1. Introduction
Candida albicans is an opportunistic fungal pathogen found on human mucosae, such as those of the oral, gastrointestinal, and genitourinary tracts [
1].
C. albicans exhibits the ability to adhere to surfaces and form biofilms, which are complex communities of microorganisms attached to biotic or abiotic surfaces and surrounded by a self-produced matrix [
2]. Under specific host conditions, especially immunodeficiency,
C. albicans causes a local infection known as candidiasis, which may spread systemically and result in candidemia, a nosocomial infection with high mortality rates [
3]. Furthermore,
C. albicans possess the ability to undergo morphological transition from commensal yeast to filamentous forms (pseudohyphae and hyphae), in a process known as polymorphism [
4]. In the hyphal form,
C. albicans is pathogenic, with the ability to invade host tissue [
5]. Therefore, polymorphism is a significant virulence factor of this opportunistic fungal species [
4,
6].
C. albicans also grows as biofilms on biomedical devices, such as catheters and dental prostheses [
7]. Oral candidiasis associated with denture use, known as denture stomatitis, is an oral infection prevalent in up to 70% of denture users, with larger proportions of females affected than males. Despite the multifactorial etiology of denture stomatitis, biofilm formation by
Candida spp. on the denture surface is considered the main etiological factor [
8]. Moreover, candidal biofilms formed in indwelling catheters act as a source of infection in candidemia, whose treatment requires both antifungal administration and timely removal of the catheter [
9].
The biofilm matrix protects microbial cells growing within the biofilm from external agents, and acts as a barrier to antimicrobials and the host’s immune cells. Thus, in a biofilm, microbial cells exhibit higher tolerance to antimicrobials [
10]. The matrix is also responsible for the three-dimensional architecture and mechanical properties of biofilms, such as their adhesive strength, cohesion, and stiffness, which enables co-aggregation of microorganisms and facilitates formation of microcolonies. The environment of biofilm modulates gene expression, metabolism, and cell signaling by molecules, influencing microbial behavior (quorum sensing) [
11]. The biofilm matrix of
C. albicans is composed of carbohydrates, such as β-glucans and mannan, as well as proteins, lipids, and extracellular DNA (eDNA) [
12], and hexosamines, uronic acid, and phosphorus [
13]. The
Candida albicans biofilm matrix, especially the polysaccharides, contributes to co-aggregation of bacteria and protects bacterial cells against antibacterial agents [
14,
15,
16,
17].
Candida albicans biofilms also produce extracellular vesicles, whose cargo (polysaccharides and proteins) forms the biofilm matrix and confers drug tolerance [
18].
Antifungal agents used to treat infections caused by
Candida spp. are divided into the following three main classes: polyenes, azoles (imidazoles and triazoles), and echinocandins [
19]. Nystatin is a topical polyene used to treat palatal mucosa affected by denture stomatitis [
20,
21]; fluconazole (triazole) and caspofungin (echinocandin) are used for the treatment of invasive candidiasis [
9]. However,
C. albicans cells growing within biofilms have been shown to exhibit 4 to 128 times lower antifungal susceptibility than their planktonic counterparts [
22]. In addition to the inherent microbial tolerance of biofilms, the widespread use and misuse of antifungals have led to the development of antifungal resistance [
6]. Such resistance leads to treatment failure with commercially available antifungal agents, and persistent infection. As a result, antimicrobial resistance remains a global threat to public health [
23]. Therefore, the development of treatments aimed at overcoming antifungal resistance and disrupting biofilm matrix on medical devices is critical.
N-acetylcysteine (NAC) is a mucolytic and thiol compound used to reduce the viscosity of pulmonary mucous in patients with chronic respiratory diseases, such as bronchitis [
24] and cystic fibrosis [
25], as well as being used to treat paracetamol (acetominophen) overdose [
26]. The sulfhydryl component of the NAC molecule breaks the disulfide bonds in mucous [
27], and also acts as a precursor for glutathione synthesis and as a reactive oxygen species (ROS) scavenger, conferring an antioxidant effect to NAC [
28]. Other studies have demonstrated antimicrobial and antibiofilm properties of NAC against pathogenic bacteria, such as
Mycobacterium tuberculosis [
29],
Klebsiella pneumoniae,
Escherichia coli,
Pseudomonas aeruginosa [
30,
31], methicillin-resistant
Staphylococcus aureus and
Staphylococcus epidermidis [
32],
Prevotella intermedia [
33],
Actinomyces naeslundii,
Lactobacillus salivarius,
Streptococcus mutans, and
Enterococcus faecalis [
34,
35]. Studies have also shown synergism between NAC and antibiotics [
31,
32,
33,
36,
37]. There have also been reports that NAC disrupts mature bacterial biofilms and reduces the matrix polysaccharides [
31,
38]. In this context, investigating new applications of existing drugs (drug repurposing) has the advantage of reducing the time and cost spent in the development of new medications, such as by negating the need for clinical safety trials [
39].
The antifungal and antibiofilm action of NAC alone or in association with antifungal agents (fluconazole, ketoconazole, and amphotericin B) against
C. albicans has also been reported [
37,
40,
41]. However, the antifungal effects of NAC against resistant strains and its effects on the components of the
C. albicans biofilm matrix, to the best of our knowledge, are still unknown. Therefore, the aim of this study was to evaluate the effect of NAC on biofilms of fluconazole-resistant (CaR) and -susceptible
C. albicans (CaS). Aside from the fungal viability and biomass, we focused on the effects of NAC on the components of the biofilm matrix: polysaccharides, proteins, lipids, and eDNA. We also evaluated the interaction of NAC with agents belonging to the following three classes of antifungals: polyenes (nystatin), azole (fluconazole), and echinocandin (caspofungin). Additionally, we analyzed biofilms treated with NAC using confocal scanning laser microscopy.
2. Materials and Methods
2.1. Strains and Culture Conditions
Standard strains of CaS (SC5314) and CaR (ATCC 96901—American Type Culture Collection, Rockville, MD, USA) were stored at −80 °C in yeast nitrogen broth (YNB; Difco, Detroit, MI, USA) with glycerol 50%; they were thawed and reactivated in Sabouraud dextrose agar with 0.05 mg/mL of chloramphenicol (SDA; Acumedia Manufacturers Inc., Lansing, MI, USA) at 37 °C for 48 h. Five colonies of each strain were transferred to YNB with 100 mM of glucose (YNBg) and incubated overnight at 37 °C. Each fungal suspension was diluted 1:10 in fresh YNBg and incubated at 37 °C until the optical density at 540 nm (OD540) reached the mid-log phase of growth (mean ± standard deviation, SD, of OD540: 0.424 ± 0.042 and 0.460 ± 0.030 arbitrary units, au, for CaS and CaR, respectively), corresponding to 6.64 × 106 ± 2.72 × 106 and 6.32 × 106 ± 1.49 × 106 colony forming units per milliliter (CFU/mL) for CaS and CaR, respectively. Then, each fungal suspension was centrifuged (10,000× g for 10 min at 4 °C) and resuspended in Roswell Park Memorial Institute (RPMI) 1640 medium—bicarbonate free (Sigma-Aldrich, St. Louis, MO, USA, product code R6504-10X1L), buffered with morpholinepropanesulfonic acid (MOPS; Sigma-Aldrich), and supplemented with 2% D-glucose (Synth, São Paulo, Brazil), pH 7.0 (RPMId). Then, the OD540 for each strain was measured (0.585 ± 0.167 and 0.498 ± 0.005 au for CaS and CaR, respectively).
2.2. Antifungal Evaluation of NAC
NAC (Sigma-Aldrich) was diluted in RPMId at 100 mg/mL, according to the manufacturer’s instructions, for use in antifungal evaluation. The minimum inhibitory concentration (MIC) and minimum fungicidal concentration (MFC) were evaluated by the microdilution method, according to the recommendations of the Clinical and Laboratory Standards Institute (CLSI) [
42] and the European Committee for Antimicrobial Susceptibility Testing (EUCAST) [
43], with some modifications. Aliquots (100 µL) of twofold serial dilutions of NAC (final concentrations of 50 to 12.5 mg/mL) in RPMId were incubated with 100 µL of each fungal suspension individually for 24 h at 37 °C in 96-well, U-bottom microtiter plates (TPP, Trasadingen, Switzerland). A temperature of 37 °C was chosen, instead of 35 °C as recommended by the CLSI, for consistency with human body temperature and the temperature used for in vitro biofilm formation [
44]. Aliquots (200 µL) of each fungal suspension with RPMId and of RPMId alone were used as controls (without NAC). For the test samples, each fungal suspension was diluted in RPMId with NAC to give a final concentration of 0.5–2.5 × 10
3 CFU/mL. After incubation, the OD
540 was measured in a spectrophotometer (FLUOstar Omega microplate reader; BMG Labtech GmbH, Offenburg, Germany), to obtain quantitative values and avoid any bias that might arise from visual inspection. The lowest concentration whose OD
540 value was similar to that observed for RPMId alone was defined as the MIC. In addition, samples were plated on SDA and incubated at 37 °C for 48 h. After incubation, the colonies were counted (CFU/mL) and the lowest NAC concentration that inhibited colony growth was defined as the MFC.
2.3. Time-Kill Curves
Time-kill curves were performed using the same protocol as for the MIC analysis above. Both strains were grown until mid-log phase, as described in
Section 2.1. Aliquots of 100 µL of NAC were added to each fungal suspension (100 µL) to achieve final NAC concentrations of 50 to 0.09 mg/mL for CaS, and of 50 to 12.5 mg/mL for CaR, using a 96-well, U-bottom microtiter plate. RPMId alone (without NAC) with the fungal inocula were used as negative controls. The final concentration of fungal suspension was 0.5–2.5 × 10
3 CFU/mL. The absorbances of the samples in the plates were measured with a spectrophotometer (at 540 nm) during 24 h of incubation at 37 °C, with readings taken every 2 h.
