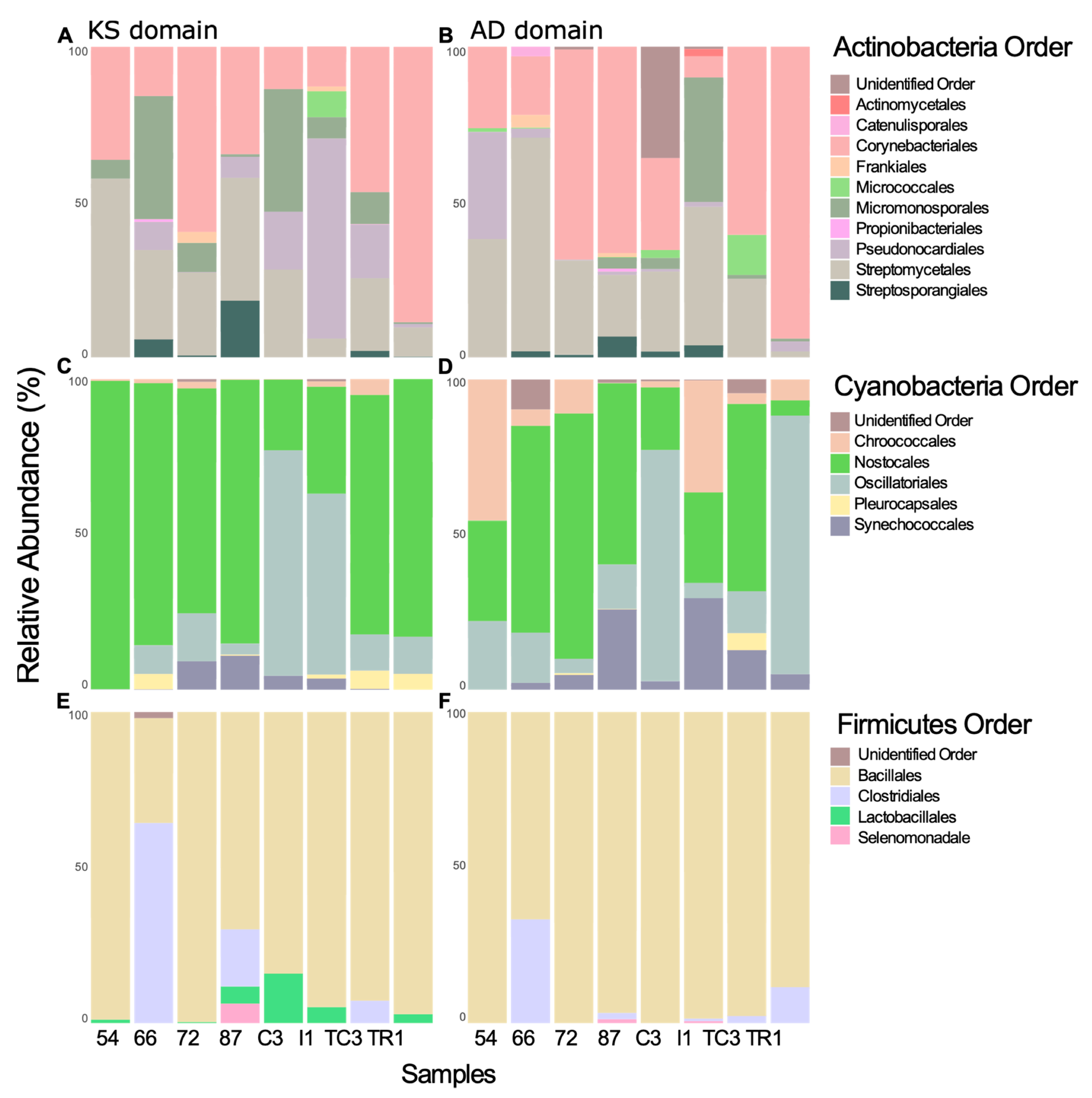
3.2. Taxonomic Provenience of the Recovered Biosynthetic Genes’ Sequences
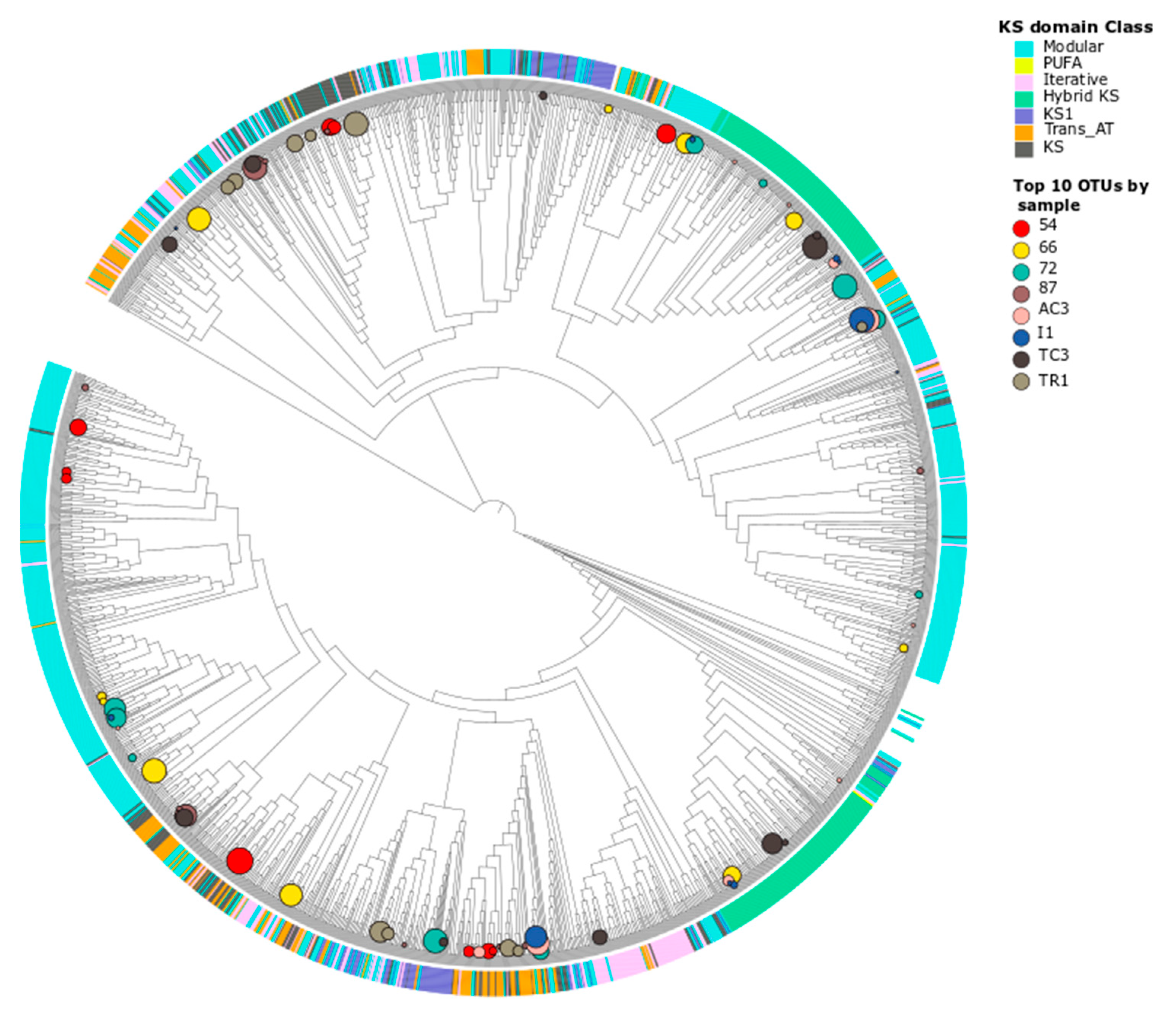
3.2.1. Taxonomic Identity of PKS Genes
3.2.2. Taxonomic Identity of NRPS Genes
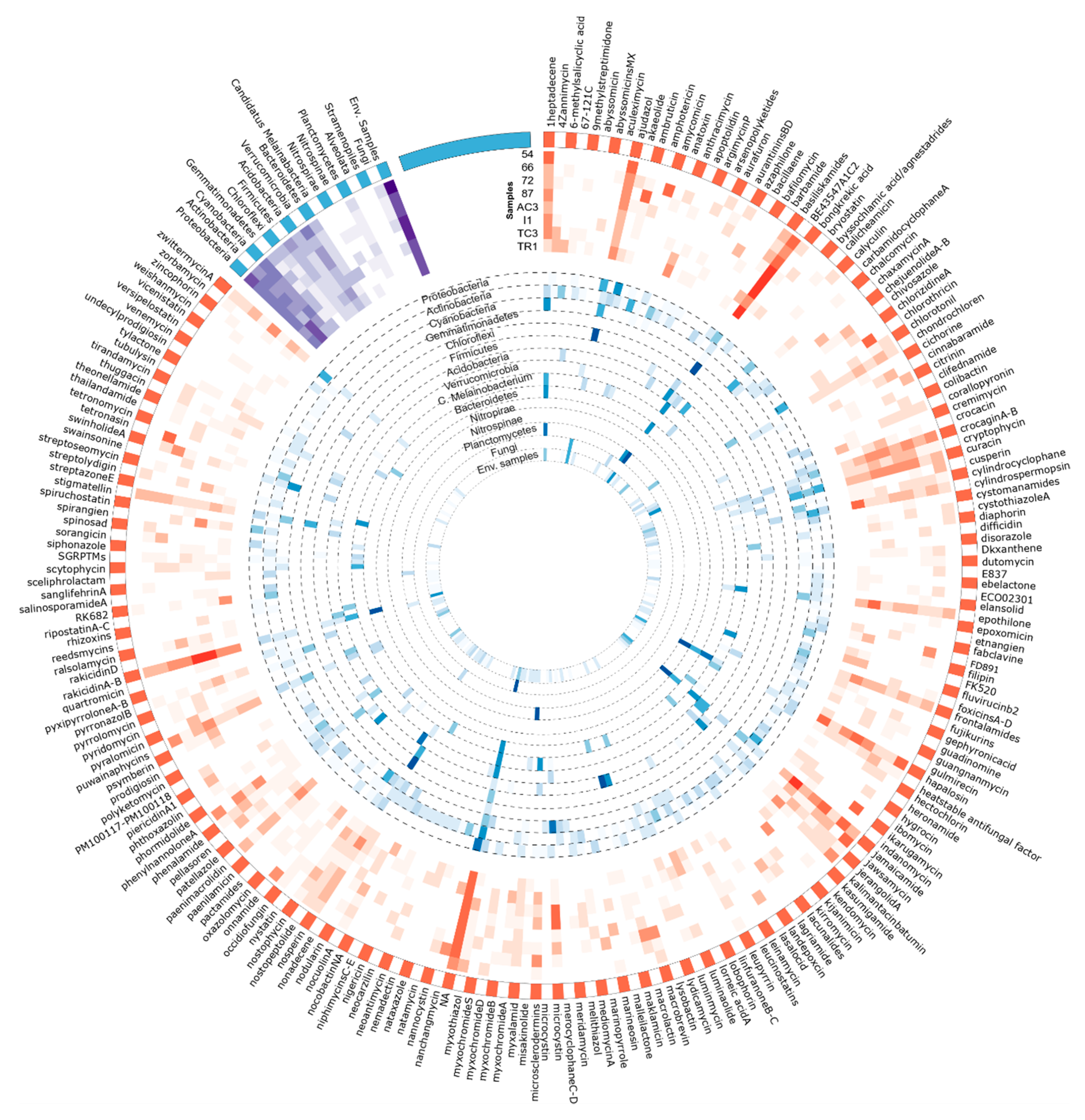
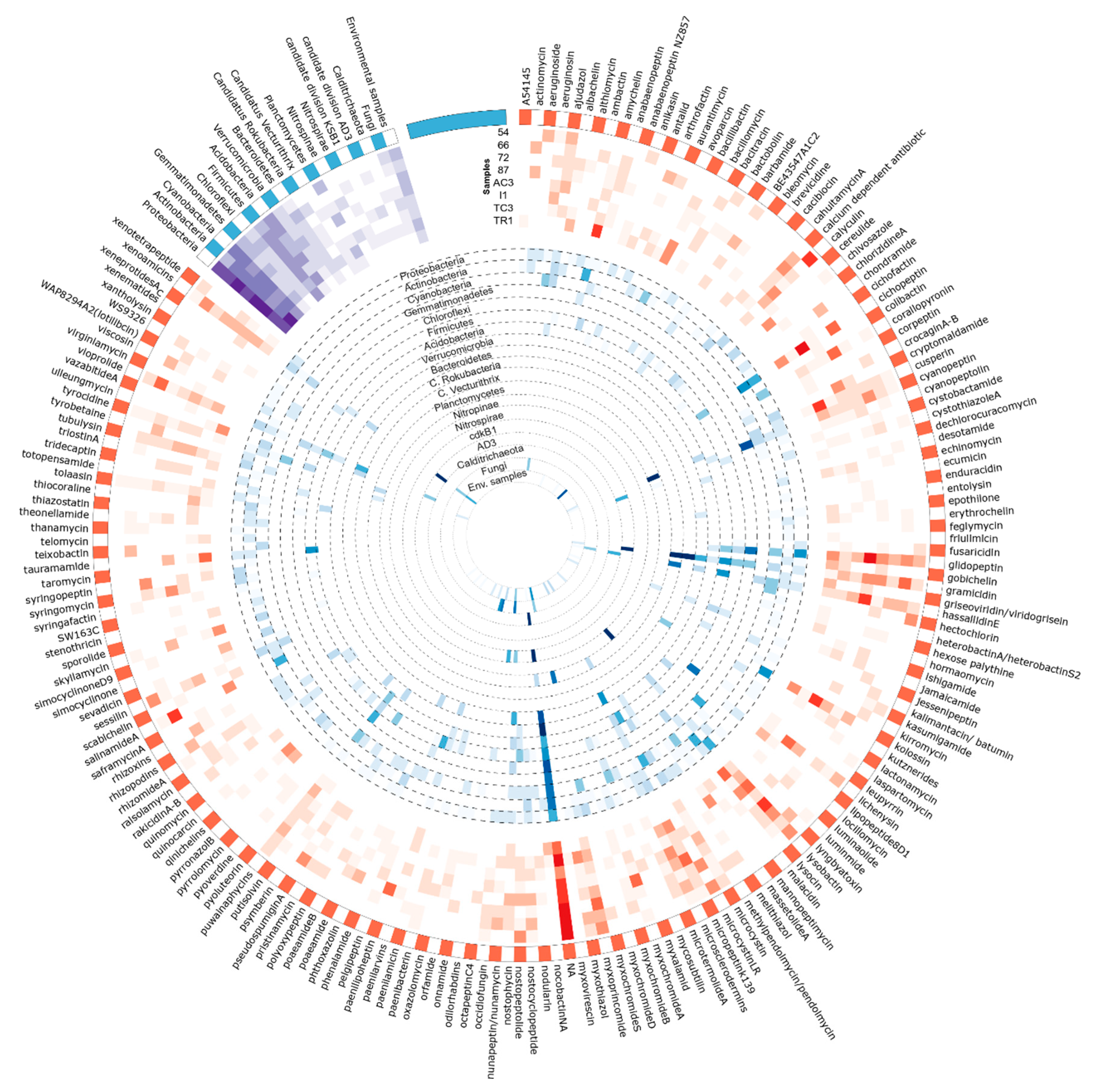
3.3. Metabolites Assignment to KS and AD Domains
3.3.1. Metabolite/BGCs Tentatively Assigned to KS Domains
3.3.2. Assigned BGC/Metabolites to AD Domain
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cragg, G.M.; Newman, D.J. Natural Products: a continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimermancic, P.; Medema, M.H.; Claesen, J.; Kurita, K.; Wieland, B.; Laura, C.; Mavrommatis, K.; Pati, A.; Godfrey, P.A.; Koehrsen, M.; et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters Peter. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sivonen, K.; Fewer, D.P. Genomic insights into the distribution, genetic diversity and evolution of polyketide synthases and nonribosomal peptide synthetases. Curr. Opin. Genet. Dev. 2015, 35, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Ayuso-Sacido, A.; Genilloud, O. New PCR primers for the screening of NRPS and PKS-I systems in actinomycetes: Detection and distribution of these biosynthetic gene sequences in major taxonomic groups. Microb. Ecol. 2005, 49, 10–24. [Google Scholar] [CrossRef]
- Schirmer, A.; Gadkari, R.; Reeves, C.D.; Ibrahim, F.; Delong, E.F.; Richard, C.; Hutchinson, C.R. Metagenomic Analysis Reveals Diverse Polyketide Synthase Gene Clusters in Microorganisms Associated with the Marine Sponge Discodermia dissoluta. Appl. Environ. Microbiol. 2005, 71, 4840–4849. [Google Scholar] [CrossRef] [Green Version]
