Genomics Reveals the Metabolic Potential and Functions in the Redistribution of Dissolved Organic Matter in Marine Environments of the Genus Thalassotalea



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, Isolation, and Physiological Test
2.2. Phylogenetic Analysis
2.3. Genome Sequencing, Assembly, and Annotation
2.4. Genomic and Phylogenomic Analyses
2.5. Data Availability
3. Results and Discussion
3.1. General Genomic Features
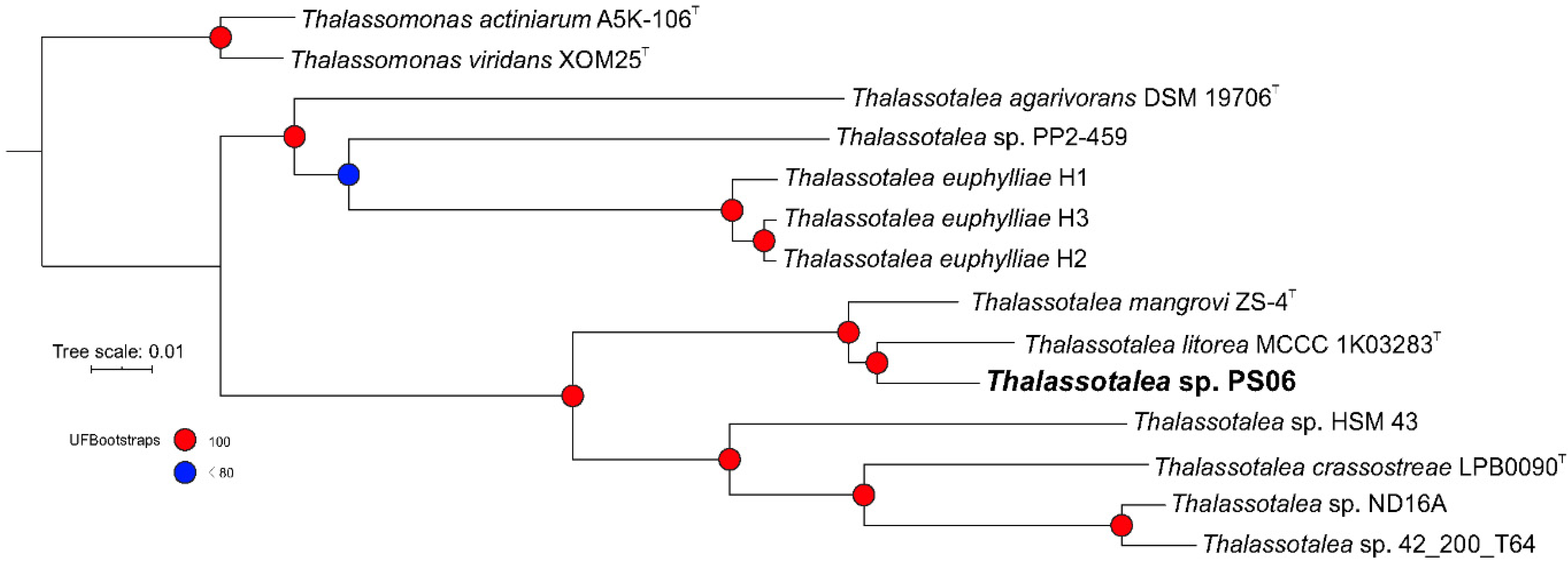
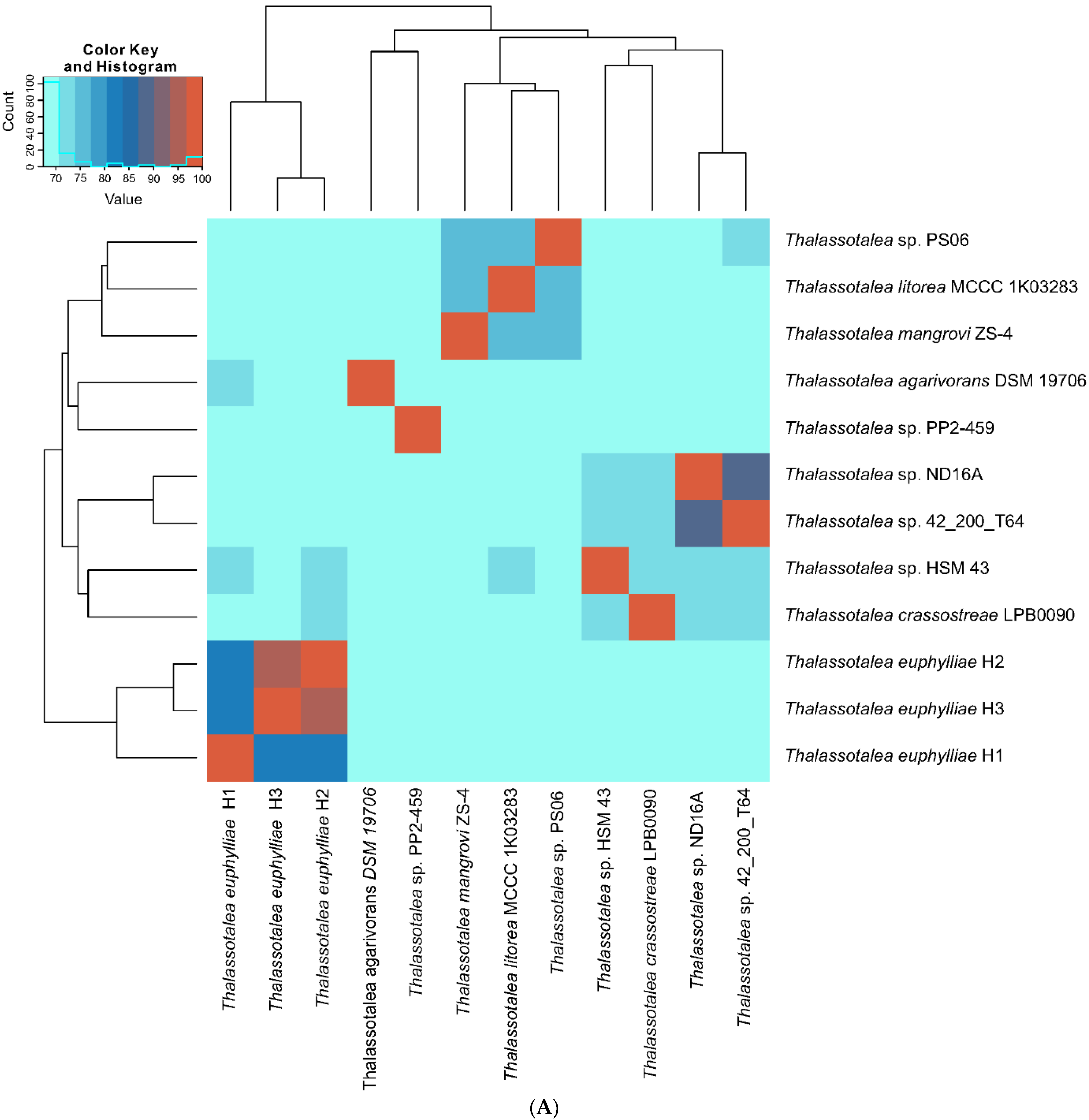
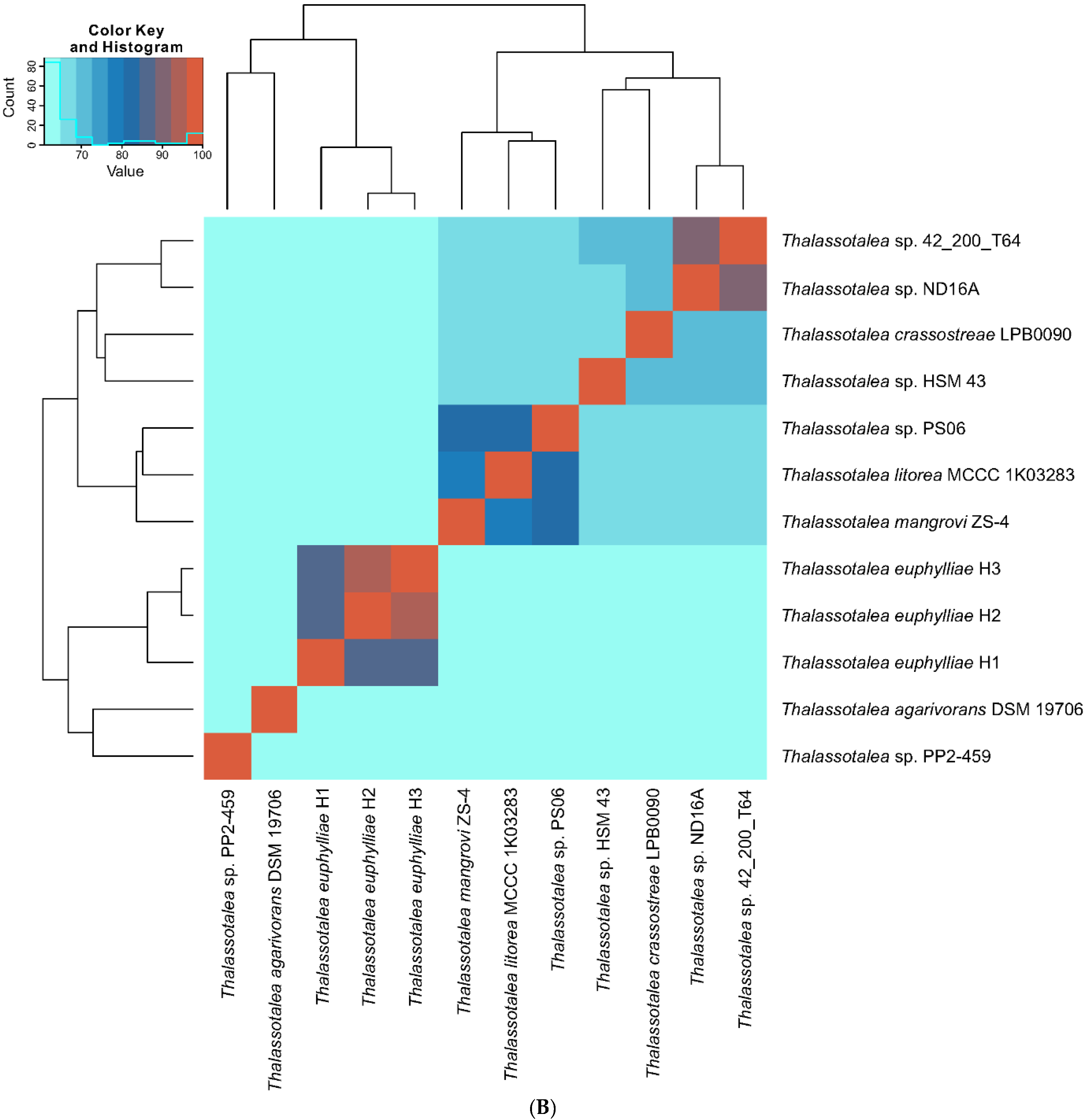
3.2. Phylogenetic and Phylogenomic Analyses
3.3. Comparative Genome Analysis of Thalassotalea Strains
3.4. Central Metabolism, Substrate Utilizations, and Respiration
3.5. Metabolic Potentials
3.6. Repertoire of Carbohydrate-Active Enzymes (CAZymes) and Degrading Activities of the Genus Thalassotalea
3.7. Environmental Stress and Adaptation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Tang, K.; Xiaochong, S.; Zhang, X.H. Description of Thalassotalea pisci.um Gen. Nov., sp. Nov., Isolated from Flounder (Paralichthys olivaceus), Reclassification of Four Species of the Genus Thalassomonas as Members of the Genus Thalassotalea Gen. Nov. and Emended Description of the Genus Thalassomonas. Int. J. Syst. Evol. Microbiol. 2014, 64, 1223–1228. [Google Scholar] [PubMed] [Green Version]
- Hou, T.T.; Liu, Y.; Zhong, Z.P.; Liu, H.C.; Liu, Z.P. Thalassotalea marina sp. Nov., Isolated from a Marine Recirculating Aquaculture System, Reclassification of Thalassomonas eurytherma as Thalassotalea eurytherma Comb. Nov. and Emended Description of the Genus Thalassotalea. Int. J. Syst. Evol. Microbiol. 2015, 65, 4710–4715. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jung, Y.T.; Kang, C.H.; Park, J.M.; Yoon, J.H. Thalassotalea ponticola sp. Nov., Isolated from Seawater, Reclassification of Thalassomonas fusca as Thalassotalea Fusca Comb. Nov. And Emended Description of the Genus Thalassotalea. Int. J. Syst. Evol. Microbiol. 2014, 64, 3676–3682. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zhang, D.; Gui, J.; Wang, J.; Zhu, X.; Lai, Q.; Wang, H.; Xu, H. Thalassotalea mangrovi sp. Nov., a Bacterium Isolated from Marine Mangrove Sediment. Int. J. Syst. Evol. Microbiol. 2019, 69, 3644–3649. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Dubinsky, E.A.; Probst, A.J.; Wang, A.J.; Sieber, C.M.K.; Tom, L.M.; Gardinali, P.R.; Banfield, J.F.; Atlas, R.M.; Andersen, G.L. Simulation of Deepwater Horizon Oil Plume Reveals Substrate Specialization within a Complex Community of Hydrocarbon Degraders. Proc. Natl. Acad. Sci. USA 2017, 114, 7432–7437. [Google Scholar] [CrossRef] [Green Version]
- Stelling, S.C.; Techtmann, S.M.; Utturkar, S.M.; Alshibli, N.K.; Brown, S.D.; Hazen, T.C. Draft Genome Sequence of Thalassotalea sp. Strain Nd16a Isolated from Eastern Mediterranean Sea Water Collected from a Depth of 1055 Meters. Genome Announc. 2014, 2, 01231-14. [Google Scholar] [CrossRef] [Green Version]
- Dethlefsen, L.; Schmidt, T.M. Performance of the Translational Apparatus Varies with the Ecological Strategies of Bacteria. J. Bacteriol. 2007, 189, 3237–3245. [Google Scholar] [CrossRef] [Green Version]