2.4. Evaluation of NAC Activity on Biofilm Formation
Aliquots (200 µL) of each fungal suspension were transferred to 96-well, flat-bottom microtiter plates (Kasvi, São José dos Pinhais, Brazil) and incubated at 37 °C for 90 min (adhesion phase). Then, the medium from each well was aspirated, and each well was washed twice with 200 µL of sterile phosphate-buffered saline (PBS—0.136 M NaCl, 1 mM KH
2PO
4, 2 mM KCl, 10 mM Na
2HPO
4, pH 7.4) to remove non-adherent cells. NAC at 100, 50, 25, and 12.5 mg/mL in RPMId was added to each well (200 µL) and the plates incubated at 37 °C for 48 h to allow biofilm formation [
44]. Control samples received only RPMId without NAC.
After 48 h, the biofilms were washed twice with 200 µL of PBS, and biofilm viability (CFU/mL) and total biomass (using the crystal violet assay) [
45] were assessed. For viability analysis, 200 µL of PBS was added into each well, biofilms were individually scraped using a pipette tip, and tenfold serial dilutions in PBS were plated in duplicate on SDA, which was incubated at 37 °C for 48 h for colony counting. For biofilm total biomass analysis, 200 µL of 80% methanol was added to each well. After 15 min, the methanol was removed; then, 200 µL of 1% crystal violet was added to each well. After 5 min, samples were washed five times with PBS, and 200 µL of 33% acetic acid was added to remove the stain. This solution was transferred to another 96-well microtiter plate, and the absorbance at 570 nm was measured [
45].
2.5. Effect of the Incubation Time with NAC on Biofilm Formation
The effect of NAC on biofilm formation was evaluated as described above, but the incubation period was reduced. The samples were evaluated for biofilm viability and total biomass after 6, 12, and 24 h of biofilm formation, with and without (control) NAC.
2.6. Evaluation of the Effect of NAC on Pre-Formed Biofilms
Biofilms of each strain were grown as described above, without NAC, for 48 h. After the first 24 h of incubation, 100 µL of each well sample was removed and 100 µL of fresh RPMId was added. After 48 h, the biofilms were washed twice with PBS, and NAC at 100, 50, 25, and 12.5 mg/mL in RPMId was added to the wells (200 µL) and incubated at 37 °C for 24 h. Controls received only RPMId without NAC. After incubation, the biofilm viability (CFU/mL) and total biomass were evaluated as described above.
2.7. Effect of NAC on the Composition of the Biofilm Matrix
In order to evaluate the effect of NAC on the composition of the biofilm matrix, biofilms of each fungal strain were grown in triplicate in 24-well microtiter plates (Kasvi) using 2.5 mL of individual fungal suspension in each well. NAC was added after the adhesion phase (50 and 12.5 mg/mL for biofilm formation experiments) or after 48 h of biofilm growth (100 mg/mL for pre-formed biofilm experiments, followed by incubation for 24 h), as described above. Biofilms were washed twice with sterile 0.89% NaCl, and after the addition of 2.5 mL of 0.89% NaCl they were scraped using a pipette tip for 1 min. The content of triplicate wells (7.5 mL) was transferred to a single Falcon tube (Kasvi): 100 µL was used for tenfold serial dilutions and plating on SDA (CFU/mL), and 100 µL was used for biofilm dry weight assessment. The remainder volume (7.3 mL) was centrifuged (5500× g; 10 min; 4 °C), and washed three times with 2 mL of sterile milliQ water. The supernatants were pooled (13.3 mL) and the pellet was resuspended in 2 mL of milliQ water. Aliquots of the supernatant were used to quantify the proteins (1.1 mL), water-soluble polysaccharides (WSPs, 3 mL), and eDNA (650 µL). The pellet suspension was used to quantify the insoluble dry weight (500 µL), proteins (550 µL), alkali-soluble polysaccharides (ASPs, 750 µL), and lipids (200 µL).
2.7.1. Biofilm Dry Weight
Aliquots (100 µL) of the biofilm scraped were transferred to microtubes that had been previously weighed. Samples were centrifuged (12,000 rpm; 15 min; 4 °C), the supernatant was discarded, and 300 µL of 99% ethanol was added. Then, samples were vortexed and kept at −80 °C for 18 h. Afterwards, the samples were centrifuged (12,000 rpm; 10 min; 4 °C), the supernatants were discarded, and the pellets were washed three times with 70% ethanol and air dried at room temperature. The microtubes were weighed again, and the difference between the weights was the biofilm dry weight [
46,
47]. To weight the samples, a precision weighing balance, with five decimal places (AUW 220D, Shimadzu, Tokyo, Japan), was used.
2.7.2. Insoluble Dry Weight
A 500 µL aliquot of the pellet suspension was transferred to a previously weighed piece of aluminum foil. Each sample was dried at 100 °C, and then weighed again. The difference between the weights was the insoluble dry weight.
2.7.3. Quantification of Soluble Proteins
A standard curve was prepared using bovine serum albumin (BSA; Sigma-Aldrich) at 0.0, 0.5, 1.0, 2.0, 4.0, 6.0, and 8.0 µg/mL in order to correlate protein concentrations with absorbance values (
Figure S1A). Aliquots of 500 µL from the biofilm supernatants and from each standard curve sample were individually mixed with 500 µL of Bradford reagent (Sigma-Aldrich) [
48] in duplicate. After 20 min mixing at 70 rpm, absorbance was measured at 595 nm.
2.7.4. Quantification of Insoluble Proteins
A 550 µL aliquot of the biofilm pellet suspension was heated at 100 °C for 90 min with mixing at 1,000 rpm, in order to extract its protein content. Then, 5 µL of the sample was added to 250 µL of Bradford reagent (Sigma-Aldrich) [
48] in duplicate. Standard curves of BSA at 0.0, 0.5, 1.0, 2.0, 4.0, 6.0, and 8.0 µg/mL and at 0.0, 0.1, 0.2, 0.4, 0.6, 0.8, 1.0, and 1.2 mg/mL were also prepared. After 20 min mixing at 70 rpm, the absorbances of the test and standard curve samples were measured at 595 nm.
2.7.5. Quantification of WSPs
A 3 mL aliquot of the biofilm supernatant was mixed with 7.5 mL of 99% ethanol. Samples were vortexed and kept at −80 °C for 18 h to precipitate the polysaccharides. Then, samples were vortexed, centrifuged (9500×
g; 20 min; 4 °C), washed three times with 70% ethanol, and air dried at room temperature. The pellets were individually resuspended in 1 mL of water, and the total carbohydrate was quantified by the phenol-sulfuric acid method [
49]. A standard curve using glucose at 0.00, 3.75, 6.25, 12.50, 25.00, 50.00, 75.00, 100.00, and 125.00 µg/mL was created (
Figure S1B). Aliquots of 200 µL of samples in triplicate and of each standard curve sample in duplicate were individually mixed with 200 µL of 5% phenol. Then, 1 mL of sulfuric acid was added to each sample. After 20 min, absorbance was measured at 490 nm.
2.7.6. Quantification of ASPs
A 750 µL aliquot of the biofilm pellet suspension was transferred to a previously weighed microtube. Samples were then centrifuged (13,000×
g; 10 min; 4 °C), the supernatants were discarded, and the pellets were dried in a sample concentrator (RVC 2-18C D, Martin Christ Gefriertrocknungsanlagen GmbH, Osterode, Germany) for 4.5 h and weighed. For ASP extraction, 300 µL of 1 N NaOH was added for each 1 mg of the sample’s dry weight. After 2 h of incubation, samples were centrifuged (13,000×
g; 10 min), and the supernatants were individually collected. Another volume of 300 µL of 1 N NaOH/(mg of dry weight) was added to the pellets, vortexed, and incubated for 2 h. After centrifugation, the supernatant was collected and pooled with the previous supernatant. The same steps described were repeated with the pellet for a third time, but instead of being incubated for 2 h, samples were vortexed and centrifuged. The last supernatant was pooled with the previous supernatants and the pellet was discarded. Thereafter, three volumes of 99% ethanol were added to each sample, which was incubated at −20 °C for 18 h for ASP precipitation. After this time, samples were centrifuged (13,000×
g; 20 min; 4 °C), the supernatants were discarded, and the pellets were washed three times with 70% ethanol as described for WSPs. The air-dried pellets were resuspended in the same volume of 1 N NaOH and used for quantification of total carbohydrate by the phenol-sulfuric acid method [
49], as described for WSPs. A standard curve with 1 N NaOH and glucose at final concentrations of 0.00, 3.75, 6.25, 12.50, 25.00, 50.00, 75.00, 100.00, and 125.00 µg/mL was prepared (
Figure S1C).
2.7.7. Quantification of eDNA
To extract eDNA from the samples, 650 µL of phenol:chloroform:isoamilic acid (25:24:1) was added to an aliquot of 650 µL of each biofilm supernatant [
50]. Then, the samples were centrifuged (10,000 rpm; 5 min; 4 °C), and the supernatants were treated with chloroform:isoamilic acid (24:1). After centrifugation, the supernatants were transferred to another tube, and three volumes of 99% ethanol and 1/10 volume of 3 M sodium acetate were added. Samples were incubated at −20 °C for 18 h for DNA precipitation. The samples were then centrifuged (13,000×
g; 20 min; 4 °C), and the pellets were washed three times with 70% ethanol, air-dried, and resuspended in Tris-EDTA (TE) buffer. The DNA concentration was measured in a nanospectrophotometer at 260 nm [
50].
