
AT Homopolymer Strings in Salmonella enterica Subspecies I Contribute to Speciation and Serovar Diversity

 , and
, and 
Abstract
:1. Introduction
Additional Background on Poultry-Associated S. enterica Serovars
2. Materials and Methods
2.1. Salmonella enterica Subspecies I Strains Analyzed for Strings of Homopolymers
2.2. Other Bacteria Analyzed for Comparison to S. enterica
2.3. Statistical Analysis of Kmer Content
2.4. Locating AT 8+Mers within Mutated Genes
3. Results
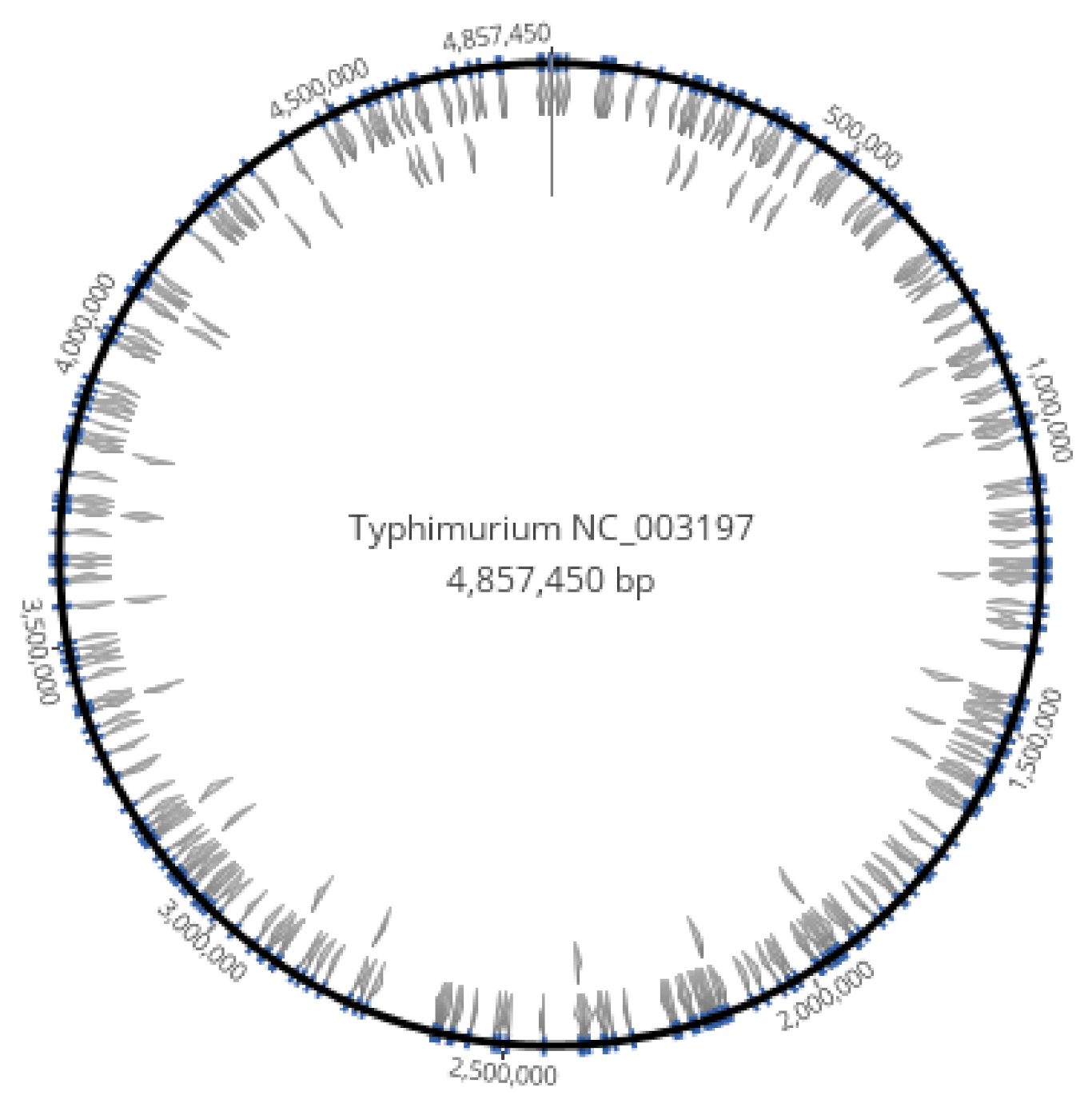
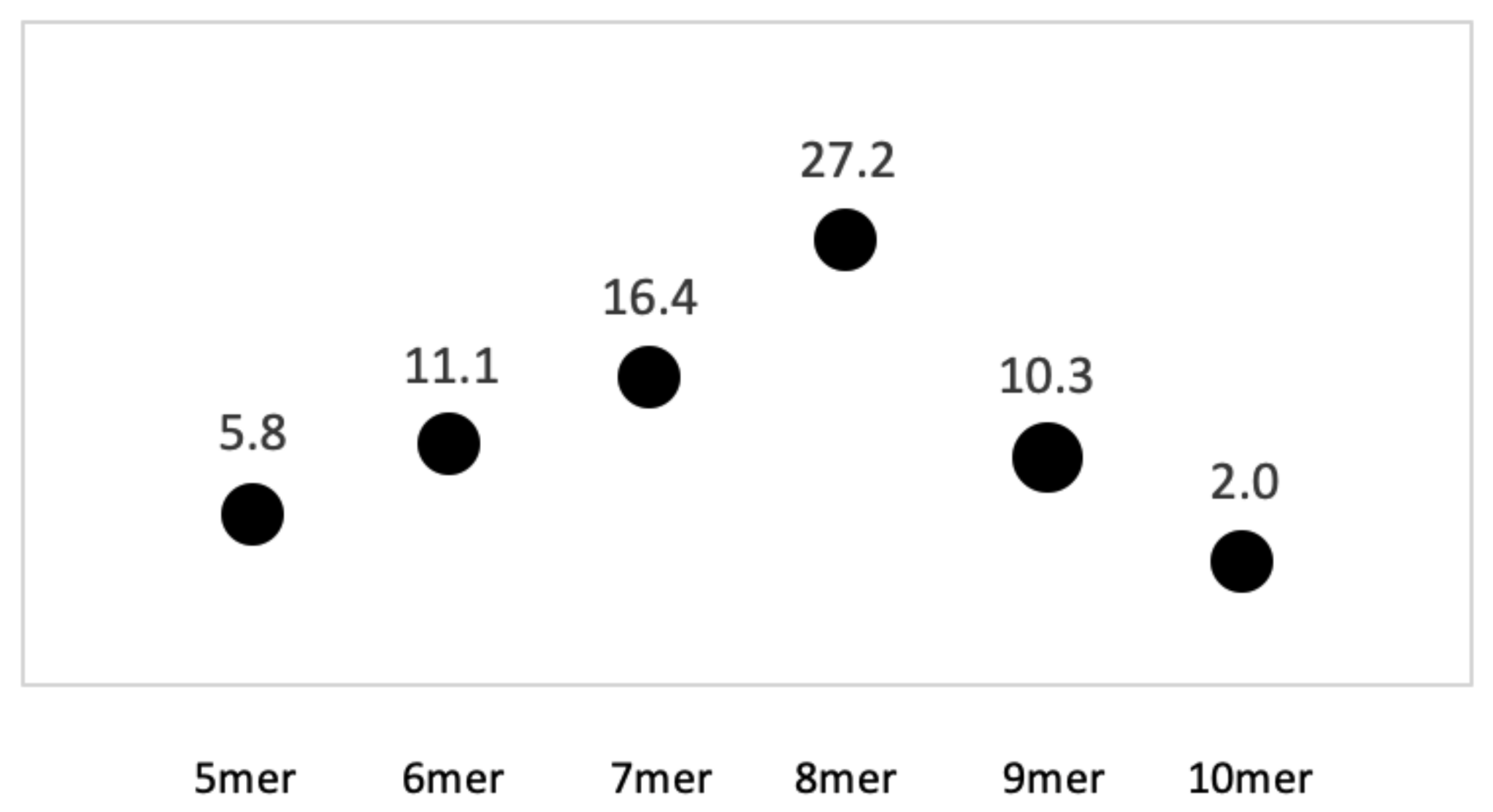
3.1. Homopolymer Strings of at Least 8 Nucleotides Were Dispersed in the Genome of S. enterica
3.2. The AT 8+mer Motif in Bacteria Was Specific to Genus and Species
3.3. The AT 8+mer Motif in the Chromosome Is Not Specific to Serovar
3.4. Characterization of the AT 8+mer Motif in Poultry-Associated Serovars of Salmonella
3.5. Location of AT 8+mers within Mutated Open Reading Frames
3.6. S. enterica Plasmids Have AT 8+mer Motifs Possibly Associated with Serovar Preference
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- CDC. An Atlas of Salmonella in the United States, 1968–2011: Laboratory-Based Enteric Disease Surveillance; Centers for Disease Control and Prevention (CDC): Atlanta, GA, USA, 2013. [Google Scholar]
- Tack, D.M.; Ray, L.; Griffin, P.M.; Cieslak, P.R.; Dunn, J.; Rissman, T.; Jervis, R.; Lathrop, S.; Muse, A.; Duwell, M.; et al. Preliminary Incidence and Trends of Infections with Pathogens Transmitted Commonly Through Food—Foodborne Diseases Active Surveillance Network, 10 U.S. Sites, 2016–2019. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 509–514. [Google Scholar] [CrossRef]
- Guard, J.; Cao, G.; Luo, Y.; Baugher, J.D.; Davison, S.; Yao, K.; Hoffmann, M.; Zhang, G.; Likens, N.; Bell, R.L.; et al. Genome sequence analysis of 91 Salmonella Enteritidis isolates from mice caught on poultry farms in the mid 1990s. Genomics 2020, 112, 528–544. [Google Scholar] [CrossRef] [PubMed]
- Morales, C.A.; Guard, J.; Sanchez-Ingunza, R.; Shah, D.H.; Harrison, M. Virulence and metabolic characteristics of Salmonella enterica serovar enteritidis strains with different sefD variants in hens. Appl. Environ. Microbiol. 2012, 78, 6405–6412. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Ingunza, R.; Guard, J.; Morales, C.A.; Icard, A.H. Reduction of Salmonella Enteritidis in the spleens of hens by bacterins that vary in fimbrial protein SefD. Foodborne Pathog. Dis. 2015, 12, 836–843. [Google Scholar] [CrossRef]
- Orsi, R.H.; Bowen, B.M.; Wiedmann, M. Homopolymeric tracts represent a general regulatory mechanism in prokaryotes. BMC Genom. 2010, 11, 102. [Google Scholar] [CrossRef] [Green Version]