- Espejo, R.T.; Plaza, N. Multiple Ribosomal Rna Operons in Bacteria; Their Concerted Evolution and Potential Consequences on the Rate of Evolution of Their 16s Rrna. Front. Microbiol. 2018, 9, 1232. [Google Scholar] [CrossRef]
- Acinas, S.G.; Marcelino, L.A.; Klepac-Ceraj, V.; Polz, M.F. Divergence and Redundancy of 16s Rrna Sequences in Genomes with Multiple Rrn Operons. J. Bacteriol. 2004, 186, 2629–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, S.; Freckelton, M.L.; Nedved, B.T.; Rice, S.A.; Hadfield, M.G. Full-Genome Sequence of Thalassotalea euphylliae H1, Isolated from a Montipora capitata Coral Located in Hawai’i. Microbiol. Resour. Announc. 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Summers, S.; Freckelton, M.L.; Nedved, B.T.; Rice, S.A.; Hadfield, M.G. Complete Genome Sequence of Thalassotalea euphylliae Strain H2. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.; Kim, E.; Shin, S.K.; Yi, H. Thalassotalea crassostreae sp. Nov., Isolated from Pacific Oyster. Int. J. Syst. Evol. Microbiol. 2017, 67, 2195–2198. [Google Scholar] [CrossRef] [PubMed]
- Sheu, D.S.; Sheu, S.Y.; Xie, P.B.; Tang, S.L.; Chen, W.M. Thalassotalea coralli sp. Nov., Isolated from the Torch Coral Euphyllia Glabrescens. Int. J. Syst. Evol. Microbiol. 2018, 68, 185–191. [Google Scholar] [CrossRef]
- Sheu, S.Y.; Liu, L.P.; Tang, S.L.; Chen, W.M. Thalassotalea euphylliae sp. Nov., Isolated from the Torch Coral Euphyllia glabrescens. Int. J. Syst. Evol. Microbiol. 2016, 66, 5039–5045. [Google Scholar] [CrossRef] [PubMed]
- Thompson, F.L.; Barash, Y.; Sawabe, T.; Sharon, G.; Swings, J.; Rosenberg, E. Thalassomonas loyana sp. Nov., a Causative Agent of the White Plague-Like Disease of Corals on the Eilat Coral Reef. Int. J. Syst. Evol. Microbiol. 2006, 56, 365–368. [Google Scholar] [CrossRef] [Green Version]
- Leal, M.C.; Sheridan, C.; Osinga, R.; Dionisio, G.; Rocha, R.J.; Silva, B.; Rosa, R.; Calado, R. Marine Microorganism-Invertebrate Assemblages: Perspectives to Solve the Supply Problem in the Initial Steps of Drug Discovery. Mar. Drugs 2014, 12, 3929–3952. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.A.; Woodcroft, B.J.; Evans, P.M.; Hugenholtz, P.; Tyson, G.W. Recovery of Nearly 8,000 Metagenome-Assembled Genomes Substantially Expands the Tree of Life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A Standardized Bacterial Taxonomy Based on Genome Phylogeny Substantially Revises the Tree of Life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A Complete Domain-to-Species Taxonomy for Bacteria Archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef]
- Reddy, C.A.; Beveridge, T.J.; Breznak, J.A.; Marzluf, G.A.; Schmidt, T.M.; Snyder, L.R. Methods for General and Molecular Microbiology, 3th ed.; American Society of Microbiology: Washington, DC, USA, 2007. [Google Scholar]
