Comprehensive Mining and Characterization of CRISPR-Cas Systems in Bifidobacterium

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
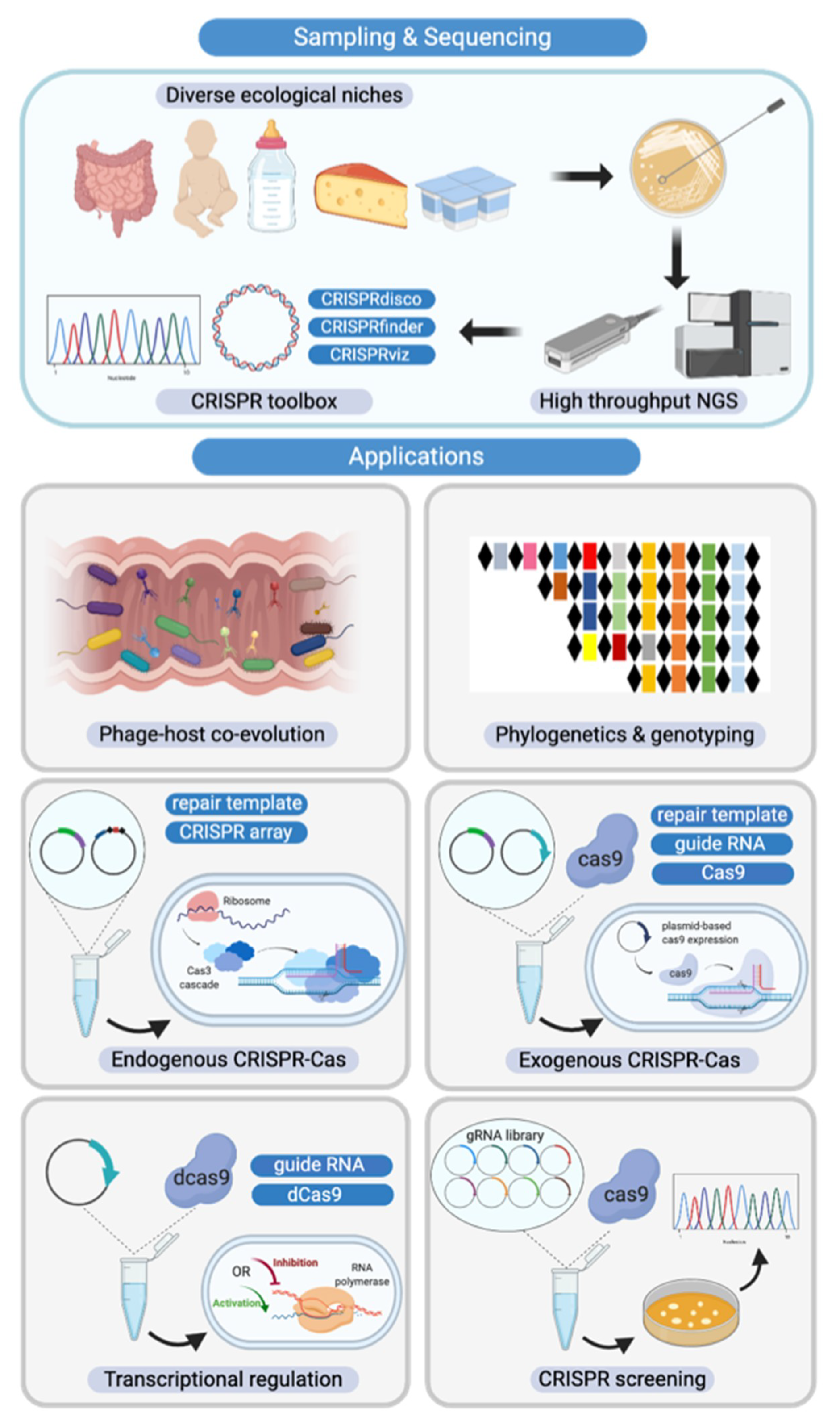
1. Introduction
2. Materials and Methods
2.1. CRISPR-Cas Systems Detection and Classification
2.2. Phylogenetic Analyses
2.3. Characterization of CRISPR-Cas Systems Analyses
3. Results
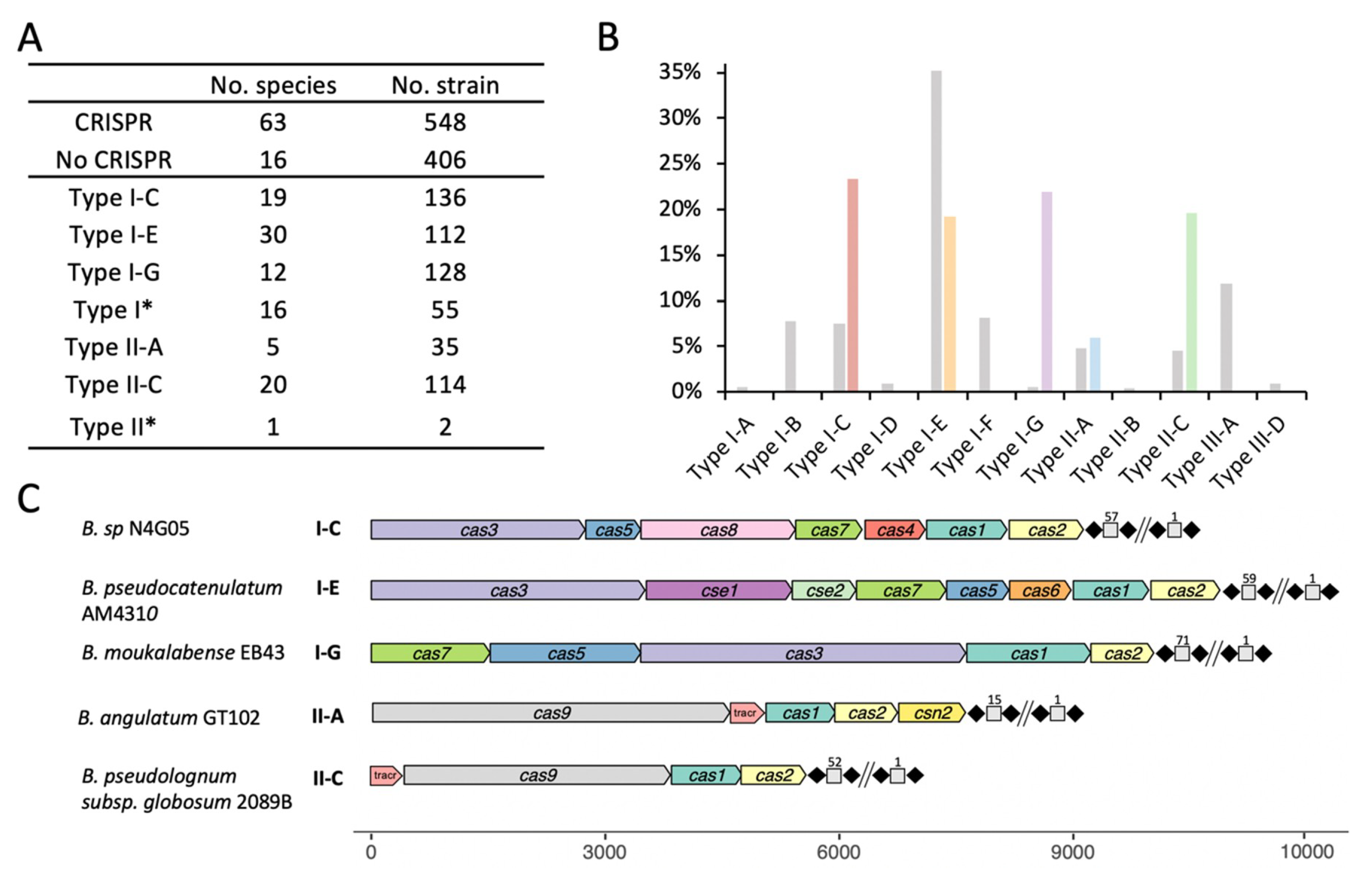
3.1. Occurrence and Diversity of CRISPR-Cas Systems in Bifidobacterium Genomes
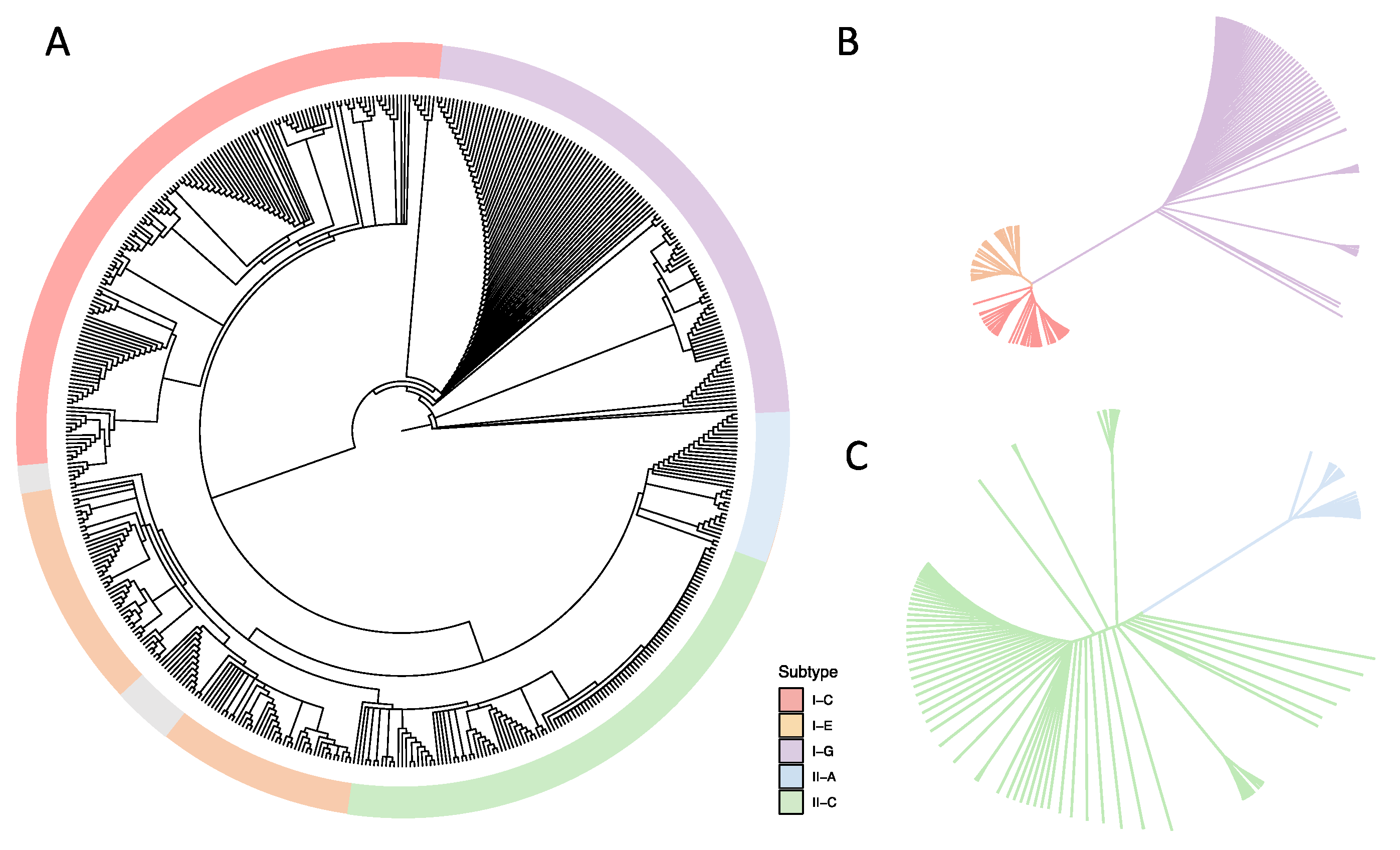
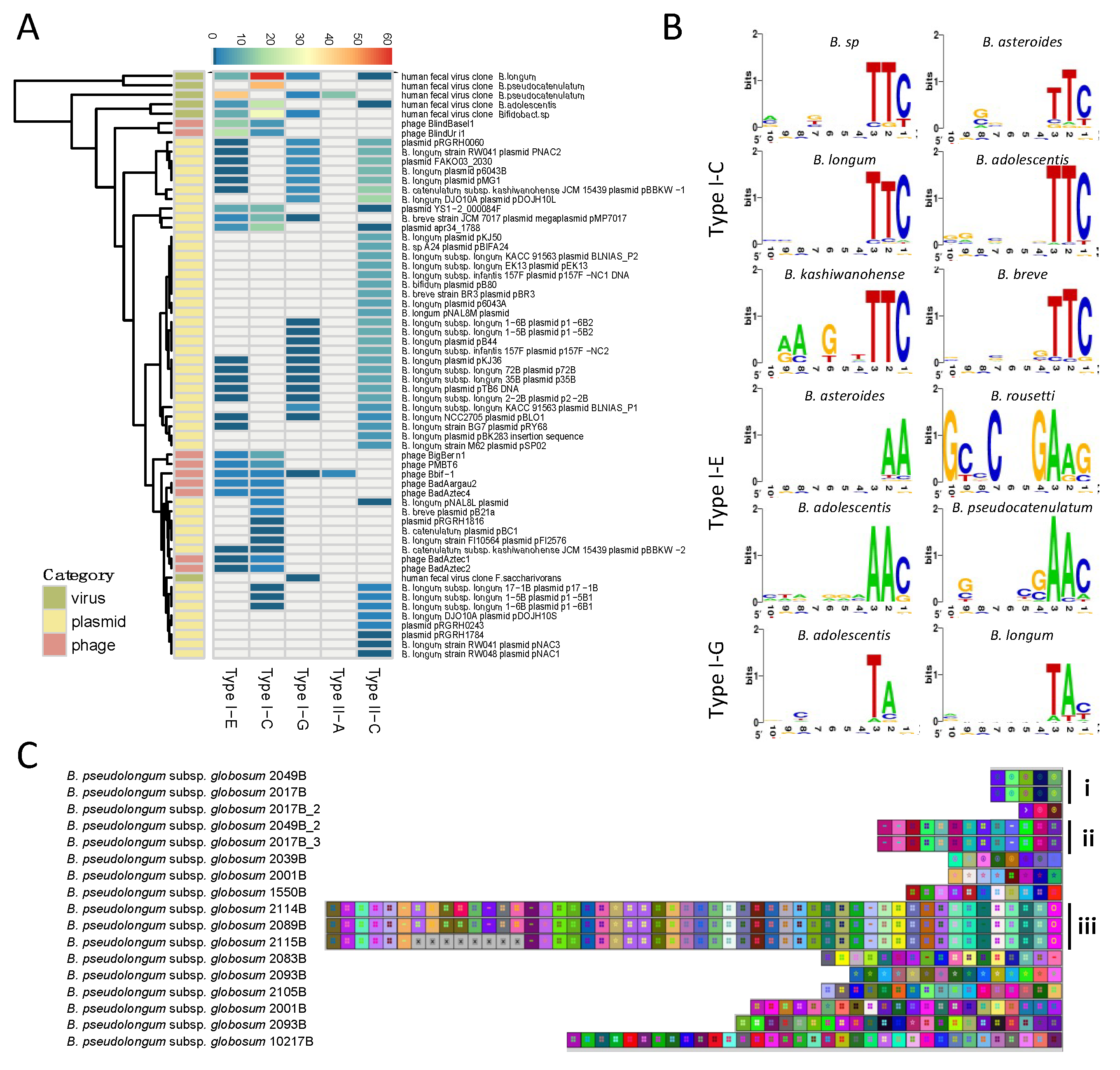
3.2. Phylogenetic Analyses
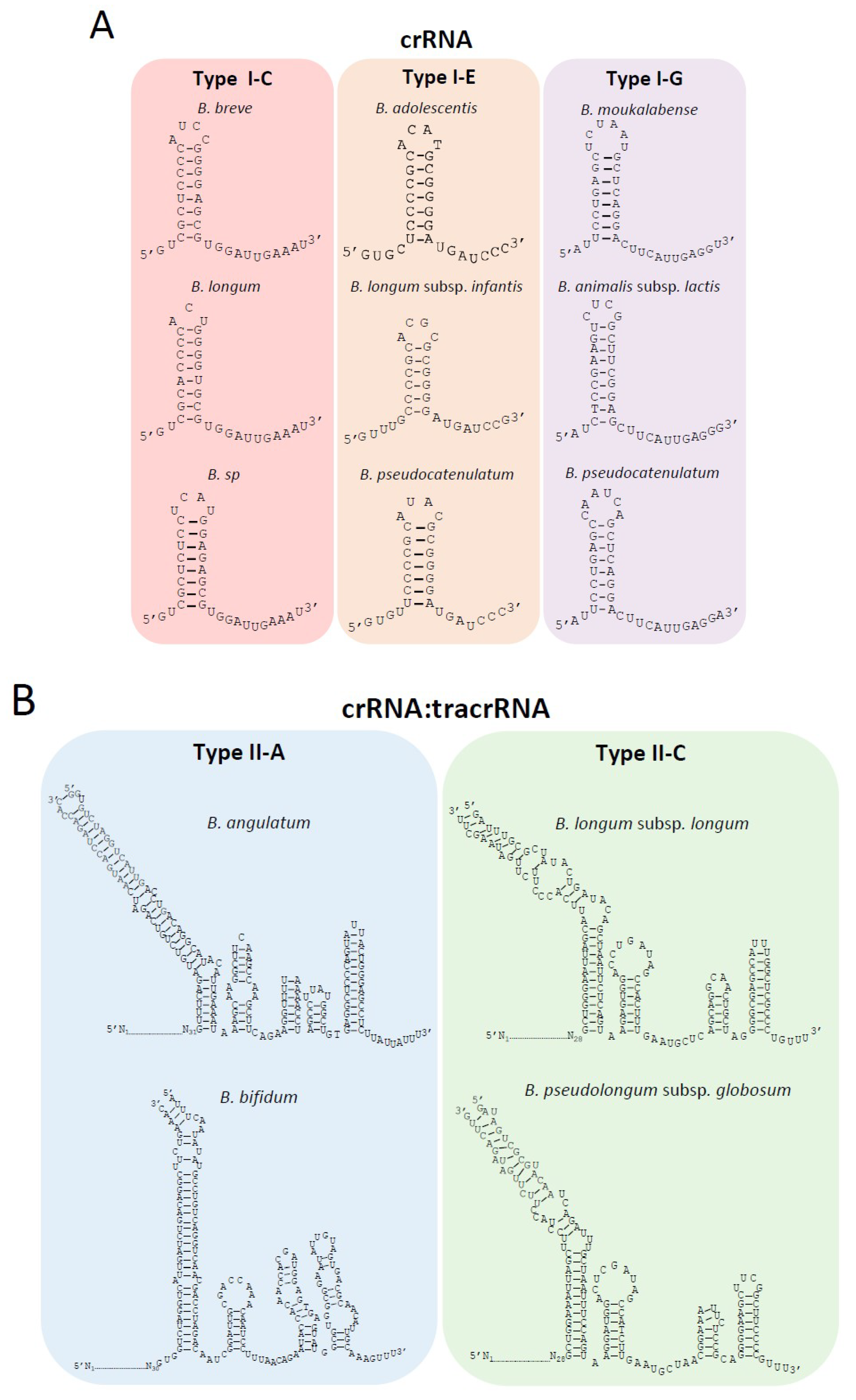
3.3. Characterization of CRISPR-Cas Systems
3.4. Spacer Homology Search and PAM Prediction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 2008, 322, 1843–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouns, S.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.; Snijders, A.P.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Hidalgo-Cantabrana, C.; Goh, Y.J.; Pan, M.; Sanozky-Dawes, R.; Barrangou, R. Genome editing using the endogenous type I CRISPR-Cas system in Lactobacillus crispatus. Proc. Natl. Acad. Sci. USA 2019, 116, 15774–15783. [Google Scholar] [CrossRef] [Green Version]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036-17. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, A.; van Sinderen, D. Bifidobacteria and their role as members of the human gut microbiota. Front. Microbiol. 2016, 7, 925. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Chen, Y.; Stanton, C.; Ross, R.P.; Lee, Y.K.; Zhao, J.; Zhang, H.; Chen, W. Bifidobacterium and lactobacillus composition at species level and gut microbiota diversity in infants before 6 weeks. Int. J. Mol. Sci. 2019, 20, 3306. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, L.; Delgado, S.; Ruas-Madiedo, P.; Sánchez, B.; Margolles, A. Bifidobacteria and their molecular communication with the immune system. Front. Microbiol. 2017, 8, 2345. [Google Scholar] [CrossRef] [Green Version]
- El Hadad, S.; Zakareya, A.; Al-Hejin, A.; Aldahlawi, A.; Alharbi, M. Sustaining exposure to high concentrations of bifidobacteria inhibits gene expression of Mouse’s mucosal immunity. Heliyon 2019, 5, e02866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tojo, R.; Suarez, A.; Clemente, M.G.; de los Reyes-Gavilan, C.G.; Margolles, A.; Gueimonde, M.; Ruas-Madiedo, P. Intestinal microbiota in health and disease: Role of bifidobacteria in gut homeostasis. World J. Gastroenterol. 2014, 20, 15163–15176. [Google Scholar] [CrossRef] [PubMed]
- Riviere, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, C.I.; Martiny, J.B.H. Evolutionary relationships among bifidobacteria and their hosts and environments. BMC Genom. 2020, 21, 26. [Google Scholar] [CrossRef] [Green Version]
- Milani, C.; Lugli, G.A.; Duranti, S.; Turroni, F.; Mancabelli, L.; Ferrario, C.; Mangifesta, M.; Hevia, A.; Viappiani, A.; Scholz, M.; et al. Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Sci. Rep. 2015, 5, 15782. [Google Scholar] [CrossRef] [Green Version]