- Owen, J.G.; Charlop-Powers, Z.; Smith, A.G.; Ternei, M.A.; Calle, P.Y.; Reddy, B.V.B.; Montiel, D.; Brady, S.F. Multiplexed metagenome mining using short DNA sequence tags facilitates targeted discovery of epoxyketone proteasome inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, 4221–4226. [Google Scholar] [CrossRef] [Green Version]
- Charlop-Powers, Z.; Owen, J.G.; Reddy, B.V.B.; Ternei, M.A.; Guimarães, D.O.; de Frias, U.A.; Pupo, M.T.; Seepe, P.; Feng, Z.; Brady, S.F. Global biogeographic sampling of bacterial secondary metabolism. Elife 2015, 4, 1–10. [Google Scholar] [CrossRef]
- Reddy, B.V.B.; Kallifidas, D.; Kim, J.H.; Charlop-Powers, Z.; Feng, Z.; Brady, S.F. Natural product biosynthetic gene diversity in geographically distinct soil microbiomes. Appl. Environ. Microbiol. 2012, 78, 3744–3752. [Google Scholar] [CrossRef] [Green Version]
- Lemetre, C.; Maniko, J.; Charlop-Powers, Z.; Sparrow, B.; Lowe, A.J.; Brady, S.F. Bacterial natural product biosynthetic domain composition in soil correlates with changes in latitude on a continent-wide scale. Proc. Natl. Acad. Sci. USA 2017, 201710262. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Li, Y.L.; Zhao, F.C. Secondary metabolites from polar organisms. Mar. Drugs 2017, 15, 28. [Google Scholar] [CrossRef] [Green Version]
- Wilson, Z.E.; Brimble, M. a Molecules derived from the extremes of life. Nat. Prod. Rep. 2009, 26, 44–71. [Google Scholar] [CrossRef] [PubMed]
- Benaud, N.; Zhang, E.; van Dorst, J.; Brown, M.V.; Kalaitzis, J.A.; Neilan, B.A.; Ferrari, B.C. Harnessing Long-Read Amplicon Sequencing to Uncover NRPS and Type I PKS Gene Sequence Diversity in Polar Desert Soils. FEMS Microbiol. Ecol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Borsetto, C.; Amos, G.C.A.; Da