- Lane, D.J.; Pace, B.; Olsen, G.J.; Stahl, D.A.; Sogin, M.L.; Pace, L.R. Rapid Determination of 16s Ribosomal Rna Sequences for Phylogenetic Analyses. Proc. Natl. Acad. Sci. USA 1985, 82, 6955–6959. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.W.; Rani, S.; Kim, S.J.; Moon, E.; Nam, S.Q.; Rhee, S.K.; Park, S.J. Halomonas Aestuarii Sp. Nov., a Moderately Halophilic Bacterium Isolated from a Tidal Flat. Int. J. Syst. Evol. Microbiol. 2017, 67, 4298–4303. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Tom, A. Bioedit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/Nt. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The Clustal_X Windows Int.erface: Flexible Strategies for Multiple Sequence Alignment Aided by Quality Analysis Tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. The Neutral Theory of Molecular Evolution and the World View of the Neutralists. Genome 1989, 31, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Evolutionary Trees from DNA Sequences: A Maximum Likelihood Approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. Mega X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. Trnascan-Se: A Program for Improved Detection of Transfer Rna Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Koh, H.W.; Hur, M.; Kang, M.S.; Ku, Y.B.; Ghai, R.; Park, S.J. Physiological and Genomic Insights into the Lifestyle of Arsenite-Oxidizing Herminiimonas Arsenitoxidans. Sci. Rep. 2017, 7, 15007. [Google Scholar] [CrossRef] [Green Version]
- Bulzu, P.A.; Andrei, A.S.; Salcher, M.M.; Mehrshad, M.; Inoue, K.; Kandori, H.; Beja, O.; Ghai, R.; Banciu, H.L. Casting Light on Asgardarchaeota Metabolism in a Sunlit Microoxic Niche. Nat. Microbiol. 2019, 4, 1129–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.J.; Lee, J.W.; Nguyen, N.L.; Rhee, S.K.; Park, S.L. The Characteristics and Comparative Analysis of Methanotrophs Reveal Genomic Insights into Methylomicrobium sp. Enriched from Marine Sediments. Syst. Appl. Microbiol. 2018, 41, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. Antismash 5.0: Updates to the Secondary Metabolite Genome Mining Pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. Dbcan: A Web Resour.ce for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2012, 40, 445–451. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. Checkm: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, K.; Jiao, N.; Liu, K.; Zhang, Y.; Li, S. Distribution and Functions of Tonb-Dependent Transporters in Marine Bacteria and Environments: Implications for Dissolved Organic Matter Utilization. PLoS ONE 2012, 7, e41204. [Google Scholar] [CrossRef] [Green Version]
- Gelfand, Y.; Rodriguez, A.; Benson, G. Trdb-the Tandem Repeats Database. Nucleic Acids Res. 2007, 35, 80–87. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. Orthovenn2: A Web Server for Whole-Genome Comparison and Annotation of Orthologous Clusters across Multiple Species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. Phaster: A Better, Faster Version of the Phast Phage Search Tool. Nucleic Acids Res. 2016, 44, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Loytynoja, A. Phylogeny-Aware Alignment with Prank. Methods Mol. Biol 2014, 1079, 155–170. [Google Scholar]