- Reich, Z.; Friedman, P.; Levin-Zaidman, S.; Minsky, A. Effects of adenine tracts on the B-Z transition. Fine tuning of DNA conformational transition processes. J. Biol. Chem. 1993, 268, 8261–8266. [Google Scholar] [CrossRef]
- Hines, E.R.; Kolek, O.I.; Jones, M.D.; Serey, S.H.; Sirjani, N.B.; Kiela, P.R.; Jurutka, P.W.; Haussler, M.R.; Collins, J.F.; Ghishan, F.K. 1,25-dihydroxyvitamin D3 down-regulation of PHEX gene expression is mediated by apparent repression of a 110 kDa transfactor that binds to a polyadenine element in the promoter. J. Biol. Chem. 2004, 279, 46406–46414. [Google Scholar] [CrossRef] [Green Version]
- Lindemose, S.; Nielsen, P.E.; Mollegaard, N.E. Polyamines preferentially interact with bent adenine tracts in double-stranded DNA. Nucleic Acids Res. 2005, 33, 1790–1803. [Google Scholar] [CrossRef] [Green Version]
- Jung, B.H.; Beck, S.E.; Cabral, J.; Chau, E.; Cabrera, B.L.; Fiorino, A.; Smith, E.J.; Bocanegra, M.; Carethers, J.M. Activin type 2 receptor restoration in MSI-H colon cancer suppresses growth and enhances migration with activin. Gastroenterology 2007, 132, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Agnoli, K.; Haldipurkar, S.S.; Tang, Y.; Butt, A.T.; Thomas, M.S. Distinct Modes of Promoter Recognition by Two Iron Starvation sigma Factors with Overlapping Promoter Specificities. J. Bacteriol. 2019, 201. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.D.; Nguyen, D.; Kunkel, T.A. Frameshift fidelity during replication of double-stranded DNA in HeLa cell extracts. Biochemistry 1993, 32, 4083–4089. [Google Scholar] [CrossRef] [PubMed]
- Traverse, C.C.; Ochman, H. Genome-Wide Spectra of Transcription Insertions and Deletions Reveal That Slippage Depends on RNA:DNA Hybrid Complementarity. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gragg, H.; Harfe, B.D.; Jinks-Robertson, S. Base composition of mononucleotide runs affects DNA polymerase slippage and removal of frameshift intermediates by mismatch repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 2002, 22, 8756–8762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guard, J.; Morales, C.A.; Fedorka-Cray, P.; Gast, R.K. Single nucleotide polymorphisms that differentiate two subpopulations of Salmonella enteritidis within phage type. BMC Res. Notes 2011, 4, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimont, P.; Weill, F.-X. Antigenic formulae of the Salmonella serovars, 9th Edition. In WHO Collaborating Centre for Reference and Research on Salmonella; World Health Organization: Paris, France, 2007. [Google Scholar]