- Bottacini, F.; Morrissey, R.; Roberts, R.J.; James, K.; van Breen, J.; Egan, M.; Lambert, J.; van Limpt, K.; Knol, J.; Motherway, M.O.; et al. Comparative genome and methylome analysis reveals restriction/modification system diversity in the gut commensal Bifidobacterium breve. Nucleic Acids Res. 2018, 46, 1860–1877. [Google Scholar] [CrossRef]
- Hidalgo-Cantabrana, C.; Goh, Y.J.; Barrangou, R. Characterization and repurposing of Type I and Type II CRISPR-Cas Systems in Bacteria. J. Mol. Biol. 2019, 431, 21–33. [Google Scholar] [CrossRef]
- Walker, J.E.; Lanahan, A.A.; Zheng, T.; Toruno, C.; Lynd, L.R.; Cameron, J.C.; Olson, D.G.; Eckert, C.A. Development of both type I-B and type II CRISPR/Cas genome editing systems in the cellulolytic bacterium Clostridium thermocellum. Metab. Eng. Commun. 2020, 10, e00116. [Google Scholar] [CrossRef]
- Fedorova, I.; Arseniev, A.; Selkova, P.; Pobegalov, G.; Goryanin, I.; Vasileva, A.; Musharova, O.; Abramova, M.; Kazalov, M.; Zyubko, T.; et al. DNA targeting by Clostridium cellulolyticum CRISPR-Cas9 Type II-C system. Nucleic Acids Res. 2020, 48, 2026–2034. [Google Scholar] [CrossRef] [Green Version]
- Millen, A.M.; Samson, J.E.; Tremblay, D.M.; Magadan, A.H.; Rousseau, G.M.; Moineau, S.; Romero, D.A. Lactococcus lactis type III-A CRISPR-Cas system cleaves bacteriophage RNA. RNA Biol. 2019, 16, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myrbraten, I.S.; Wiull, K.; Salehian, Z.; Havarstein, L.S.; Straume, D.; Mathiesen, G.; Kjos, M. CRISPR