- Criscuolo, A.; Gribaldo, S. Bmge (Block Mapping and Gathering with Entropy): A New Software for Selection of Phylogenetic Informative Regions from Multiple Sequence Alignments. BMC Evol. Biol 2010, 10, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susko, E.; Roger, A.J. On Reduced Amino Acid Alphabets for Phylogenetic Inference. Mol. Biol Evol. 2007, 24, 2139–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. Iq-Tree: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, S.L. Modelfinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, S.L. Ufboot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA Hybridization Values and Their Relationship to Whole-Genome Sequence Similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, K.T.; Rossello-Mora, R.; Amann, R. Uncultivated Microbes in Need of Their Own Taxonomy. ISME J. 2017, 11, 2399–2406. [Google Scholar] [CrossRef]
- Kang, H.; Kim, H.; Nam, Y.I.; Joung, Y.; Jang, T.Y.; Joh, K. Thalassotalea litorea sp. Nov., Isolated from Seashore Sand. Int. J. Syst. Evol. Microbiol. 2017, 67, 2268–2273. [Google Scholar] [CrossRef]
- Kapli, P.; Yang, Z.; Telford, M.J. Phylogenetic Tree Building in the Genomic Age. Nat. Rev. Genet. 2020, 21, 428–444. [Google Scholar] [CrossRef]
- Zhu, Q.; Mai, U.; Pfeiffer, W.; Janssen, S.; Asnicar, F.; Sanders, J.G.; Belda-Ferre, P.; Al-Ghalith, G.A.; Kopylova, E.; McDonald, D.; et al. Phylogenomics of 10,575 Genomes Reveals Evolutionary Proximity between Domains Bacteria and Archaea. Nat. Commun. 2019, 10, 5477. [Google Scholar] [CrossRef] [Green Version]
- Almpanis, A.; Swain, M.; Gatherer, D.; McEwan, N. Correlation between Bacterial G+C Content, Genome Size and the G+C Content of Associated Plasmids and Bacteriophages. Microb. Genom. 2018, 4, e000168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Song, W.; Yu, M.; Lin, X. Comparative Genomics Analysis of Five Psychrobacter Strains Isolated from World-Wide Habitats Reveal High Intra-Genus Variations. Extremophiles 2017, 21, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Yamada, E.W.; Jakoby, W.B. Aldehyde Oxidation. V. Direct Conversion of Malonic Semialdehyde to Acetyl-Coenzyme, A. J. Biol Chem. 1960, 235, 589–594. [Google Scholar] [PubMed]
- De Ley, J.; Doudoroff, M. The Metabolism of D-Galactose in Pseudomonas saccharophila. J. Biol Chem. 1957, 227, 745–757. [Google Scholar] [PubMed]
- Gobet, A.; Barbeyron, T.; Matard-Mann, M.; Magdelenat, G.; Vallenet, D.; Duchaud, E.; Michel, G. Evolutionary Evidence of Algal Polysaccharide Degradation Acquisition by Pseudoalteromonas carrageenovora 9t to Adapt to Macroalgal Niches. Front. Microbiol. 2018, 9, 2740. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, L.; Pathiraja, D.; Bech, P.K.; Schultz-Johansen, M.; Hennessy, R.; Teze, D.; Choi, I.C.; Stougaard, P. A Multifunctional Polysaccharide Utilization Gene Cluster in Colwellia echini Encodes Enzymes for the Complete Degradation of K-Carrageenan, I-Carrageenan, and Hybrid B/K-Carrageenan. MSphere 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Nishikawa, H.; Gao, Y.; Sawa, Y.; Shibata, H.; Yabuta, Y.; Maruta, T.; Shigeoka, S. The Pathway Via D-Galacturonate/L-Galactonate Is Significant for Ascorbate Biosynthesis in Euglena gracilis. J. Biol Chem. 2008, 283, 31133–31141. [Google Scholar] [CrossRef] [Green Version]