Rocha, U.N.; Mitchell, A.L.; Finn, R.D.; Laidi, R.F.; Vallin, C.; Pearce, D.A.; Newsham, K.K.; Wellington, E.M.H. Microbial community drivers of PK / NRP gene diversity in selected global soils. Microbiome 2019, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambrechts, S.; Willems, A.; Tahon, G. Uncovering the Uncultivated Majority in Antarctic Soils: Toward a Synergistic Approach. Front. Microbiol. 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, K.G.; Steen, A.D.; Ladau, J.; Yin, J.; Crosby, L. Phylogenetically Novel Uncultured Microbial Cells Dominate Earth Microbiomes. mSystems 2018, 3, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulschen, A.A.; Bendia, A.G.; Fricker, A.D.; Pellizari, V.H.; Galante, D.; Rodrigues, F. Isolation of uncultured bacteria from antarctica using long incubation periods and low nutritional media. Front. Microbiol. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pudasaini, S.; Wilson, J.; Ji, M.; van Dorst, J.; Snape, I.; Palmer, A.S.; Burns, B.P.; Ferrari, B.C. Microbial diversity of browning Peninsula, Eastern Antarctica revealed using molecular and cultivation methods. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Michaud, A.B.; Šabacká, M.; Priscu, J.C. Cyanobacterial diversity across landscape units in a polar desert: Taylor Valley, Antarctica. FEMS Microbiol. Ecol. 2012, 82, 268–278. [Google Scholar] [CrossRef]
- Pushkareva, E.; Pessi, I.S.; Wilmotte, A.; Namsaraev, Z.; Elster, J.; Mano, M.-J. Cyanobacteria inhabiting biological soil crusts of a polar desert: Sør Rondane Mountains, Antarctica. Syst. Appl. Microbiol. 2018, 41, 363–373. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, L.; Chang, X.; Jiang, F.; Gao, X.; Yao, Y.; Li, C.; Cao, S.; Zhou, Q.; Peng, F. Small-Scale Soil Microbial Community Heterogeneity Linked to Landform Historical Events on King George Island, Maritime Antarctica. Front. Microbiol. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Amaro, E.; Padeiro, A.; Mão de Ferro, A.; Mota, A.M.; Leppe, M.; Verkulich, S.; Hughes, K.A.; Peter, H.U.; Canário, J. Assessing trace element contamination in Fildes Peninsula (King George Island) and Ardley Island, Antarctic. Mar. Pollut. Bull. 2015, 97, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.F.; Zhang, T.; Zhang, F.; Wang, E.T.; He, J.F.; Ding, H.; Zhang, B.T.; Liu, J.; Ran, X.B.; Zang, J.Y. Diversity and structure of soil bacterial communities in the Fildes Region (maritime Antarctica) as revealed by 454 pyrosequencing. Front. Microbiol. 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Rocha, G.; Muñoz-Cartes, G.; Canales-Aguirre, C.B.; Lima, C.A.; Domínguez-Yévenes, M.; Bello-Toledo, H.; Hernández, C.E. Diversity structure of culturable bacteria isolated from the Fildes Peninsula (King George Island, Antarctica): A phylogenetic analysis perspective. PLoS ONE 2017, 12, e0179390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padeiro, A.; Amaro, E.; Dos Santos, M.M.C.; Araújo, M.F.; Gomes, S.S.; Leppe, M.; Verkulich, S.; Hughes, K.A.; Peter, H.U.; Canário, J. Trace element contamination and availability in the Fildes Peninsula, King George Island, Antarctica. Environ. Sci. Process. Impacts 2016, 18, 648–657. [Google Scholar] [CrossRef]
- Magalhäes, C.M.; Wiebe, W.J.; Joye, S.B.; Bordalo, A.A. Inorganic nitrogen dynamics in intertidal rocky biofilms and sediments of the Douro River estuary (Portugal). Estuaries 2005, 28, 592–607. [Google Scholar] [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing 2019. Available online: https://www.R-project.org/ (accessed on 18 February 2020).