Interference for Rapid Knockdown of Essential Cell Cycle Genes in Lactobacillus plantarum. mSphere 2019, 4, e00007-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawley, A.B.; Henriksen, E.D.; Stout, E.; Brandt, K.; Barrangou, R. Characterizing the activity of abundant, diverse and active CRISPR-Cas systems in lactobacilli. Sci. Rep. 2018, 8, 11544. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo-Cantabrana, C.; Crawley, A.B.; Sanchez, B.; Barrangou, R. Characterization and Exploitation of CRISPR Loci in Bifidobacterium longum. Front. Microbiol. 2017, 8, 1851. [Google Scholar] [CrossRef] [PubMed]
- Briner, A.E.; Lugli, G.A.; Milani, C.; Duranti, S.; Turroni, F.; Gueimonde, M.; Margolles, A.; van Sinderen, D.; Ventura, M.; Barrangou, R. Occurrence and Diversity of CRISPR-Cas Systems in the Genus Bifidobacterium. PLoS ONE 2015, 10, e0133661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2015, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Nethery, M.A.; Barrangou, R. CRISPR Visualizer: Rapid identification and visualization of CRISPR loci via an automated high-throughput processing pipeline. RNA Biol. 2019, 16, 577–584. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Burstein, D.; Harrington, L.B.; Strutt, S.C.; Probst, A.J.; Anantharaman, K.; Thomas, B.C.; Doudna, J.A.; Banfield, J.F. New CRISPR-Cas systems from uncultivated microbes. Nature 2017, 542, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolde, R. Pheatmap: Pretty Heatmaps. R package 0.7.7. Available online: http://cran.r-project.org/web/packages/pheatmap/ (accessed on 8 April 2020).