- Kuivanen, J.; Penttila, M.; Richard, P. Metabolic Engineering of the Fungal D-Galacturonate Pathway for L-Ascorbic Acid Production. Microb. Cell Fact. 2015, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Durand, A.; Bourbon, M.L.; Steunou, A.S.; Khalfaoui-Hassani, B.; Legrand, C.; Guitton, A.; Astier, C.; Ouchane, S. Biogenesis of the Bacterial Cbb3 Cytochrome C Oxidase: Active Subcomplexes Support a Sequential Assembly Model. J. Biol Chem. 2018, 293, 808–818. [Google Scholar] [CrossRef] [Green Version]
- Batista, K.A.; Bataus, L.A.M.; Ivan, T.N.C.; Fernandes, K.F. Development of Culture Medium Using Extruded Bean as a Nitrogen Source for Yeast Growth. J. Microbiol. Methods 2013, 92, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Nancib, N.; Branlant, C.; Boudrant, J. Metabolic Roles of Peptone Yeast Extract for the Culture of a Recombinant Strain Ofescherichia Coli. J. Ind. Microbiol. 1991, 8, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Plugge, C.M. Anoxic Media Design, Preparation, Considerations. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2005; pp. 3–16. [Google Scholar]
- Mathuriya, A.S.; Yakhmi, J.V. Polyhydroxyalkanoates: Biodegradable Plastics Their Applications. In Handbook of Ecomaterials; Martínez, L.M.T., Kharissova, O.V., Kharisov, B.I., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 2873–2900. [Google Scholar]
- Jendrossek, D. Microbial Degradation of Polyesters. In Biopolyesters; Steinbüchel, W.B.A., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 293–325. [Google Scholar]
- Simon, R.D.; Weathers, P. Determination of the Structure of the Novel Polypeptide Containing Aspartic Acid and Arginine Which Is Found in Cyanobacteria. Biochim. Biophys. Acta Protein Struct. 1976, 420, 165–176. [Google Scholar] [CrossRef]
- Krehenbrink, M.; Oppermann-Sanio, F.B.; Steinbuchel, A. Evaluation of Non-Cyanobacterial Genome Sequences for Occurrence of Genes Encoding Proteins Homologous to Cyanophycin Synthetase and Cloning of an Active Cyanophycin Synthetase from Acinetobacter Sp. Strain Dsm 587. Arch. Microbiol. 2002, 177, 371–380. [Google Scholar] [CrossRef]
- Ziegler, K.; Deutzmann, R.; Lockau, W. Cyanophycin Synthetase-Like Enzymes of Non-Cyanobacterial Eubacteria: Characterization of the Polymer Produced by a Recombinant Synthetase of Desulfitobacterium hafniense. Z. Nat. C 2002, 57, 522–529. [Google Scholar] [CrossRef]
- Obst, M.; Steinbuchel, A. Microbial Degradation of Poly(Amino Acid)S. Biomacromolecules 2004, 5, 1166–1176. [Google Scholar] [CrossRef]
- Obst, M.; Krug, A.; Luftmann, H.; Steinbuchel, A. Degradation of Cyanophycin by Sedimentibacter hongkongensis Strain Ki and Citrobacter amalonaticus Strain G Isolated from an Anaerobic Bacterial Consortium. Appl. Environ. Microbiol. 2005, 71, 3642–3652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obst, M.; Oppermann-Sanio, F.B.; Luftmann, H.; Steinbuchel, A. Isolation of Cyanophycin-Degrading Bacteria, Cloning Characterization of an Extracellular Cyanophycinase Gene (Cphe) from Pseudomonas anguilliseptica Strain Bi. J. Biol Chem. 2002, 277, 25096–25105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapébie, P.; Lombard, V.; Drula, E.; Terrapon, N.; Henrissat, B. Bacteroidetes Use Thousands of Enzyme Combinations to Break Down Glycans. Nat. Commun. 2019, 10, 2043. [Google Scholar] [CrossRef]