2.7.8. Quantification of Lipids
A 200 µL aliquot of the biofilm pellet suspension was centrifuged (13,000×
g; 10 min; 4 °C), the supernatant was removed, and the pellet was resuspended in 100 µL of 99% ethanol. Then, samples were vortexed, and the lipids were quantified by sulfo-phospho-vanillin reaction [
51]. A standard curve using olive oil diluted in 99% ethanol at 0, 0.375, 0.750, 1.125, 2.250, 4.500, 7.500, and 9.750 mg/mL was prepared (
Figure S1D). Aliquots of 20 µL of each standard curve sample in duplicate were evaporated by incubation at 70 °C for 15 min. Then, 200 µL of sulfuric acid was added to each experimental sample and to each standard curve sample and vortexed. The test tubes were placed in boiling water for 10 min, and cooled in cold water for 5 min. Afterwards, 10 mL of the phospho-vanillin reagent was added to each tube, which was vortexed and incubated in boiling water for 15 min. The samples were cooled, and after 30 min absorbance was measured at 540 nm.
2.8. Interaction of NAC with Antifungal Agents
The checkerboard microdilution method was used to evaluate the interaction of NAC with the antifungal agents nystatin (polyene), fluconazole (azole), and caspofungin (echinocandin) against planktonic cultures. Antifungal agents were purchased from Sigma-Aldrich and diluted (stock solutions) in dimethylsulfoxide (DMSO; for fluconazole and nystatin) or RPMId (caspofungin). Each drug solution was diluted 40× in the culture medium (RPMId) (work solution), so the final DMSO concentration was 2.5%. Twofold serial dilutions of NAC (50 µL) (in RPMId) and antifungal agents (50 µL) were distributed along the rows and columns, respectively, of a 96-well, U-bottom microtiter plate (Kasvi). The final concentrations of the drugs for both strains are shown in
Table 1. Aliquots of 100 µL of CaS or CaR were added at the final concentration of 0.5–2.5×10
3 CFU/mL. The control consisted of fungal inoculum with no drug (only the vehicle solution). After 24 h of incubation at 37 °C, the OD
540 was measured; the control and samples whose OD
540 values were lower than the control were diluted and plated on SDA for colony counting. The fractional inhibitory concentration index (FICI) was determined by the sum of the FICI of each agent (FICI = FICI
NAC + FICI
antifungal agent). The FICI of each agent was calculated by dividing the MIC of the agent in combination by the MIC of the agent alone (FICI
A = MIC
A in the presence of B/ MIC
A alone). The interpretation of the FICI value was as follows: FICI < 0.5, synergism; 0.5 ≤ FICI ≤ 4.0, no interaction; and FICI > 4.0, antagonism [
52]. Due to shortcomings of the FICI method [
53], Bliss independence analysis [
53,
54,
55] was performed, which is based on the idea that each drug acts independently following the probability of independent events. This model is described by the equation: E
IND = E
A + E
B − E
A × E
B for a combination of drug A at concentration
a with drug B at concentration
b. E
A is the percentage of growth inhibition observed for drug A alone at concentration
a, E
B is the percentage of growth inhibition observed for drug B alone at concentration
b, and E
IND is the expected percentage of growth inhibition of a non-interactive association of drug A at
a with drug B at
b. The difference (ΔE = E
OBS − E
IND) between the observed percentage of growth inhibition (E
OBS) and the expected one (E
IND) defined the interaction of drugs for each concentration as follows: Bliss synergism, when ΔE and its 95% confidence interval (CI) were > 0; Bliss antagonism, when ΔE and its 95% CI were < 0; and Bliss independence, when 95% CI of ΔE overlapped 0 [
53,
54,
55]. The experiments were performed in three independent occasions, FICI and Bliss independence analyses were performed for each combination of drugs for each occasion with the OD
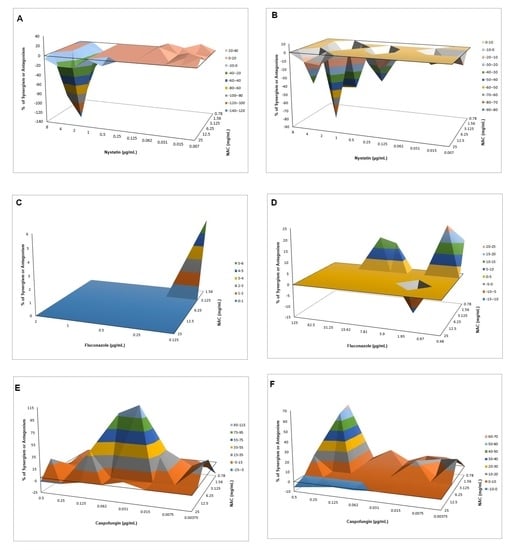
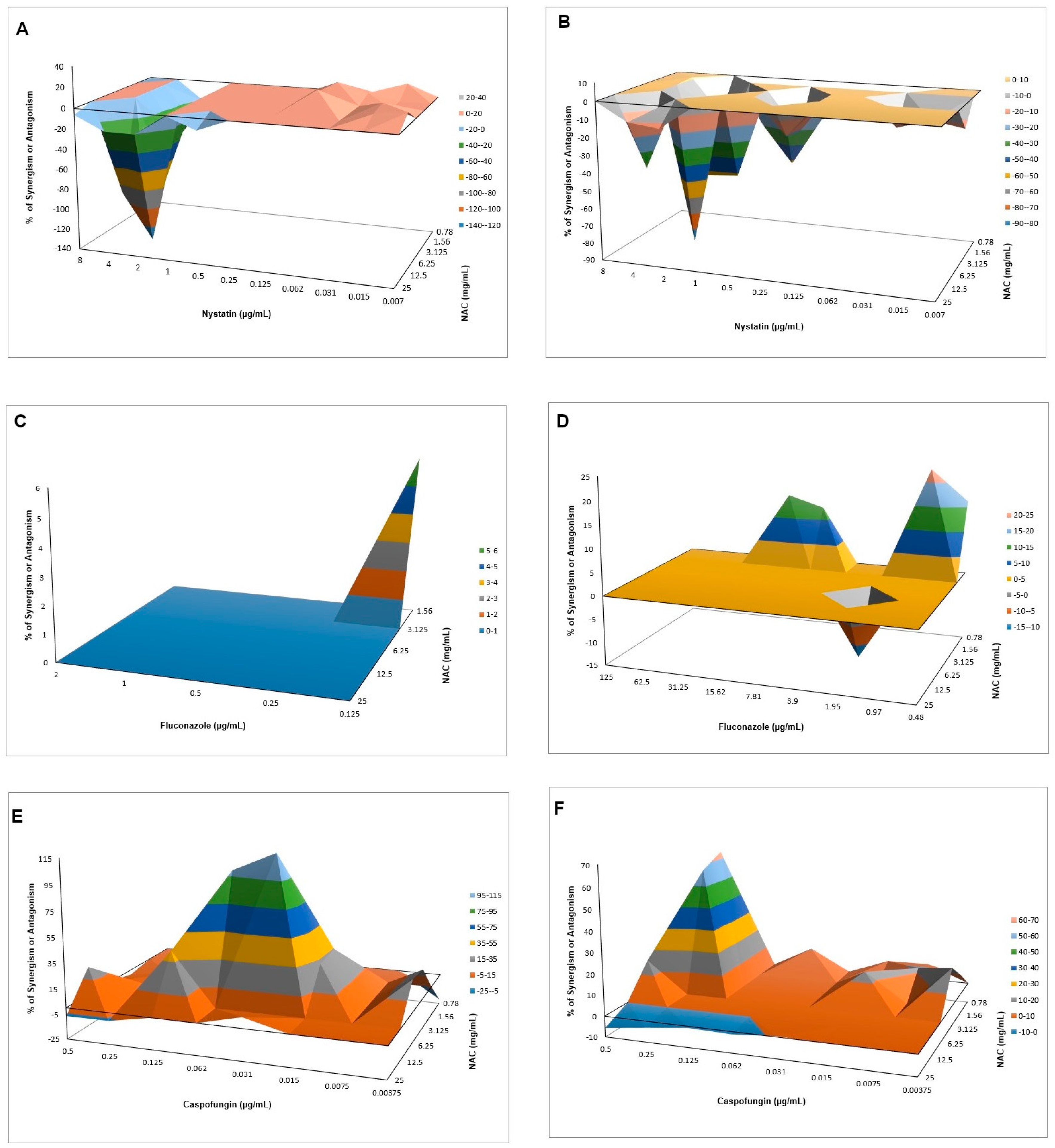
540 values, and mean values of ΔE were used to produce a three-dimensional, interaction surface plot, where peaks above the 0 correspond to synergism and valleys below the 0 correspond to antagonism, while the 0 plane itself indicates no statistically significant interaction.