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Nethery, M.A.; Barrangou, R. Predicting and visualizing features of CRISPR-Cas systems. Methods Enzymol. 2019, 616, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Briner, A.E.; Henriksen, E.D.; Barrangou, R. Prediction and Validation of Native and Engineered Cas9 Guide Sequences. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot086785. [Google Scholar] [CrossRef] [Green Version]
- Zadeh, J.N.; Steenberg, C.D.; Bois, J.S.; Wolfe, B.R.; Pierce, M.B.; Khan, A.R.; Dirks, R.M.; Pierce, N.A. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 2011, 32, 170–173. [Google Scholar] [CrossRef]
- Crawley, A.B.; Henriksen, J.R.; Barrangou, R. CRISPRdisco: An Automated Pipeline for the Discovery and Analysis of CRISPR-Cas Systems. CRISPR J. 2018, 1, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Nunez, J.K.; Kranzusch, P.J.; Noeske, J.; Wright, A.V.; Davies, C.W.; Doudna, J.A. Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014, 21, 528–534. [Google Scholar] [CrossRef]
- Toms, A.; Barrangou, R. On the global CRISPR array behavior in class I systems. Biol. Direct 2017, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Deveau, H.; Barrangou, R.; Garneau, J.E.; Labonte, J.; Fremaux, C.; Boyaval, P.; Romero, D.A.; Horvath, P.; Moineau, S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008, 190, 1390–1400. [Google Scholar] [CrossRef] [Green Version]