- Wickham, H. ggplot2—Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 9780387981406. Available online: https://www.biblio.com/9780387981406 (accessed on 18 February 2020).
- Pebesma, E. Simple features for R: Standardized support for spatial vector data. R J. 2018, 10, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Dunnington, D. ggspatial: Spatial Data Framework for ggplot2 2018. Available online: https://CRAN.R-project.org/package=ggspatial (accessed on 18 February 2020).
- South, A. rnaturalearth: World Map Data from Natural Earth 2017. Available online: https://cran.r-project.org/web/packages/rnaturalearth/index.html (accessed on 18 February 2020).
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L.; Owens, S.; Gilbert, J.A.; Wall, D.H.; Caporaso, J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef] [Green Version]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Sousa, A.G.G.; Tomasino, M.P.; Duarte, P.; Fernández-Méndez, M.; Assmy, P.; Ribeiro, H.; Surkont, J.; Leite, R.B.; Pereira-Leal, J.B.; Torgo, L.; et al. Diversity and Composition of Pelagic Prokaryotic and Protist Communities in a Thin Arctic Sea-Ice Regime. Microb. Ecol. 2019, 78, 388–408. [Google Scholar] [CrossRef]
- Charlop-Powers, Z.; Pregitzer, C.C.; Lemetre, C.; Ternei, M.A.; Maniko, J.; Hover, B.M.; Calle, P.Y.; McGuire, K.L.; Garbarino, J.; Forgione, H.M.; et al. Urban park soil microbiomes are a rich reservoir of natural product biosynthetic diversity. Proc. Natl. Acad. Sci. USA 2016, 113, 14811–14816. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R.; Al, W.E.T. Naıve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Oksanen, A.J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; Hara, R.B.O.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘ vegan’. 2015. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 18 February 2020).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; De Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C.; et al. Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Fewer, D.P.; Holm, L.; Rouhiainen, L.; Sivonen, K. Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc. Natl. Acad. Sci. USA 2014, 111, 9259–9264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rego, A.; Raio, F.; Martins, T.P.; Ribeiro, H.; Sousa, A.G.G.; Séneca, J.; Baptista, M.S.; Lee, C.K.; Cary, S.C.; Ramos, V.; et al. Actinobacteria and Cyanobacteria Diversity in Terrestrial Antarctic Microenvironments Evaluated by Culture-Dependent and Independent Methods. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Charlop-Powers, Z.; Owen, J.G.; Reddy, B.V.B.; Ternei, M.A.; Brady, S.F. Chemical-biogeographic survey of secondary metabolism in soil. Proc. Natl. Acad. Sci. USA 2014, 111, 3757–3762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenke-Kodama, H.; Dittmann, E. Evolution of metabolic diversity: Insights from microbial polyketide synthases. Phytochemistry 2009, 70, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Podell, S.; Penn, K.; Badger, J.H.; Allen, E.; Jensen, P.R. The natural product domain seeker NaPDoS: A phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ridley, C.P.; Lee, H.Y.; Khosla, C. Evolution of polyketide synthases in bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 4595–4600. [Google Scholar] [CrossRef] [Green Version]
- Crits-Christoph, A.; Diamond, S.; Butterfield, C.N.; Thomas, B.C.; Banfield, J.F. Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature 2018, 558, 440–444. [Google Scholar] [CrossRef]
- Demay, J.; Bernard, C.; Reinhardt, A.; Marie, B. Natural products from cyanobacteria: Focus on beneficial activities. Mar. Drugs 2019, 17, 320. [Google Scholar] [CrossRef] [Green Version]
- Taton, A.; Grubisic, S.; Ertz, D.; Hodgson, D.A.; Piccardi, R.; Biondi, N.; Tredici, M.R. Polyphasic Study of Antarctic Cyanobacterial Strains. J. Phycol. 2006, 42, 1257–1270. [Google Scholar] [CrossRef]
- Subramani, R.; Sipkema, D. Marine rare actinomycetes: A promising source of structurally diverse and unique novel natural products. Mar. Drugs 2019, 17, 249. [Google Scholar] [CrossRef] [Green Version]