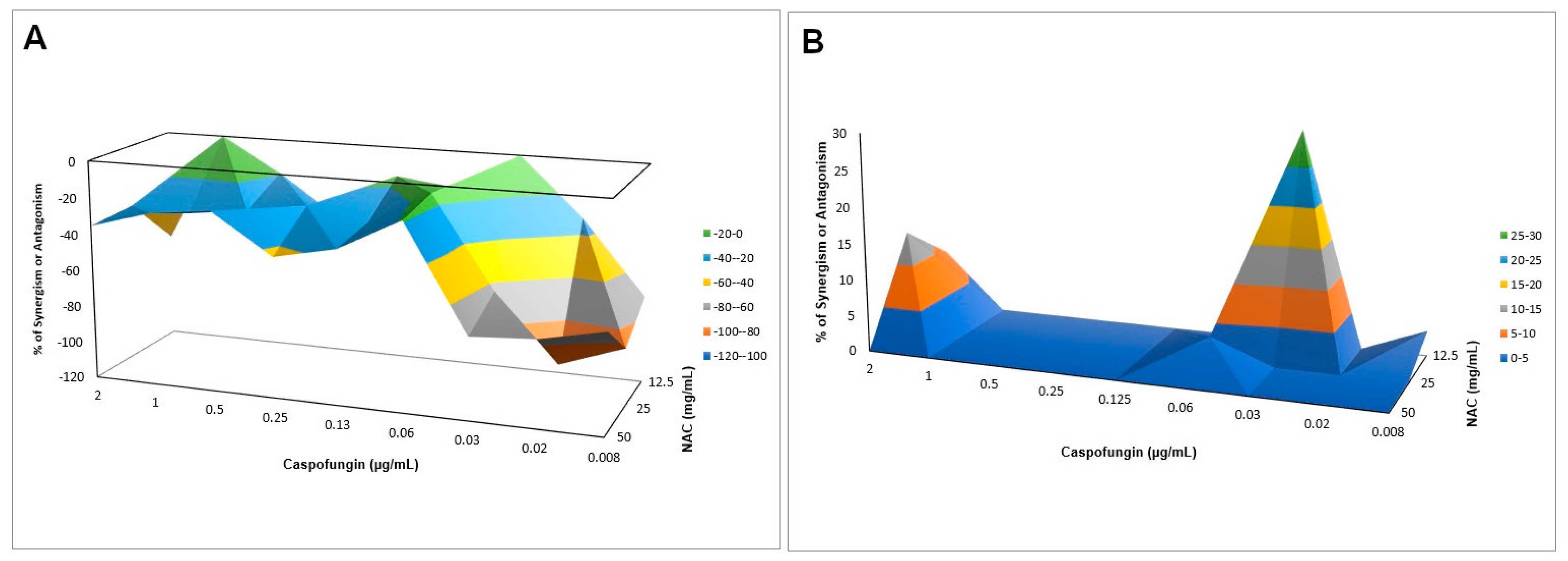
Interaction of NAC with Caspofungin against Biofilms
Biofilms from each strain were cultured for 48 h, as described before (
Section 2.6), then the checkerboard assay was performed as described above. Only caspofungin was evaluated, since it was the only antifungal agent that showed synergism with NAC against planktonic cultures (see Results section). For both strains, the final concentrations of NAC (100 µL) were 50, 25, 12.5, and 0 mg/mL, and of caspofungin (100 µL) were 2, 1, 0.5, 0.25, 0.13, 0.06, 0.03, 0.02, 0.01, and 0 µg/mL. Controls received only the drug vehicle solution. After incubation at 37 °C for 24 h, samples were washed twice with PBS and the metabolic activity was evaluated by the 2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino) carbonyl]-2H-tetrazolium hydroxide (XTT; Sigma-Aldrich) colorimetric assay. The XTT was prepared using ultrapure water at a concentration of 1 mg/mL and kept at −70 °C until needed. A menadione solution (Sigma-Aldrich) was prepared in acetone at 0.007 g/mL just prior to each experiment. The XTT solution prepared in all experiments consisted of PBS with 200 mM of glucose, XTT, and the menadione solution in the following proportions: 158 µL, 40 µL, and 2 µL, respectively. An aliquot of 200 µL of the XTT solution was added into each well with the biofilms, and incubated at 37 °C for 3 h. After this time, aliquots of 100 µL of the XTT solution degradation product (supernatant) were transferred to the wells of another 96-well, flat-bottom plate and absorbance was measured at 492 nm. Bliss independence analysis was performed as described above and illustrated using interaction surface plots.
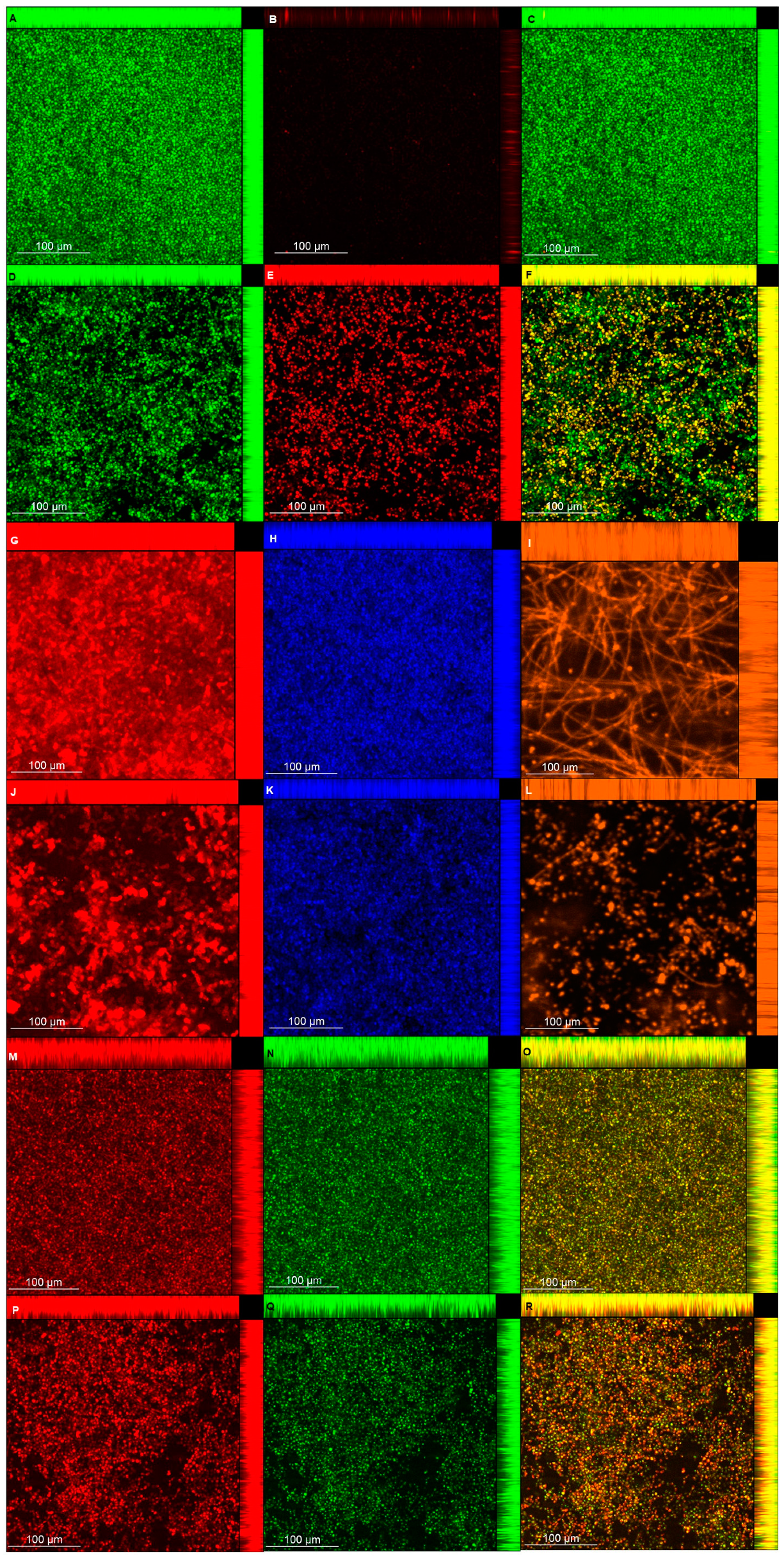
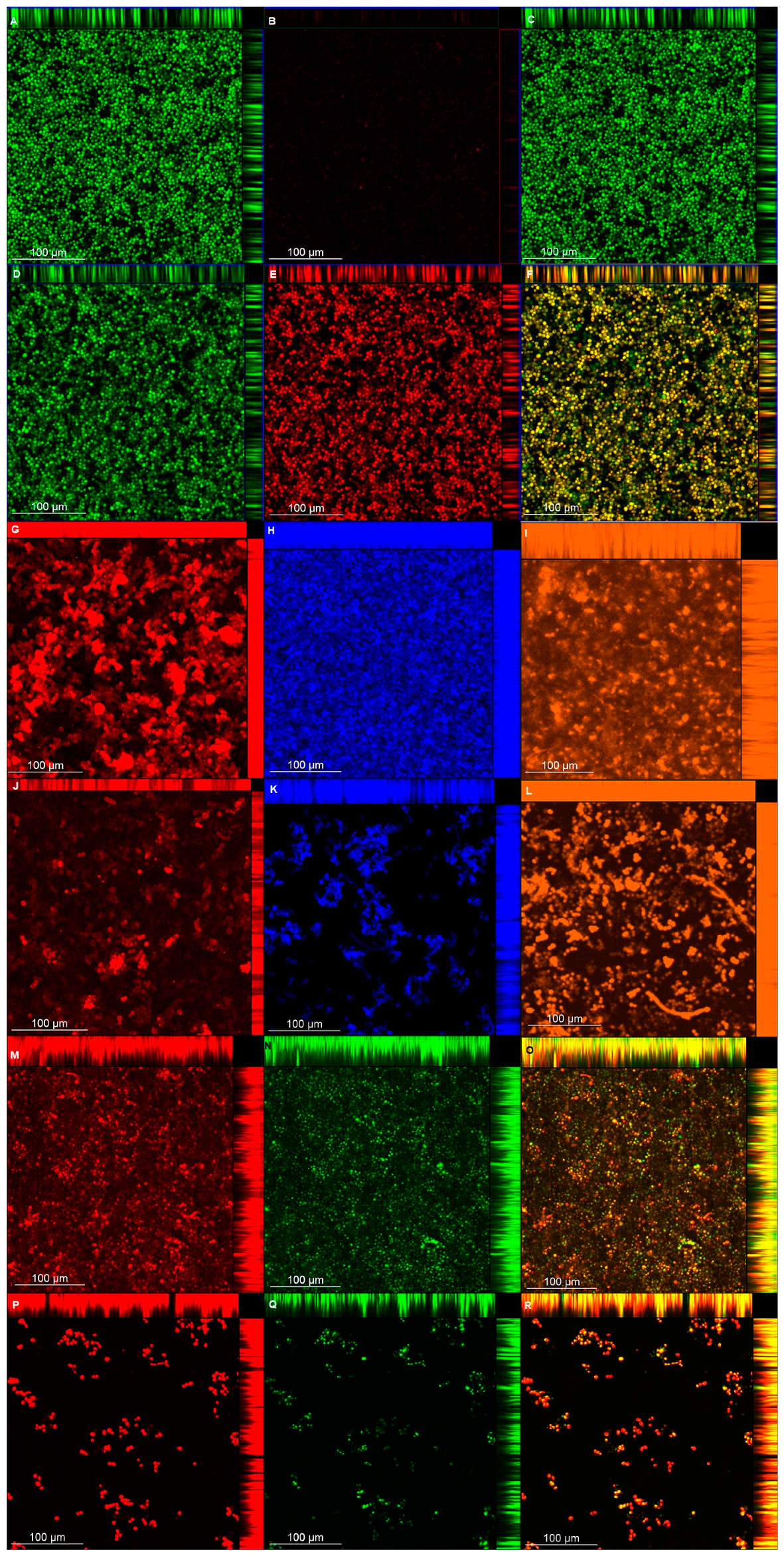
2.9. Confocal Laser Scanning Microscopy (CLSM)
Biofilm viability and matrix composition were also analyzed with a confocal laser microscope (LSM 800 with Airyscan; Carl Zeiss, Germany). Biofilms were grown in duplicate in 24-well, flat-bottom microtiter plates (Kasvi), as described above for pre-formed biofilms, treated with or without (control) NAC at 100 mg/mL for 24 h, washed twice with PBS, stained according to the manufacturers’ instructions for each fluorochrome, and then samples were washed once with PBS before being observed by confocal laser scanning microscopy (CLSM).