- Horvath, P.; Romero, D.A.; Coute-Monvoisin, A.C.; Richards, M.; Deveau, H.; Moineau, S.; Boyaval, P.; Fremaux, C.; Barrangou, R. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 2008, 190, 1401–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojica, F.J.M.; Diez-Villasenor, C.; Garcia-Martinez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.J.; Mtui, D.; Oswald, B.P.; Van Leuven, J.T.; Vallender, E.J.; Schultz-Darken, N.; Ross, C.N.; Tardif, S.D.; Austad, S.N.; Forney, L.J. Comparative genomics of Bifidobacterium species isolated from marmosets and humans. Am. J. Primatol. 2019, 81, e983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [Green Version]
- Horvath, P.; Coute-Monvoisin, A.C.; Romero, D.A.; Boyaval, P.; Fremaux, C.; Barrangou, R. Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int. J. Food Microbiol. 2009, 131, 62–70. [Google Scholar] [CrossRef]
- Barrangou, R.; Dudley, E.G. CRISPR-Based Typing and Next-Generation Tracking Technologies. Annu. Rev. Food Sci. Technol. 2016, 7, 395–411. [Google Scholar] [CrossRef]
- Fukiya, S.; Sakanaka, M.; Yokota, A. Chapter 15—Genetic Manipulation and Gene Modification Technologies in Bifidobacteria. In The Bifidobacteria and Related Organisms; Mattarelli, P., Biavati, B., Holzapfel, W.H., Wood, B.J.B., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 243–259. [Google Scholar] [CrossRef]