- Aleti, G.; Sessitsch, A.; Brader, G. Genome mining: Prediction of lipopeptides and polyketides from Bacillus and related Firmicutes. Comput. Struct. Biotechnol. J. 2015, 13, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Tahon, G.; Willems, A. Isolation and characterization of aerobic anoxygenic phototrophs from exposed soils from the Sør Rondane Mountains, East Antarctica. Syst. Appl. Microbiol. 2017, 40, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Vollmers, J.; Frentrup, M.; Rast, P.; Jogler, C.; Kaster, A.K. Untangling genomes of novel Planctomycetal and Verrucomicrobial species from monterey bay kelp forest metagenomes by refined binning. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrat, R.R.C.; Ionescu, D.; Dávila, A.M.R.; Grossart, H.P. Recovering genomics clusters of secondary metabolites from lakes using genome-resolved metagenomics. Front. Microbiol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Undabarrena, A.; Beltrametti, F.; Claverías, F.P.; González, M.; Moore, E.R.B.; Seeger, M.; Cámara, B. Exploring the diversity and antimicrobial potential of marine actinobacteria from the comau fjord in Northern Patagonia, Chile. Front. Microbiol. 2016, 7, 1–16. [Google Scholar] [CrossRef]
- Graça, A.P.; Calisto, R.; Lage, O.M. Planctomycetes as Novel Source of Bioactive Molecules. Front. Microbiol. 2016, 7, 1241. [Google Scholar] [CrossRef]
- Sayari, M.; van der Nest, M.A.; Steenkamp, E.T.; Soal, N.C.; Wilken, P.M.; Wingfield, B.D. Distribution and Evolution of Nonribosomal Peptide Synthetase Gene Clusters in the Ceratocystidaceae. Genes (Basel) 2019, 10, 328. [Google Scholar] [CrossRef] [Green Version]
- Clardy, J.; Fischbach, M.A.; Walsh, C.T. New antibiotics from bacterial natural products. Nat. Biotechnol. 2006, 24, 1541–1550. [Google Scholar] [CrossRef]
- Kim, D.; Lee, E.J.; Lee, J.; Leutou, A.S.; Shin, Y.H.; Choi, B.; Hwang, J.S.; Hahn, D.; Choi, H.; Chin, J.; et al. Antartin, a cytotoxic zizaane-type Sesquiterpenoid from a Streptomyces sp. Isolated from an antarctic marine sediment. Mar. Drugs 2018, 16, 130. [Google Scholar] [CrossRef] [Green Version]
- Bringmann, G.; Lang, G.; Maksimenka, K.; Hamm, A.; Gulder, T.A.M.; Dieter, A.; Bull, A.T.; Stach, J.E.M.; Kocher, N.; Müller, W.E.G.; et al. Gephyromycin, the first bridged angucyclinone, from Streptomyces griseus strain NTK 14. Phytochemistry 2005, 66, 1366–1373. [Google Scholar] [CrossRef]
- Bruntner, C.; Binder, T.; Pathom-aree, W.; Goodfellow, M.; Bull, A.T.; Potterat, O.; Puder, C.; Hörer, S.; Schmid, A.; Bolek, W.; et al. Frigocyclinone, a novel angucyclinone antibiotic produced by a Streptomyces griseus strain from Antarctica. J. Antibiot. (Tokyo) 2005, 58, 346–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhakal, D.; Rayamajhi, V.; Mishra, R.; Sohng, J.K. Bioactive molecules from Nocardia: diversity, bioactivities and biosynthesis. J. Ind. Microbiol. Biotechnol. 2019, 46, 385–407. [Google Scholar] [CrossRef] [PubMed]
- Babalola, O.O.; Kirby, B.M.; Le Roes-Hill, M.; Cook, A.E.; Cary, S.C.; Burton, S.G.; Cowan, D.A. Phylogenetic analysis of actinobacterial populations associated with Antarctic Dry Valley mineral soils. Environ. Microbiol. 2009, 11, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Charlop-Powers, Z.; Milshteyn, A.; Brady, S.F. Metagomic small molecule discovery methods. Curr. Opin. Microbiol. 2014, 19, 70–75. [Google Scholar] [CrossRef] [Green Version]
- Villadsen, N.L.; Jacobsen, K.M.; Keiding, U.B.; Weibel, E.T.; Christiansen, B.; Vosegaard, T.; Bjerring, M.; Jensen, F.; Johannsen, M.; Tørring, T.; et al. Synthesis of ent-BE-43547A 1 reveals a potent hypoxia-selective anticancer agent and uncovers the biosynthetic origin of the APD-CLD natural products. Nat. Chem. 2017, 9, 264–272. [Google Scholar] [CrossRef] [Green Version]
- McBrien, K.D.; Berry, R.L.; Lowe, S.E.; Neddermann, K.M.; Bursuker, I.; Steven, S.H.; Klohr, S.E.; Leet, J.E. Rakicidins, New Cytotoxic Lipopeptides from Micromonospora sp. Fermentation, Isolation and Characterization. J. Antibiot. (Tokyo) 1995, 48, 1446–1452. [Google Scholar] [CrossRef] [Green Version]
- Edwards, D.J.; Marquez, B.L.; Nogle, L.M.; McPhail, K.; Goeger, D.E.; Robberts, M.A.; Gerwick, W.H. Structure and Biosynthesis of the Jamaicamides, New Mixed Polyketide-Peptide Neurotoxins from the Marine Cyanobacterium Lyngbya majuscula. Chem. Biol. 2004, 11, 817–833. [Google Scholar] [CrossRef] [Green Version]
- Stratmann, K.; Burgoyne, D.L.; Moore, R.E.; Patterson, G.M.L.; Smith, C.D. Hapalosin, a Cyanobacterial Cyclic Depsipeptide with Multidrug-Resistance Reversing Activity. J. Org. Chem. 1994, 59, 7219–7226. [Google Scholar] [CrossRef]