For viability analysis, samples were stained with the Live/Dead BacLight Bacterial Viability kit (Molecular Probes, Invitrogen Corp., Carlsbad, CA, USA). Syto9 and propidium iodide (excitation/emission at 488/488–550 nm and 488/656–700 nm, respectively) were diluted 1:1000 and incubated with the biofilm for 45 min in the dark. For polysaccharides (α-D-mannosyl and α-D-glucosyl) [
56], concanavalin A-Alexa Fluor 594 (Molecular Probes) (561/600–700 nm) at 75 µg/mL was incubated with the biofilms for 30 min. For β-glucans [
57], Calcofluor White (Sigma-Aldrich) (405/450 nm) at 0.5 g/L in 10% KOH was incubated with the biofilms for 10 min. For proteins [
58], biofilms were incubated with FilmTracer Sypro Ruby (Molecular Probes) (488/576–700 nm) in the dark for 30 min. For lipids [
59], Nile Red (Sigma-Aldrich) at 50 µg/mL prepared with 10 mL of 25% DMSO/H
2O was incubated with the biofilm for 10 min (488/563 nm for neutral lipids and 561/613–700 nm for polar lipids). In order to distinguish the red fluorescence of Nile Red from the polar lipids, the green color was chosen for neutral lipids as an alternative to orange color.
For each image, biofilm thickness was determined with z-stack readings at 1 µm. Quantification of each matrix component was estimated by the fluorescence intensity generated by each fluorochrome. For each biofilm sample (n = 2), 5 random fields were measured, totaling 10 fields for both duplicates.
2.10. Statistical Analysis
The MIC/MFC and biofilm assays were performed in quadruplicate on three different occasions (n = 12 for each group). The values of CFU/mL were log10 transformed and outliers were removed. For the matrix composition of biofilms, the sample size (α = 5%, power = 80%) was calculated using MedCalc software (version 19.1; MedCalc Software, Mariakerke, Belgium), resulting in n = 6. For each assay, data obtained were rated according to their original volume (biofilm supernatant or biofilm pellet suspension). Biofilm dry weight was used to normalize the data from WSPs, soluble proteins, and eDNA, while insoluble dry weight was used to normalize the data from ASPs, lipids, and insoluble proteins. Normal distribution and homoscedasticity of data were evaluated by Shapiro–Wilk and Levene tests, respectively. Data were analyzed by two-way ANOVA (with strain and treatment as independent variables). For homoscedastic data, a post-hoc Tukey test was used. When data were heteroscedastic, they were evaluated by a post-hoc Games–Howell test. The level of significance was 5% and SPSS software (version 20.0; SPSS Inc., Chicago, IL, USA) was used. Data from the checkerboard assays and CLSM were analyzed as previously described.
4. Discussion
Our investigation demonstrated that NAC showed the same MIC (25 mg/mL) for CaS and CaR, an important finding in the current battle against antimicrobial resistance. Although we did not evaluate the target of NAC in the fungal cell to explain its fungicidal action, one of the limitations of our study, we suggest that NAC may act on the polysaccharides of the fungal cell wall, which is composed of mannan, glucan and chitin [
4]. Such hypothesis is supported by our findings from the matrix components assays (reduction of soluble polysaccharides) and may also explain the same susceptibility of both fluconazole-resistant and -susceptible strains to NAC. Although other studies have shown an antifungal effect of NAC against
C. albicans, only clinical isolates were used [
37,
40,
41] and only one study reported that the strains were susceptible to ketoconazole [
41]. Therefore, the present investigation may be the first to report the antifungal effect of NAC on a resistant strain of
C. albicans. Moreover, this investigation evaluated reference strains since, as was observed by Alnuaimi et al. (2013) [
60], reference strains have higher biofilm growth activity than clinical isolates, which is important for an in vitro evaluation before an in vivo evaluation. The studies mentioned above showed NAC MIC values of 40, 20, and 16.5 mg/mL against clinical isolates of
C. albicans using the macrodilution and agar dilution methods [
40,
41]. The differences between the results of our investigation and those of the mentioned studies [
35,
36] may be ascribed to the different stock concentrations of NAC and the strains used. In all these investigations, the MIC values were close to the NAC stock concentrations used, which demonstrates that high concentrations are needed for a fungistatic effect. However, a fungicidal effect (MFC) was not observed in our investigation, since none of the concentrations of NAC inhibited colony growth. Another study showed that NAC at 160 mg/mL inhibited 99.9% of fungal growth [
41]. However, inhibition of microbial growth should be evaluated by logarithm, since only 1 log of reduction is the same as a 99% decrease. According to the American Society for Microbiology, an agent is considered as antimicrobial when it reduces the microbial growth by at least 3 log [
61]. In the present investigation, significant reductions from 1.81 to 4.06 log were observed. Therefore, although an MFC was not found, NAC was effective in reducing the fungal viability of both strains.
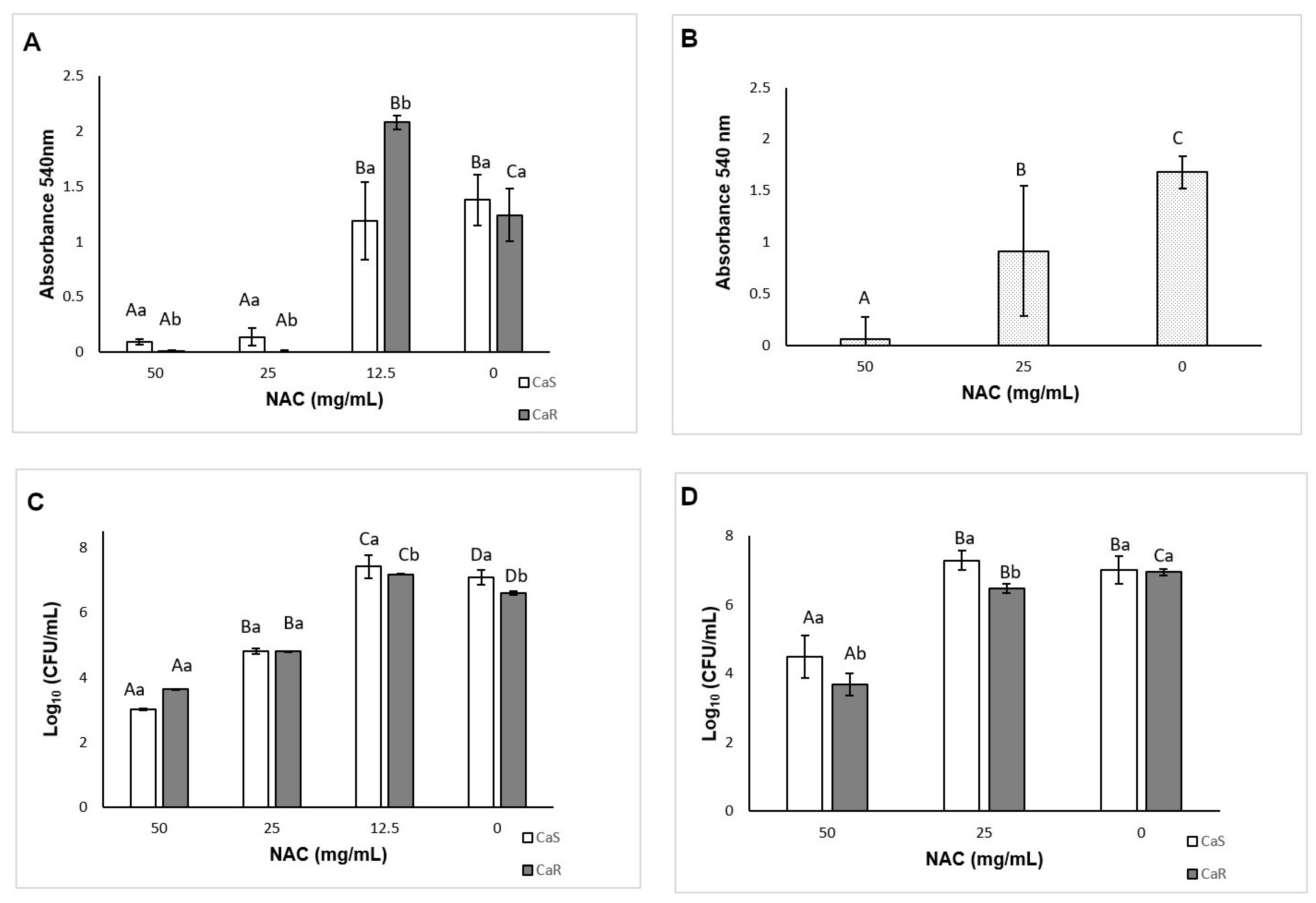
The time-kill curves showed that NAC at the MIC or higher concentrations reduced the fungal growth after 24 h; this was more evident after 6–10 h. Another study also verified that NAC at 80 mg/mL decreased
C. albicans viability after 24 h, while lower concentrations resulted in an increase of fungal viability after 2–4 h [
40]. The present investigation also found an increase of fungal growth when NAC was used at subMIC levels. NAC is a precursor of glutathione, an essential metabolite for
C. albicans; at low concentrations NAC may provide glutathione to
C. albicans, which is metabolized via γ-glutamylcysteine synthetase—GCS1 [
62]. Thus, the results suggest that NAC at subMIC levels may stimulate fungal growth, but this in vitro finding cannot be extrapolated to the clinical scenario.