- Guard-Bouldin, J.; Gast, R.K.; Humphrey, T.J.; Henzler, D.J.; Morales, C.; Coles, K. Subpopulation characteristics of egg-contaminating Salmonella enterica serovar Enteritidis as defined by the lipopolysaccharide O chain. Appl. Environ. Microbiol. 2004, 70, 2756–2763. [Google Scholar] [CrossRef] [Green Version]
- Gantois, I.; Ducatelle, R.; Pasmans, F.; Haesebrouck, F.; Van Immerseel, F. The Salmonella Enteritidis lipopolysaccharide biosynthesis gene rfbH is required for survival in egg albumen. Zoonoses Public Health 2009, 56, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.T.; Liebana, E.; Henzler, D.J.; Guard-Petter, J. Lipopolysaccharide O-chain microheterogeneity of Salmonella serotypes Enteritidis and Typhimurium. Environ. Microbiol. 2001, 3, 332–342. [Google Scholar] [CrossRef]
- Huang, K.; Fresno, A.H.; Skov, S.; Olsen, J.E. Dynamics and Outcome of Macrophage Interaction Between Salmonella Gallinarum, Salmonella Typhimurium, and Salmonella Dublin and Macrophages From Chicken and Cattle. Front. Cell. Infect. Microbiol. 2019, 9, 420. [Google Scholar] [CrossRef]
- Foley, S.L.; Nayak, R.; Hanning, I.B.; Johnson, T.J.; Han, J.; Ricke, S.C. Population dynamics of Salmonella enterica serotypes in commercial egg and poultry production. Appl. Environ. Microbiol. 2011, 77, 4273–4279. [Google Scholar] [CrossRef] [Green Version]
- Branchu, P.; Bawn, M.; Kingsley, R.A. Genome Variation and Molecular Epidemiology of Salmonella enterica Serovar Typhimurium Pathovariants. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- McMillan, E.A.; Gupta, S.K.; Williams, L.E.; Jove, T.; Hiott, L.M.; Woodley, T.A.; Barrett, J.B.; Jackson, C.R.; Wasilenko, J.L.; Simmons, M.; et al. Antimicrobial Resistance Genes, Cassettes, and Plasmids Present in Salmonella enterica Associated With United States Food Animals. Front. Microbiol. 2019, 10, 832. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.T.; Porwollik, S.; Long, F.; Cheng, P.; Wollam, A.; Bhonagiri-Palsikar, V.; Hallsworth-Pepin, K.; Clifton, S.W.; Weinstock, G.M.; McClelland, M. Evolutionary Genomics of Salmonella enterica Subspecies. mBio 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Achtman, M.; Hale, J.; Murphy, R.A.; Boyd, E.F.; Porwollik, S. Population structures in the SARA and SARB reference collections of Salmonella enterica according to MLST, MLEE and microarray hybridization. Infect. Genet. Evol. 2013, 16, 314–325. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, C.; Woodward, M.J. Cloning, DNA nucleotide sequence and distribution of the gene encoding the SEF14 fimbrial antigen of Salmonella enteritidis. J. Gen. Microbiol. 1993, 139, 1477–1485. [Google Scholar] [CrossRef] [Green Version]