- Chernysheva, N.; Bystritskaya, E.; Stenkova, A.; Golovkin, I.; Nedashkovskaya, O.; Isaeva, M. Comparative Genomics and Cazyme Genome Repertoires of Marine Zobellia amurskyensis Kmm 3526t and Zobellia laminariae Kmm 3676t. Mar. Drugs 2019, 17, 661. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.Y.; Huang, X.; Cheong, K.L. Recent Advances in Marine Algae Polysaccharides: Isolation, Structure, and Activities. Mar. Drugs 2017, 15, 388. [Google Scholar] [CrossRef] [Green Version]
- Siegl, A.; Kamke, J.; Hochmuth, T.; Piel, J.; Richter, M.; Liang, C.L.; Dandekar, T.; Hentschel, U. Single-Cell Genomics Reveals the Lifestyle of Poribacteria, a Candidate Phylum Symbiotically Associated with Marine Sponges. ISME J. 2011, 5, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Page, R.D.M.; Holmes, E.C. Molecular Evolution: A Phylogenetic Approach; Wiley: Hoboke, NJ, USA, 2009. [Google Scholar]
- Alex, A.; Antunes, A. Genus-Wide Comparison of Pseudovibrio Bacterial Genomes Reveal Diverse Adaptations to Different Marine Invertebrate Hosts. PLoS ONE 2018, 13, e0194368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garron, M.L.; Cygler, M. Structural and Mechanistic Classification of Uronic Acid-Containing Polysaccharide Lyases. Glycobiology 2010, 20, 1547–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, L.C.; Weiler, E.B.; Berger, W. Engineering Broad-Spectrum Digestion of Polyuronides from an Exolytic Polysaccharide Lyase. Biotechnol. Biofuels 2016, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Frobel, J.; Rose, P.; Muller, M. Twin-Arginine-Dependent Translocation of Folded Proteins. Philos Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 1029–1046. [Google Scholar] [CrossRef] [Green Version]
- Frain, K.M.; Robinson, C.; Van Dijl, M. Transport of Folded Proteins by the Tat System. Protein J. 2019, 38, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Padan, E.; Bibi, E.; Ito, M.; Krulwich, T.A. Alkaline Ph Homeostasis in Bacteria: New Insights. Biochim. Biophys. Acta 2005, 1717, 67–88. [Google Scholar] [CrossRef] [Green Version]
- Bremer, E.; Kramer, R. Responses of Microorganisms to Osmotic Stress. Annu Rev. Microbiol. 2019, 73, 313–334. [Google Scholar] [CrossRef]
- Eisenreich, W.; Bacher, A.; Arigoni, D.; Rohdich, F. Biosynthesis of Isoprenoids Via the Non-Mevalonate Pathway. Cell Mol. Life Sci. 2004, 61, 1401–1426. [Google Scholar] [CrossRef]
- Pan, J.J.; Solbiati, J.O.; Ramamoorthy, G.; Hillerich, B.S.; Seidel, R.D.; Cronan, J.E.; Almo, C.S.; Poulter, C.D. Biosynthesis of Squalene from Farnesyl Diphosphate in Bacteria: Three Steps Catalyzed by Three Enzymes. ACS Cent. Sci. 2015, 1, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Aono, R.; Masahiro, I.; Keith, N.J.; Horikoshi, K. A High Cell Wall Negative Charge Is Necessary for the Growth of the Alkaliphile Bacillus lentus C-125 at Elevated Ph. Microbiology 1995, 141, 2955–2964. [Google Scholar] [CrossRef] [Green Version]