- Kunze, B.; Jansen, R.; Hofle, G.; Reichenbach, H. Ajudazols, New Inhibitors Mitochondrial of the from Electron Transport. J. Antibiot. 2004, 57, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Gottardi, E.M.; Krawczyk, J.M.; Von Suchodoletz, H.; Schadt, S.; Mühlenweg, A.; Uguru, G.C.; Pelzer, S.; Fiedler, H.P.; Bibb, M.J.; Stach, J.E.M.; et al. Abyssomicin Biosynthesis: Formation of an Unusual Polyketide, Antibiotic-Feeding Studies and Genetic Analysis. ChemBioChem 2011, 12, 1401–1410. [Google Scholar] [CrossRef] [Green Version]
- Chang, Z.; Flatt, P.; Gerwick, W.H.; Nguyen, V.A.; Willis, C.L.; Sherman, D.H. The barbamide biosynthetic gene cluster: A novel marine cyanobacterial system of mixed polyketide synthase (PKS)-non-ribosomal peptide synthetase (NRPS) origin involving an unusual trichloroleucyl starter unit. Gene 2002, 296, 235–247. [Google Scholar] [CrossRef]
- Kirst, H.A.; Michel, K.H.; Mynderase, J.S.; Chio, E.H.; Yao, R.C.; Nakasukasa, W.M.; Boeck, L.D.; Occlowitz, J.L.; Paschal, J.W.; Deeter, J.B.; et al. Discovery, Isolation, and Structure Elucidation of a Family of Structurally Unique, Fermentation-Derived Tetracyclic Macrolides. In Synthesis and Chemistry of Agrochemicals III; American Chemical Society: Washington, DC, USA, 1992; pp. 214–225. [Google Scholar]
- Tomita, K.; Oda, N.; Hoshino, Y.; Ohkusa, N.; Chikazawa, H. Fluvirucins A1, A2, B1, B2, B3, B4 and B5, new antibiotics active against influenza a virus. IV. Taxonomy on the producing organisms. J. Antibiot. (Tokyo) 1991, 44, 940–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Wang, M.; Wu, C.; Tan, Y.; Li, J.; Hao, X.; Duan, Y.; Guan, Y.; Shang, X.; Wang, Y.; et al. Identification and Proposed Relative and Absolute Configurations of Niphimycins C-E from the Marine-Derived Streptomyces sp. IMB7-145 by Genomic Analysis. J. Nat. Prod. 2018, 81, 178–187. [Google Scholar] [CrossRef]
- Berger, J.; Rachlin, A.I.; Scott, W.E.; Sternbach, L.H.; Goldberg, M.W. The Isolation of Three New Crystalline Antibiotics from Streptomyces. J. Am. Chem. Soc. 1951, 73, 5295–5298. [Google Scholar] [CrossRef]
- Robbins, N.; Spitzer, M.; Wang, W.; Waglechner, N.; Patel, D.J.; O’Brien, J.S.; Ejim, L.; Ejim, O.; Tyers, M.; Wright, G.D. Discovery of Ibomycin, a Complex Macrolactone that Exerts Antifungal Activity by Impeding Endocytic Trafficking and Membrane Function. Cell Chem. Biol. 2016, 23, 1383–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.J.; Pauwelyn, E.; Ongena, M.; Debois, D.; Leclère, V.; Jacques, P.; Bleyaert, P.; Höfte, M. Characterization of cichopeptins, new phytotoxic cyclic lipodepsipeptides produced by Pseudomonas cichorii SF1-54 and their role in bacterial midrib rot disease of lettuce. Mol. Plant-Microbe Interact. 2015, 28, 1009–1022. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhou, H.; Chen, H.; Jing, X.; Zheng, W.; Li, R.; Sun, T.; Liu, J.; Fu, J.; Huo, L.; et al. Discovery of recombinases enables genome mining of cryptic biosynthetic gene clusters in Burkholderiales species. Proc. Natl. Acad. Sci. USA 2018, 115, E4255–E4263. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Nakaya, S.; Ohashi, Y.; Hirata, H.; Fujii, K.; Harada, K. WAP-8294A2, a novel anti-MRSA antibiotic produced by Lysobacter sp. J. Am. Chem. Soc. 1997, 119, 6680–6681. [Google Scholar] [CrossRef]
- Gräfe, U.; Schlegel, R.; Ritzau, M.; Ihn, W.; Dornberger, K.; Stengel, C.; Fleck, W.F.; Gutsche, W.; Härtl, A.; Paulus, E.F. Aurantimycins, new depsipeptide antibiotics from Streptomyces aurantiacus IMET 43917. Production, isolation, structure elucidation, and biological activity. J. Antibiot. 1995, 48, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Robbel, L.; Knappe, T.A.; Linne, U.; Xie, X.; Marahiel, M.A. Erythrochelin-a hydroxamate-type siderophore predicted from the genome of Saccharopolyspora erythraea. FEBS J. 2010, 277, 663–676. [Google Scholar] [CrossRef]
- Stintzi, A.; Pierre, C.; Hohnadel, D.; Meyer, J.; Dean, C.; Poole, K.; Kourambas, S.; Krishnapillai, V. Novel pyoverdine biosynthesis gene(s) of Pseudomonas aeruginosa PA0. Microbiology 1996, 142, 1181–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laycock, M.V.; Hildebrand, P.D.; Thibault, P.; Walter, J.A.; Wright, J.L.C. Viscosin, a potent peptidolipid biosurfactant and phytopathogenic mediator produced by a pectolytic strain of Pseudomonas fluorescens. J. Agric. Food Chem. 2005, 39, 483–489. [Google Scholar] [CrossRef]