- Clouthier, S.C.; Collinson, S.K.; Kay, W.W. Unique fimbriae-like structures encoded by sefD of the SEF14 fimbrial gene cluster of Salmonella enteritidis. Mol. Microbiol. 1994, 12, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.D.; Schmieder, R.; Silva, G.G.; Busch, J.; Cassman, N.; Dutilh, B.E.; Green, D.; Matlock, B.; Heffernan, B.; Olsen, G.J.; et al. Genomic Comparison of the Closely-Related Salmonella enterica Serovars Enteritidis, Dublin and Gallinarum. PLoS ONE 2015, 10, e0126883. [Google Scholar] [CrossRef]
- Sayers, E.W.; Agarwala, R.; Bolton, E.E.; Brister, J.R.; Canese, K.; Clark, K.; Connor, R.; Fiorini, N.; Funk, K.; Hefferon, T.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2019, 47, D23–D28. [Google Scholar] [CrossRef] [Green Version]
- McClelland, M.; Sanderson, K.E.; Spieth, J.; Clifton, S.W.; Latreille, P.; Courtney, L.; Porwollik, S.; Ali, J.; Dante, M.; Du, F.; et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 2001, 413, 852–856. [Google Scholar] [CrossRef] [Green Version]
- Thomson, N.R.; Clayton, D.J.; Windhorst, D.; Vernikos, G.; Davidson, S.; Churcher, C.; Quail, M.A.; Stevens, M.; Jones, M.A.; Watson, M.; et al. Comparative genome analysis of Salmonella Enteritidis PT4 and Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome Res. 2008, 18, 1624–1637. [Google Scholar] [CrossRef] [Green Version]
- Allard, M.W.; Luo, Y.; Strain, E.; Pettengill, J.; Timme, R.; Wang, C.; Li, C.; Keys, C.E.; Zheng, J.; Stones, R.; et al. On the evolutionary history, population genetics and diversity among isolates of Salmonella Enteritidis PFGE pattern JEGX01.0004. PLoS ONE 2013, 8, e55254. [Google Scholar] [CrossRef]
- Du, D.; van Veen, H.W.; Murakami, S.; Pos, K.M.; Luisi, B.F. Structure, mechanism and cooperation of bacterial multidrug transporters. Curr. Opin. Struct. Biol. 2015, 33, 76–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiney, D.G.; Fang, F.C.; Krause, M.; Libby, S. Plasmid-mediated virulence genes in non-typhoid Salmonella serovars. FEMS Microbiol. Lett. 1994, 124, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fardsanei, F.; Nikkhahi, F.; Bakhshi, B.; Salehi, T.Z.; Tamai, I.A.; Dallal, M.M.S. Molecular characterization of Salmonella enterica serotype Enteritidis isolates from food and human samples by serotyping, antimicrobial resistance, plasmid profiling, (GTG)5-PCR and ERIC-PCR. New Microbes New Infect. 2016, 14, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Jelesic, Z.; Kulauzov, M.; Kozoderovic, G. Analysis of the plasmid profile of various Salmonella serotypes. Med. Pregl. 2000, 53, 564–567. [Google Scholar]
- Santangelo, T.J.; Cubonova, L.; Skinner, K.M.; Reeve, J.N. Archaeal intrinsic transcription termination in vivo. J. Bacteriol. 2009, 191, 7102–7108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.; Matsumoto, Y.; Takashima, Y.; Otsuka, H. Mutation hot spots in the canine herpesvirus thymidine kinase gene. Virus Genes 2005, 31, 107–111. [Google Scholar] [CrossRef]