- Melton, E.D.; Sorokin, D.Y.; Overmars, L.; Lapidus, A.L.; Pillay, M.; Ivanova, N.; Del Rio, T.G.; Kyrpides, N.; Woyke, T.; Muyzer, G. Draft Genome Sequence of Dethiobacter alkaliphilus Strain Aht1(T), a Gram-Positive Sulfidogenic Polyextremophile. Stand. Genom. Sci. 2017, 12, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangermani, M.; Hug, I.; Sauter, N.; Pfohl, T.; Jenal, U. Tad Pili Play a Dynamic Role in Caulobacter crescentus Surface Colonization. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goosens, V.J.; Busch, A.; Georgiadou, M.; Castagnini, M.; Forest, K.T.; Waksman, G.; Pelicic, V. Reconstitution of a Minimal Machinery Capable of Assembling Periplasmic Type Iv Pili. Proc. Natl. Acad. Sci. USA 2017, 114, E4978–E4986. [Google Scholar] [CrossRef] [Green Version]
- Shu, H.Y.; Fung, C.P.; Liu, Y.M.; Wu, K.M.; Chen, Y.T.; Li, L.H.; Liu, T.T.; Kirby, R.; Tsai, S.F. Genetic Diversity of Capsular Polysaccharide Biosynthesis in Klebsiella pneumoniae Clinical Isolates. Microbiology 2009, 155, 4170–4183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Zhou, K.; Aertsen, A.; Michiels, C.W. The Role of Variable DNA Tandem Repeats in Bacterial Adaptation. FEMS Microbiol. Rev. 2014, 38, 119–141. [Google Scholar] [CrossRef] [Green Version]
- Grissa, I.; Vergnaud, G.; Pourcel, C. The Crisprdb Database and Tools to Display Crisprs and to Generae Dictionaries of Spacers and Repeats. BMC Bioinform. 2007, 8, 172. [Google Scholar] [CrossRef] [Green Version]
- Nasko, D.J.; Ferrell, B.D.; Moore, R.M.; Bhavsar, J.D.; Polson, S.W.; Wommack, K.E. Crispr Spacers Indicate Preferential Matching of Specific Virioplankton Genes. MBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Zhang, Y.; Liu, Z.; Dong, Z.; Xie, C.; Bravo, A.; Soberon, M.; Mahillon, J.; Sun, M.; Peng, D. The Crispr-Cas Systems Were Selectively Inactivated During Evolution of Bacillus Cereus Group for Adaptation to Diverse Environments. ISME J. 2020, 14, 1479–1493. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Function Categories | Lek Cluster Number | Substrates | Thalassotalea sp. ND16A | Thalassotalea crassostreae LPB0090 | Thalassotalea sp. PP2-459 | Thalassotalea sp. 42_200_T64 | Thalassotalea euphylliae H1 | Thalassotalea euphylliae H3 | Thalassotalea euphylliae H2 | Thalassotalea sp. HSM 43 | Thalassotalea mangrovi ZS-4 | Thalassotalea litorea MCCC 1K03283 | Thalassotalea sp. PS06 | Thalassotalea agarivorans DSM 19706 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group I: Dissolved organic matter transporters | Cluster_3090 | Chito-oligosaccharides, phytate, maltodextrin, maltose, chitin, xylan, xylose, pectin | 13 | 10 | 14 | 7 | 7 | 4 | 6 | 10 | 9 | 13 | 8 | 11 |
| Cluster_720 | Digested proteins, starch/malto-oligo-saccharides, chondroitin sulfate/hyaluronic acid | - | - | - | - | - | - | - | - | - | - | - | - | |
| Cluster_427 | Arabinose | 7 | 4 | 7 | 5 | 10 | 7 | 7 | 6 | 9 | 5 | 7 | 4 | |
| Cluster_952 | Sucrose | - | - | 1 | - | - | - | - | - | - | - | - | - | |
| Group II: Siderophores/Vitamins transporters | Cluster_3303 | Ferric-citrate | - | - | 1 | - | - | 1 | 1 | - | - | 1 | - | - |