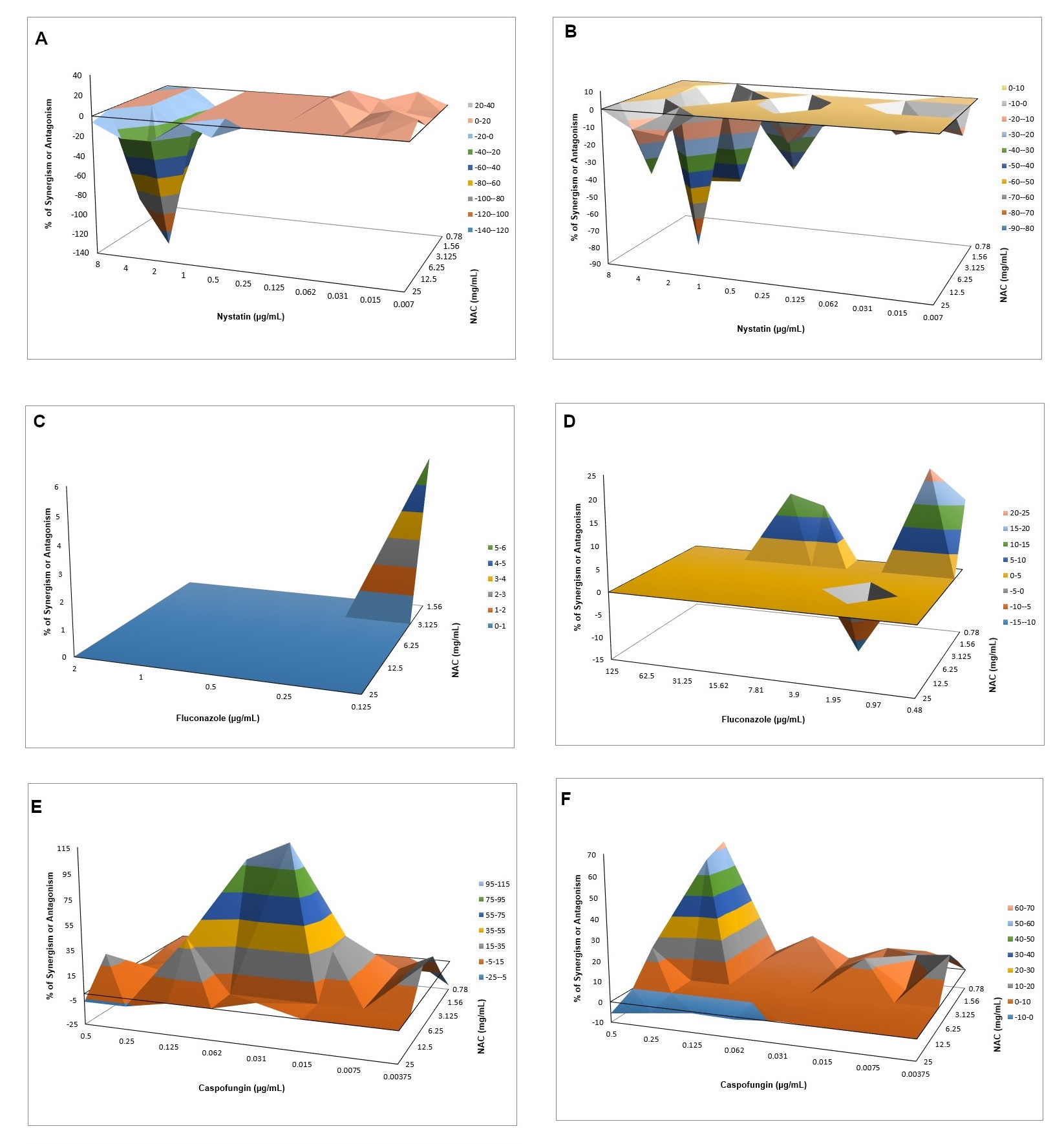
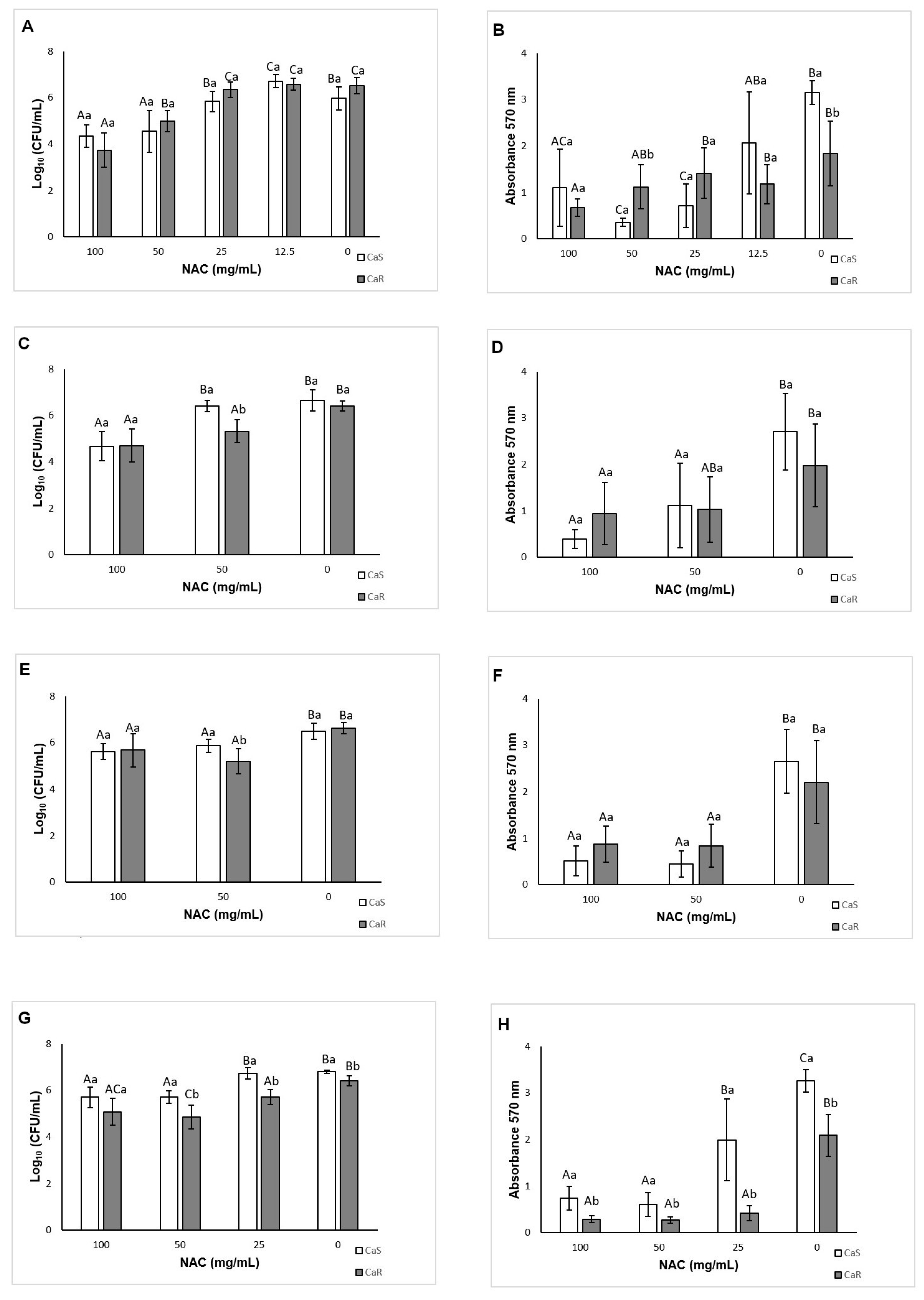
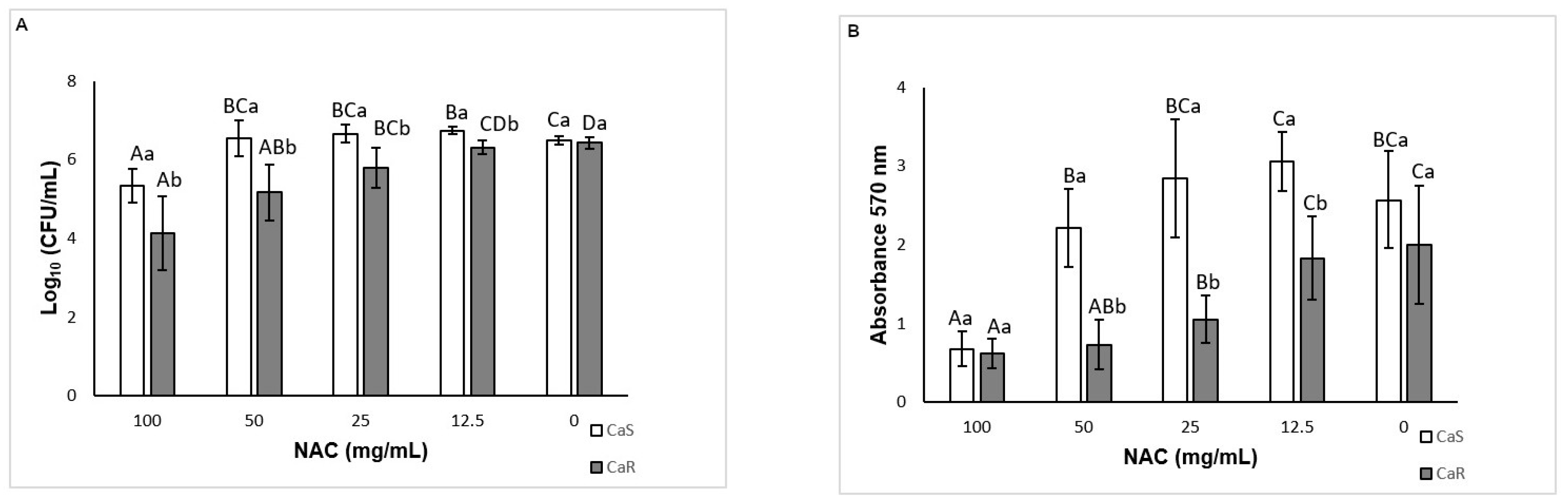
As a preventive strategy to control biofilm development, NAC was added during biofilm formation. NAC at concentrations higher than the MIC inhibited biofilm formation for both strains tested, reducing both the viability and the biomass of the biofilms. This result was not surprising, since the MIC assay demonstrated that these concentrations were fungistatic. Another study showed a reduction of biofilm formation in clinical
C. albicans isolates using NAC at subMIC [
41]. However, in this study, NAC at subMIC (12.5 mg/mL) showed a similar result to the control biofilm (without NAC) for CaR, while NAC at 12.5 mg/mL increased biofilm viability for CaS. These findings confirm that NAC at low concentrations may have an opposite effect to an antimicrobial agent. When the incubation time was evaluated, NAC at the MIC (25 mg/mL) reduced biofilm formation of CaR at the early stage (6 h), while NAC at concentrations higher than the MIC decreased the biofilm formation at all stages (6, 12, and 24 h) of both strains, except for the viability of CaS biofilms incubated with 50 mg/mL NAC for 24 h. Although NAC at the MIC was not evaluated for the intermediate–late stages (12 and 24 h), the findings observed in the incubation time assay corroborated those found for 48 h biofilm formation, in which only NAC at 100 and 50 mg/mL reduced biofilm formation.