- Koscielniak, D.; Wons, E.; Wilkowska, K.; Sektas, M. Non-programmed transcriptional frameshifting is common and highly RNA polymerase type-dependent. Microb. Cell Fact. 2018, 17, 184. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.; Puente, J.L.; Calva, E. Salmonella virulence plasmid: Pathogenesis and ecology. Pathog. Dis. 2017, 75, ftx070. [Google Scholar] [CrossRef]
- Brandis, G.; Cao, S.; Hughes, D. Co-evolution with recombination affects the stability of mobile genetic element insertions within gene families of Salmonella. Mol. Microbiol. 2018, 108, 697–710. [Google Scholar] [CrossRef] [Green Version]
- Brandis, G.; Cao, S.; Hughes, D. Measuring Homologous Recombination Rates between Chromosomal Locations in Salmonella. Bio. Protoc. 2019, 9, e3159. [Google Scholar] [CrossRef]
- Achtman, M.; Zhou, Z.; Alikhan, N.F.; Tyne, W.; Parkhill, J.; Cormican, M.; Chiou, C.S.; Torpdahl, M.; Litrup, E.; Prendergast, D.M.; et al. Genomic diversity of Salmonella enterica -The UoWUCC 10K genomes project. Wellcome Open Res. 2020, 5, 223. [Google Scholar] [CrossRef]
- Alikhan, N.F.; Zhou, Z.; Sergeant, M.J.; Achtman, M. A genomic overview of the population structure of Salmonella. PLoS Genet. 2018, 14, e1007261. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.J.; Cho, S.; Boxrud, D.; Rankin, S.; Downe, F.; Lovchik, J.; Gibson, J.; Erdman, M.; Saeed, A.M. Single-nucleotide polymorphism typing analysis for molecular subtyping of Salmonella Tennessee isolates associated with the 2007 nationwide peanut butter outbreak in the United States. Gut Pathog. 2017, 9, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.R.; Brown, E.; Keys, C.; Strain, E.; Luo, Y.; Muruvanda, T.; Grim, C.; Beaubrun, J.J.-G.; Jarvis, K.; Ewing, L.; et al. Whole Genome DNA Sequence Analysis of Salmonella subspecies enterica serotype Tennessee obtained from related peanut butter foodborne outbreaks. PLoS ONE 2016, 11, e0146929. [Google Scholar] [CrossRef] [PubMed]
- Guard, J.; Shah, D.; Morales, C.A.; Call, D. Evolutionary trends associated with niche specialization as modeled by whole genome analysis of egg-contaminating Salmonella enterica serovar Enteritidis. In Salmonella: From Genome to Function; Porwollik, S., Ed.; Caister Academic Press: San Diego, CA, USA, 2011; pp. 91–106. [Google Scholar]
- Zhou, Z.; McCann, A.; Litrup, E.; Murphy, R.; Cormican, M.; Fanning, S.; Brown, D.; Guttman, D.S.; Brisse, S.; Achtman, M. Neutral genomic microevolution of a recently emerged pathogen, Salmonella enterica serovar Agona. PLoS Genet. 2013, 9, e1003471. [Google Scholar] [CrossRef] [Green Version]