| Cluster_410 | Aerobactin, alcaligin, anguibactin, catecholates, chrysobactin, coprogen, ferrioxamine B, rhodoturolic acid, desferrioxamine, ferric malleobactin, ferric ornibactin, ferrichrome, hexylsulfate, pseudobactin A, pseudobactin M114, pyochelin, pyoverdine, rhizobactin 1021, thiamin, vibriobactin, yersiniabactin | 7 | 8 | 10 | 4 | 17 | 16 | 14 | 9 | 13 | 10 | 11 | 8 | |
| Cluster_973 | Vitamin B12, catecholates, enterobactin, 2,3-dihydroxybenzoylserine(DHBS) | 2 | 3 | 6 | 5 | 6 | 5 | 4 | 2 | 1 | 2 | 1 | 3 | |
| Cluster_325 | Vitamin B12 | - | - | - | 1 | - | - | - | - | - | - | 1 | - | |
| Cluster_180 | Fibronectin, thiamin | - | - | - | 1 | - | - | - | - | - | - | - | - | |
| Cluster_2835 | Thiamin | - | - | - | - | 1 | - | - | - | - | - | - | - | |
| Group III: Heme/Hemophores/Iron(heme)-binding transporters | Cluster_1609 | Heme | - | - | 2 | - | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Cluster_1856 | Heme | 1 | - | 2 | - | 4 | 3 | 3 | 1 | 2 | 2 | - | - | |
| Group IV: Metal transporters | Cluster_767 | Copper, Copper chelate | - | - | 1 | - | 1 | 1 | 1 | - | - | - | - | - |
| Cluster_987 | Nickel, Cobalt | - | - | 1 | - | - | - | - | - | - | - | 1 | - |
| Strain | Transporters by TCDB a | CAZy b | Peptidases by MEROPS c | TBDT d |
|---|---|---|---|---|
| Thalassotalea sp. ND16A | 215 | 117 | 75 | 30 |
| Thalassotalea crassostreae LPB0090 | 180 | 138 | 51 | 25 |
| Thalassotalea sp. PP2-459 | 206 | 118 | 80 | 45 |
| Thalassotalea sp. 42_200_T64 | 180 | 107 | 58 | 23 |
| Thalassotalea euphylliae H1 | 199 | 121 | 63 | 47 |
| Thalassotalea euphylliae H3 | 196 | 112 | 65 | 38 |
| Thalassotalea euphylliae H2 | 193 | 114 | 65 | 37 |
| Thalassotalea sp. HSM 43 | 205 | 145 | 74 | 29 |
| Thalassotalea mangrovi ZS-4 | 189 | 132 | 62 | 35 |
| Thalassotalea litorea MCCC 1K03283 | 190 | 147 | 65 | 34 |
| Thalassotalea sp. PS06 | 176 | 128 | 67 | 30 |
| Thalassotalea agarivorans DSM 19706 | 171 | 126 | 65 | 27 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Cha, I.-T.; Lee, K.-E.; Lee, E.-Y.; Park, S.-J. Genomics Reveals the Metabolic Potential and Functions in the Redistribution of Dissolved Organic Matter in Marine Environments of the Genus Thalassotalea. Microorganisms 2020, 8, 1412. https://doi.org/10.3390/microorganisms8091412
Kim M, Cha I-T, Lee K-E, Lee E-Y, Park S-J. Genomics Reveals the Metabolic Potential and Functions in the Redistribution of Dissolved Organic Matter in Marine Environments of the Genus Thalassotalea. Microorganisms. 2020; 8(9):1412. https://doi.org/10.3390/microorganisms8091412
Chicago/Turabian StyleKim, Minji, In-Tae Cha, Ki-Eun Lee, Eun-Young Lee, and Soo-Je Park. 2020. "Genomics Reveals the Metabolic Potential and Functions in the Redistribution of Dissolved Organic Matter in Marine Environments of the Genus Thalassotalea" Microorganisms 8, no. 9: 1412. https://doi.org/10.3390/microorganisms8091412
APA StyleKim, M., Cha, I. -T., Lee, K. -E., Lee, E. -Y., & Park, S. -J. (2020). Genomics Reveals the Metabolic Potential and Functions in the Redistribution of Dissolved Organic Matter in Marine Environments of the Genus Thalassotalea. Microorganisms, 8(9), 1412. https://doi.org/10.3390/microorganisms8091412