- Zhang, J.; Zong, W.; Hong, W.; Zhang, Z.T.; Wang, Y. Exploiting endogenous CRISPR-Cas system for multiplex genome editing in Clostridium tyrobutyricum and engineer the strain for high-level butanol production. Metab. Eng. 2018, 47, 49–59. [Google Scholar] [CrossRef]
- Goh, Y.J.; Klaenhammer, T.R. Genetic mechanisms of prebiotic oligosaccharide metabolism in probiotic microbes. Annu. Rev. Food Sci. Technol. 2015, 6, 137–156. [Google Scholar] [CrossRef]
- Sela, D.A. Bifidobacterial utilization of human milk oligosaccharides. Int. J. Food Microbiol. 2011, 149, 58–64. [Google Scholar] [CrossRef]
- James, K.; O’Connell Motherway, M.; Penno, C.; O’Brien, R.L.; van Sinderen, D. Bifidobacterium breve UCC2003 employs multiple transcriptional regulators to control metabolism of particular human milk oligosaccharides. Appl. Environ. Microbiol. 2018, 84, e02774-17. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chen, T.; Liu, Y.; Lv, X.; Li, J.; Du, G.; Ledesma-Amaro, R.; Liu, L. CRISPRi allows optimal temporal control of N-acetylglucosamine bioproduction by a dynamic coordination of glucose and xylose metabolism in Bacillus subtilis. Metab. Eng. 2018, 49, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.L.; Mullis, A.S.; Leenay, R.T.; Beisel, C.L. Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression. Nucleic Acids Res. 2015, 43, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.; Su, T.; Qi, Q.; Liang, Q. Easy regulation of metabolic flux in Escherichia coli using an endogenous type I-E CRISPR-Cas system. Microb. Cell