- Sangal, V.; Harbottle, H.; Mazzoni, C.J.; Helmuth, R.; Guerra, B.; Didelot, X.; Paglietti, B.; Rabsch, W.; Brisse, S.; Weill, F.X.; et al. Evolution and population structure of Salmonella enterica serovar Newport. J. Bacteriol. 2010, 192, 6465–6476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.J.; Andam, C.P. Distinct but Intertwined Evolutionary Histories of Multiple Salmonella enterica Subspecies. mSystems 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, D.F.; Zhou, X.; Xu, L.; Zhang, L.; Shi, X. Comprehensive Analysis Reveals Two Distinct Evolution Patterns of Salmonella Flagellin Gene Clusters. Front. Microbiol. 2017, 8, 2604. [Google Scholar] [CrossRef]
- Richards, A.K.; Hopkins, B.A.; Shariat, N.W. Conserved CRISPR arrays in Salmonella enterica serovar Infantis can serve as qPCR targets to detect Infantis in mixed serovar populations. Lett. Appl. Microbiol. 2020, 71, 138–145. [Google Scholar] [CrossRef]
- Shariat, N.; Dudley, E. CRISPR Typing of Salmonella Isolates. Methods Mol. Biol. 2021, 2182, 39–44. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Phylum | Genus Species 1 | Other Genome Information | Number of Genomes Analyzed | Characteristics | Genome Size (bp) | Common Denominator (nt) 2 | Expected Number of 8+kmers | Observed AT 8+mers | Observed GC 8+mers | Observed vs. Expecterd AT 8+mers 3 | Observed vs. Expected GC 8+mers 3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Proteobacteria | Salmonella enterica | Typhimurium | 12 | Average | 4,890,448 | 16,299 | 300.0 | 332.6 | 17.2 | 1.11 | 0.06 |
| stdev | 50,356 | --- | 3.1 | 13.0 | 3.6 | --- | --- | ||||
| Proteobacteria | Salmonella enterica | Enteritidis | 12 | Average | 4,686,462 | 16,299 | 287.5 | 323.7 | 21.5 | 1.13 | 0.07 |
| stdev | 20,384 | --- | 1.3 | 10.5 | 4.2 | --- | --- | ||||
| Proteobacteria | Salmonella enterica | Typhi | 12 | Average | 4,770,414 | 16,299 | 292.7 | 316.9 | 29.5 | 1.08 | 0.10 |
| stdev | 60,270 | --- | 3.7 | 5.9 | 3.2 | --- | --- | ||||
| Proteobacteria | Salmonella enterica | mixed | 12 | Average | 4,713,701 | 16,299 | 289.2 | 315.6 | 17.2 | 1.09 | 0.06 |
| stdev | 80,652 | --- | 4.9 | 13.9 | 4.4 | --- | --- | ||||
| Proteobacteria | Escherichia coli | --- | 12 | Average | 5,087,133 | 16,299 | 312.1 | 281.9 | 18.3 | 0.90 | 0.06 |
| stdev | 262,098 | --- | 16.1 | 30.2 | 6.5 | --- | --- | ||||
| Proteobacteria | Proteus mirabilis | --- | 3 | Average | 4,124,431 | 16,299 | 253.0 | 712.7 | 15.7 | 2.82 | 0.06 |
| stdev | 83,305 | --- | 5.1 | 42.0 | 2.1 | --- | --- | ||||
| Proteobacteria | Shigella sonnei | --- | 3 | Average | 4,929,599 | 16,299 | 302.4 | 261.3 | 11.7 | 0.86 | 0.04 |