- Groupe, V.; Pugh, L.H.; Weiss, D.; Kochi, M. Observations on Antiviral Activity of Viscosin. Exp. Biol. Med. 1951, 78, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Oberer, L.; Ino, T.; König, W.A.; Busch, M.; Weckesser, J. Cyanopeptolins, new depsipeptides from the cyanobacterium Microcystis sp. PCC 7806. J. Antibiot. (Tokyo) 1993, 46, 1550–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mareš, J.; Hájek, J.; Urajová, P.; Kopecký, J.; Hrouzek, P. A hybrid non-ribosomal peptide/polyketide synthetase containing fatty-acyl ligase (FAAL) synthesizes the β-amino fatty acid lipopeptides puwainaphycins in the Cyanobacterium Cylindrospermum alatosporum. PLoS ONE 2014, 9, e111904. [Google Scholar] [CrossRef] [Green Version]
- Herfindal, L.; Myhren, L.; Kleppe, R.; Krakstad, C.; Selheim, F.; Jokela, J.; Sivonen, K.; Døskeland, S.O. Nostocyclopeptide-M1: A potent, nontoxic inhibitor of the hepatocyte drug transporters OATP1B3 and OATP1B1. Mol. Pharm. 2011, 8, 360–367. [Google Scholar] [CrossRef]
- Imai, Y.; Meyer, K.J.; Iinishi, A.; Favre-Godal, Q.; Green, R.; Manuse, S.; Caboni, M.; Mori, M.; Niles, S.; Ghiglieri, M.; et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576, 459–464. [Google Scholar] [CrossRef]
- Hover, B.M.; Kim, S.H.; Katz, M.; Charlop-Powers, Z.; Owen, J.G.; Ternei, M.A.; Maniko, J.; Estrela, A.B.; Molina, H.; Park, S.; et al. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat. Microbiol. 2018, 3, 415–422. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AD Domain | KS Domain | |||
|---|---|---|---|---|
| Mantel Statistic r | Significance | Mantel Statistic r | Significance | |
| 16S rRNA | 0.6787 | 0.002* | 0.7535 | 0.001* |
| Phylum | ||||
| Proteobacteria | 0.6616 | 0.007* | 0.3687 | 0.053 |
| Bacteroidetes | 0.5234 | 0.018* | 0.3285 | 0.059 |
| Acidobacteria | 0.652 | 0.003* | 0.4483 | 0.039* |
| Actinobacteria | 0.5297 | 0.016* | 0.5561 | 0.014* |
| Verrucomicrobia | 0.6682 | 0.001* | 0.5808 | 0.004* |
| Chloroflexi | 0.6192 | 0.01 | 0.6378 | 0.005* |
| Gemmatimonadetes | 0.4463 | 0.031* | 0.3671 | 0.054 |
| Planctomycetes | 0.2158 | 0.121 | 0.4116 | 0.022* |
| Cyanobacteria | 0.2158 | 0.102 | 0.1221 | 0.23 |
| Distribution in Each Sample (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| MiBIG Metabolite/BGC | 54 | 66 | 72 | 87 | AC3 | I1 | TC3 | TR1 | Total Distribution (%) |
| BE-43547 A1-C2 | 12.2 | 3.6 | 9.5 | 17.3 | 19.0 | 22.2 | 5.7 | 18.7 | 13.3 |
| NA | 5.0 | 6.9 | 7.5 | 10.7 | 12.5 | 9.6 | 11.1 | 12.3 | 9.8 |
| Rakicidin A/Rakicidin B | 2.3 | 1.9 | 4.0 | 3.4 | 14.1 | 16.5 | 2.7 | 2.5 | 6.0 |
| Jamaicamide | 2.1 | 8.6 | 0.7 | 12.0 | 3.3 | 1.4 | 13.3 | 0.8 | 5.0 |
| Hapalosin | 11.4 | 0.7 | 0.3 | 5.1 | 8.8 | 2.5 | 0.6 | 11.0 | 4.3 |
| Ajudazol | 10.6 | 5.7 | 3.5 | 3.9 | 1.8 | 1.1 | 2.4 | 5.4 | 3.7 |
| Distribution in Each Sample (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| MiBIG Metabolite | 54 | 66 | 72 | 87 | AC3 | I1 | TC3 | TR1 | Total Distribution (%) |
| NA | 40.7 | 24.3 | 28.9 | 17.4 | 14.8 | 4.3 | 33.3 | 10.0 | 20.5 |
| Cichopeptin | 0.0 | 0.0 | 9.1 | 0.0 | 0.1 | 29.8 | 0.0 | 2.7 | 7.5 |
| Glidopeptin | 2.0 | 4.3 | 1.4 | 3.6 | 30.4 | 2.3 | 0.8 | 4.4 | 6.0 |
| Lysobactin | 6.0 | 0.3 | 5.0 | 18.7 | 0.7 | 10.5 | 1.2 | 0.2 | 5.4 |
| Kolossin | 0.0 | 0.0 | 1.5 | 0.1 | 0.9 | 20.6 | 0.1 | 3.1 | 4.9 |
| Calyculin | 21.7 | 0.0 | 0.3 | 0.0 | 4.3 | 2.2 | 0.0 | 0.2 | 3.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rego, A.; Sousa, A.G.G.; Santos, J.P.; Pascoal, F.; Canário, J.; Leão, P.N.; Magalhães, C. Diversity of Bacterial Biosynthetic Genes in Maritime Antarctica. Microorganisms 2020, 8, 279. https://doi.org/10.3390/microorganisms8020279
Rego A, Sousa AGG, Santos JP, Pascoal F, Canário J, Leão PN, Magalhães C. Diversity of Bacterial Biosynthetic Genes in Maritime Antarctica. Microorganisms. 2020; 8(2):279. https://doi.org/10.3390/microorganisms8020279
Chicago/Turabian StyleRego, Adriana, António G. G. Sousa, João P. Santos, Francisco Pascoal, João Canário, Pedro N. Leão, and Catarina Magalhães. 2020. "Diversity of Bacterial Biosynthetic Genes in Maritime Antarctica" Microorganisms 8, no. 2: 279. https://doi.org/10.3390/microorganisms8020279
APA StyleRego, A., Sousa, A. G. G., Santos, J. P., Pascoal, F., Canário, J., Leão, P. N., & Magalhães, C. (2020). Diversity of Bacterial Biosynthetic Genes in Maritime Antarctica. Microorganisms, 8(2), 279. https://doi.org/10.3390/microorganisms8020279