Fact. 2016, 15, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selle, K.; Klaenhammer, T.R.; Barrangou, R. CRISPR-based screening of genomic island excision events in bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 8076–8081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canez, C.; Selle, K.; Goh, Y.J.; Barrangou, R. Outcomes and characterization of chromosomal self-targeting by native CRISPR-Cas systems in Streptococcus thermophilus. FEMS Microbiol. Lett. 2019, 366, fnz105. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, M.; Nethery, M.A.; Hidalgo-Cantabrana, C.; Barrangou, R. Comprehensive Mining and Characterization of CRISPR-Cas Systems in Bifidobacterium. Microorganisms 2020, 8, 720. https://doi.org/10.3390/microorganisms8050720
Pan M, Nethery MA, Hidalgo-Cantabrana C, Barrangou R. Comprehensive Mining and Characterization of CRISPR-Cas Systems in Bifidobacterium. Microorganisms. 2020; 8(5):720. https://doi.org/10.3390/microorganisms8050720
Chicago/Turabian StylePan, Meichen, Matthew A. Nethery, Claudio Hidalgo-Cantabrana, and Rodolphe Barrangou. 2020. "Comprehensive Mining and Characterization of CRISPR-Cas Systems in Bifidobacterium" Microorganisms 8, no. 5: 720. https://doi.org/10.3390/microorganisms8050720
APA StylePan, M., Nethery, M. A., Hidalgo-Cantabrana, C., & Barrangou, R. (2020). Comprehensive Mining and Characterization of CRISPR-Cas Systems in Bifidobacterium. Microorganisms, 8(5), 720. https://doi.org/10.3390/microorganisms8050720