| stdev | 90,607 | --- | 5.6 | 8.4 | 3.5 | --- | --- | ||||
| Proteobacteria | Yersinia pseudotuberculosis | --- | 3 | Average | 4,802,245 | 16,299 | 294.6 | 429.3 | 120.3 | 1.46 | 0.41 |
| stdev | 118,706 | --- | 7.3 | 2.3 | 3.8 | --- | --- | ||||
| Proteobacteria | Vibrio vulnificus | chromosome I | 3 | Average | 3,330,104 | 16,299 | 204.3 | 180.0 | 10.3 | 0.88 | 0.05 |
| stdev | 79,423 | --- | 4.9 | 14.0 | 4.9 | --- | --- | ||||
| Proteobacteria | Vibrio vulnificus | chromosome II | 3 | Average | 1,756,668 | 16,299 | 107.8 | 90.0 | 3.3 | 0.83 | 0.03 |
| stdev | 87,177 | --- | 5.3 | 7.9 | 3.1 | --- | --- | ||||
| Firmicutes | Staphylococcus aureus | --- | 3 | Average | 2,948,373 | 16,299 | 180.9 | 108.3 | 0.0 | 0.60 | 0.00 |
| stdev | 114,371 | --- | 7.0 | 10.6 | 0.0 | --- | --- | ||||
| Firmicutes | Streptococcus pyogenes | --- | 3 | Average | 1,895,707 | 16,299 | 116.3 | 263.7 | 0.3 | 2.27 | 0.00 |
| stdev | 42,370 | --- | 2.6 | 15.4 | 0.6 | --- | --- | ||||
| Firmicutes | Enterococcus faecalis | --- | 3 | Average | 3,090,387 | 16,299 | 189.6 | 649.7 | 2.0 | 3.42 | 0.01 |
| stdev | 117,259 | --- | 7.2 | 84.1 | 3.5 | --- | --- | ||||
| Firmicutes | Bacillus anthracis | --- | 3 | Average | 5,228,732 | 16,299 | 320.8 | 432.0 | 1.3 | 1.35 | 0.00 |
| stdev | 1349 | --- | 0.1 | 11.5 | 0.6 | --- | --- | ||||
| Firmicutes | Bacillus cereus | --- | 3 | Average | 5,406,060 | 16,299 | 331.7 | 700.3 | 13.0 | 2.11 | 0.04 |
| stdev | 16,615 | --- | 1.0 | 53.7 | 6.1 | --- | --- |
| STM Gene Accession | SEEG Gene Accession | SEN Gene Accession | AT 8+mer Sequence | Common Name of Gene | Description of Target Gene | Gene Function | Biological Process |
|---|---|---|---|---|---|---|---|
| no homolog | SEEG9184_21515 | SEN_RS22080 | conserved (3 locations) | sefC | SEEG pseudogene | Has 3 AT 8mers in sequence; outer membrane fimbrial protein SefC, pseudo in SG, which has an extra A/T to make a 7mer | adhesion |
| STM0071 | SEEG9184_20585 | SEN_RS00360 | conserved | caiC | SEEG pseudogene | crotonobetaine/carnitine-CoA ligase | transporter |
| STM0858 | SEEG9184_16520 | SEN_RS04155 | conserved | unnamed | SEEG pseudogene | electron transfer flavoprotein-ubiquinone oxidoreductase | transporter |
| STM2020 | SEEG9184_10295 | SEN_RS10515 | conserved | cbiO | SEEG pseudogene | cobalt transport atp-binding protein CbiO: B12 synthesis associated? Truncated, maybe shorter product? | transporter |
| STM2241 | SEEG9184_09260 | SEN_RS11570 | conserved | sspH2 | SEEG pseudogene | E3 ubiquitin-protein ligase; induced by the SPI-2 regulatory ssrA/B | virulence factor |
| STM2274 | SEEG9184_09070 | SEN_RS11735 | conserved | unnamed | SEEG pseudogene | MFS transporter | transporter |