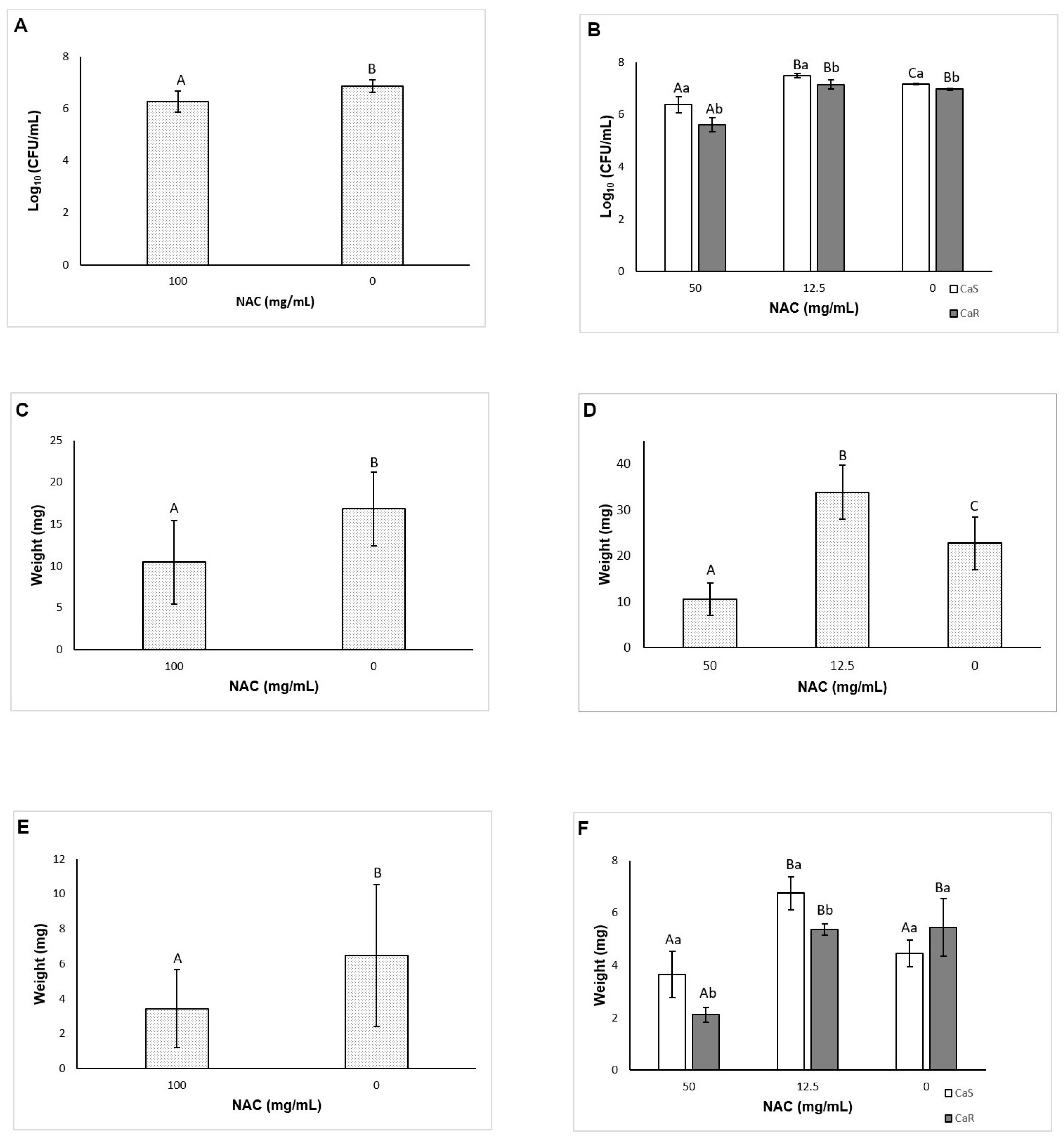
As NAC is a mucolytic agent, it was tested against pre-formed biofilms as a therapeutic strategy for disrupting biofilms. Only NAC at 100 mg/mL reduced the viability and biomass of CaS biofilm, while other concentrations (50, and 25 mg/mL, as well as 100 mg/mL) could also decrease the viability and biomass of CaR biofilm. Therefore, NAC was more effective for pre-formed biofilms of CaR. Other studies also have observed a reduction of the viability and biomass of pre-formed biofilms after treatment with NAC [
37,
41]. However, these studies did not evaluate resistant strains. The present investigation also demonstrated a significant increase in the biofilm viability when NAC was used at 12.5 mg/mL. However, clinical isolates may have a different susceptibility response. Therefore, since we did not evaluate clinical isolates, it may be a limitation of our study.
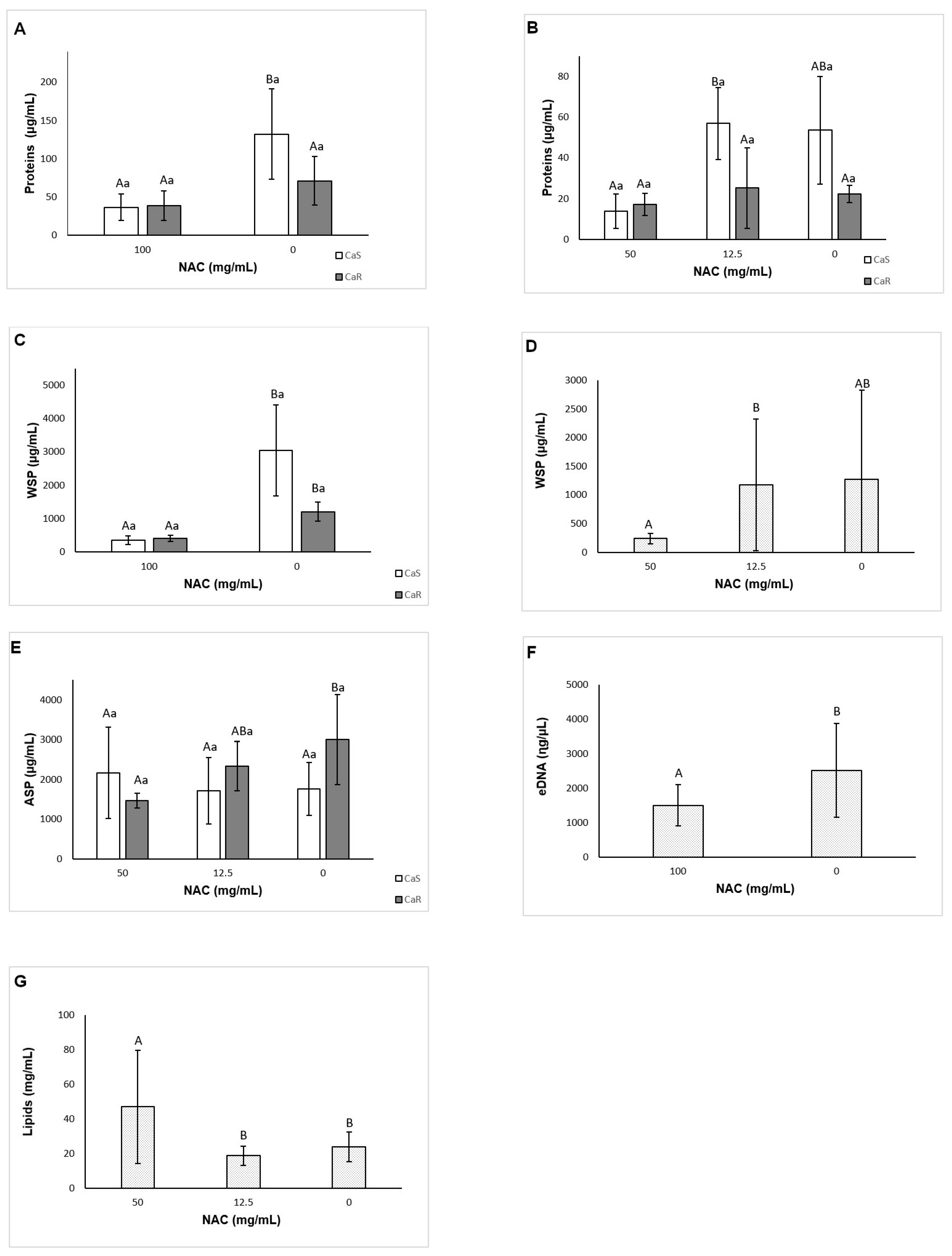
A novel aspect of this investigation is the evaluation of the effect of NAC on the matrix composition of fungal biofilms. Before the quantification of the matrix components, the viability of biofilms treated (pre-formed biofilms) and grown with NAC (biofilm formation in the presence of NAC) was evaluated. The reductions observed for pre-formed biofilms treated with NAC at 100 mg/mL was lower than those observed in the previous assay using 96-well microtiter plate (see
Section 3.5). Probably the larger area of 24-well microtiter plate and volume of fungal inoculum used in the matrix assays could have improved the biofilm grown and reduced its susceptibility to NAC. On the other hand, for biofilm formation, the reductions observed in the presence of NAC at 50 mg/mL and the increase of CaS with NAC at 12.5 mg/mL agreed with those found the previous assay (see
Section 3.3). The results also showed that NAC reduced the soluble components of the biofilm matrix (soluble proteins for CaS, and WSPs and eDNA for both strains) for pre-formed biofilms, but not for biofilm formation. Other investigations have evaluated the effect of NAC only on the extracellular polysaccharides of the biofilm supernatant (WSPs) of bacterial strains [
31,
38]. In those studies, NAC inhibited WSP production at lower concentrations (0.5 and 1 mg/mL) than those needed for viability inhibition [
31,
38]. In this study, only concentrations of NAC higher than the MIC were used to evaluate the matrix components, since NAC at subMIC did not reduce biofilm formation and pre-formed biofilms in the previous assays. Thus, NAC at subMIC would be effective in reducing the WSPs only for bacterial biofilms. Although NAC reduced the viability, the biofilm dry weight, and the insoluble dry weight of both pre-formed biofilms and of biofilms during their formation in the present investigation, it was able to reduce the soluble components of the matrix only for pre-formed biofilm. NAC had no effect on the soluble components during biofilm formation. This result suggests that NAC is able to disrupt pre-formed biofilms by acting on their soluble components, but does not interfere in the synthesis of the matrix components during biofilm formation.
NAC was ineffective in reducing most water-insoluble components (ASPs and lipids), with the exception of the ASPs of CaR during biofilm formation. A previous study evaluated the effect of an antifungal agent (fluconazole) on the biofilm matrix of the same resistant strain used in the present investigation. Using the same methodology to quantify the matrix compounds, the authors observed reduction of WSPs and biomass (total dry weight) for all strains evaluated, and reduction of ASPs only for CaS [
63]. Another investigation demonstrated higher values of ASPs than WSPs from biofilms of CaS treated with fluconazole and nystatin [
64]. All these findings demonstrate that the insoluble components of the biofilm matrix may be less susceptible to antibiofilm approaches.
The CLSM analysis corroborated, in part, the biochemical quantification assays of the matrix components. Biofilms treated with NAC showed a lower thickness than the control biofilms, except for those of CaS stained with Live/Dead (for viability analysis). This unexpected result may be ascribed to the dense cells observed for biofilms of CaS (
Figure 9) compared with those of CaR (
Figure 10), which may hinder the penetration of NAC into the CaS biofilm. Moreover, the fluorescence intensity results for biofilms treated with NAC also showed lower values than for the control biofilms, except for polysaccharides (β-glucans), proteins, and neutral lipids of CaS biofilms. These exceptions may be justified by the insolubility of these components in water, as observed in the matrix-component quantification assays. However, beyond the lower thickness of samples treated with NAC, the images clearly show more “black spaces” in the samples treated with NAC compared with controls. In addition, hyphae were observed only in biofilms stained with Sypro Ruby (proteins). Since the biofilms were grown with an RPMI-based medium, it was expected that we would observe a predominance of hyphae in the samples. It is likely that the supplementation of the medium with glucose and the staining procedure used for the other chromophores may have influenced the yeast morphology of
C. albicans. Another study also used Calcofluor White and Sypro Ruby to evaluate, respectively, the architecture and the permeability of the biofilms of two types of
C. albicans: a heterozygous strain of the mating type locus (MTL; a/α cells) and an MTL-homozygous strain (a/a cells). Although biofilms formed by both kind of cells showed similar architecture, a/α biofilms showed pathogenic traits of impermeability, impenetrability by human phagocytic leukocytes, and fluconazole-resistance, while a/a biofilms did not show these traits [
65].
According to Nett et al. (2007) [
66] and Zarnowski et al. (2014) [
12], the WSP comprises the α-mannans, and the ASP comprises 1,6-β-glucan and 1,3-β-glucan. Mannan is indicated as the most abundant polysaccharide in the fungal biofilm matrix, comprising approximately 87% of the polysaccharide, followed by 1,6-β-glucan at 13%, and a tiny percentage of 1,3-β-glucan, which together give rise to the mannan-glucan complex (MGC) [
12]. Another investigation suggested that the stability of each matrix polysaccharide (α-mannans, 1,6-β-glucan, and 1,3-β-glucan) is responsible for maintaining the MGC, which is crucial for biofilm resistance [
67]. Moreover, the 1,3-β-glucan in the biofilm is also responsible for drug sequestration and fluconazole resistance [
66,
68]. Consequently, a reduction in any polysaccharide of the matrix may increase the susceptibility of the biofilm to antifungal agents. When the enzyme 1,3-β-glucanase was used against
C. albicans, a reduction in the viability of the biofilm but not the planktonic culture was observed [
66]. Moreover, other enzymes promoted detachment of
C. albicans biofilms from a polystyrene surface, such as proteinase K, chitinase, DNAse I, β-
N-acetylglucosaminidase, and lyticase, while lipase type II, phospholipase A2, and protease type XIV did not [
13]. Lyticase, which also hydrolases 1,3-β-glucan, caused the greatest reduction of the biofilms [
13].
When associated with antifungal agents, NAC showed greater synergism with caspofungin for planktonic cultures, which may be explained by the similar mechanism of action of the two drugs. Since caspofungin inhibits 1,3-β-D-glucan synthase, an enzyme necessary for the synthesis of an important fungal cell-wall polysaccharide [
69], and NAC is reported to reduce the polysaccharides of biofilm matrix [
31,
38], as observed in the quantification of WSPs of pre-formed biofilms in the present investigation, the association of both drugs may have potentiated the antifungal effect. However, this association showed antagonism for CaS biofilms, which suggests that the biofilm alters the action of this combination of drugs, although higher decrease of metabolic activity of CaS was observed when the drugs were associated than that observed with NAC alone. Moreover, antagonism was also observed for NAC associated with nystatin, suggesting that the mechanism of action of one or both drugs may be hindered when the drugs are combined. Although the combination of NAC with fluconazole showed synergism at some concentrations, this result should be viewed with caution, since low mean values of ΔE (percentage growth inhibition) were observed (only 6% for CaS and 12% to 23% for CaR), which may be not enough to properly treat an infection caused by
C. albicans.
In another study [
37], NAC had a synergistic effect with amphotericin B and fluconazole against biofilms of
C. albicans. However, the authors did not evaluate planktonic cultures and the synergism was evaluated by the median effect, which employs equations to calculate the combination indices. Furthermore, NAC and fluconazole did not show synergism when the combination ratio was 1000:1, evaluated by an inhibitory effect of 90% [
37]. In the present investigation, we did not evaluate the interaction of NAC with another polyene (nystatin) and fluconazole in biofilms, because the FICI showed no synergism of these drugs in the planktonic assays. Another study demonstrated that the association of NAC with ketoconazole promoted higher inhibition of biofilm adherence and pre-formed biofilms than the drugs alone [
41]. Synergism was also observed when fluconazole and amphotericin B were associated with the enzyme 1,3-β-glucanase against
C. albicans biofilm [
66]. However, the mentioned studies did not use the Bliss independence method for synergism evaluation.
It is important to note that NAC is a drug commercially available for clinical use, especially for respiratory illnesses, and, therefore, it is safe. A report reviewing the multiple clinical applications of NAC pointed out that even doses up to 1200 mg are well tolerated and have unusual side effects [
70]. This information gave us confidence regarding the use of NAC in our research, because the highest concentration tested was 100 mg/mL. For this reason, we did not evaluate the cytotoxic effect of NAC, which is another limitation of our study. However, as already mentioned, the clinical use of NAC is well established, and consequently it does not show side effects when properly used.
In conclusion, NAC showed a fungistatic effect against CaS and CaR. However, a subMIC level (12.5 mg/mL) resulted in the opposite effect (an increase of fungal growth). The antibiofilm effect of NAC was due to its fungistatic action, since only concentrations greater than the MIC reduced biofilm formation, pre-formed biofilms, biofilm thickness, and the matrix composition, especially the water-soluble components of pre-formed biofilms. NAC also showed synergism with caspofungin for planktonic cultures, but not for biofilms of CaS. However, clinical isolates were not used in this investigation, which may have different virulence factors and susceptibility to NAC. Therefore, our in vitro findings cannot be extrapolated to the clinical scenario. Future studies are needed to elucidate the fungistatic mechanism of action of NAC against C. albicans, and to evaluate the efficacy of NAC against other non-albicans Candida species and the infections caused by them.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}