| STM2691 | SEEG9184_07115 | SEN_RS13585 | conserved | unnamed | SEEG pseudogene | type I secretion system permease/ATPase: TolC family OMP | virulence factor |
| STM3658 | SEEG9184_00930 | SEN_RS18105 | conserved | yiaH | SEEG pseudogene | acetyltransferase | biosynthesis |
| STM1054 | SEEG9184_09275 | SEN_RS23040 | conserved | unnamed | SEN pseudogene, SEEG 78 bp tRNA region | Gifsy-2 prophage protein in STM: GC rich region has a 7 bp deletion in SEN in a guanidine rich fragment, causing a frameshift; homology in SEEG to tRNA-pro | phage associated |
| STM4039 | SEEG9184_23265 | SEN_RS24515 | conserved | unnamed | SEN, SEEG pseudogene | HO protein | inner membrane protein |
| STM1666 | no homolog | SEN_RS07090 | conserved | unnamed | STM pseudogene, SEEG absent | STM has in-frame stop following codon 24; SEN, hypothetical protein | unknown |
| STM1550 | no homolog | SEN_RS07790 | Deletion of 154 bp in SEN | unnamed | SEN pseudogene, SEEG absent | type II toxin-antitoxin system mRNA interferase toxin | cellular detoxification |
| STM0341 | SEEG9184_19090&19095 | SEN_RS01660 | SEEG 1 bp deletion | unnamed | SEEG pseudogene or split into two genes | STM and SEN, putative inner membrane protein; SEEG, 2 transmembrane regulators | inner membrane protein |
| STM1130 | SEEG9184_15700 | SEN_RS05135 | SEEG 1 bp deletion | unnamed | SEEG pseudogene | N-acetylneuraminic acid mutarotase | metabolism |
| STM1602 | SEEG9184_13085 | SEN_RS07535 | SEEG 1 bp deletion | sifB | SEEG pseudogene | effector protein SifB | virulence factor |
| STM1698 | SEEG9184_13605 | SEN_RS06925 | SEEG 1 bp deletion | steC | SEEG pseudogene | secreted effector kinase SteC | virulence factor |
| STM1869 | SEEG9184_14565 | SEN_RS06020 | SEEG 1 bp deletion | unnamed | SEEG pseudogene | HO protein | phage associated |
| STM1939 | SEEG9184_15200 | SEN_RS05535 | SEEG 1 bp deletion | unnamed | SEEG pseudogene | putative glucose-6-phosphate dehydrogenase | metabolism |
| STM2129 | SEEG9184_09800 | SEN_RS11065 | SEEG 1 bp deletion | yegB | SEEG pseudogene | multidrug transporter subunit MdtD | transporter |
| no homolog | SEEG9184_21510 | SEN_RS22085 | SEEG 1 bp substitution | sefD | SEEG pseudogene | adhesin and master global regulator of phase transition; often mutated in SEN due to 1bp del in adenine homopolymer 8mer | regulon, adhesion |
| STM1941 | SEEG9184_15210 | SEN_RS05525 | SEEG, SEN 1 bp deletion | unnamed | SEN, SEEG pseudogene | HO protein | inner membrane protein |
| STM3674 | SEEG9184_00845 | SEN_RS18190 | SEEG, SEN 1 bp substitution | lyxK | SEEG pseudogene | carbohydrate kinase | metabolism |
| Serovar | Typhimurium | Enteritidis | Gallinarum | Other Information | ||
|---|---|---|---|---|---|---|
| Accession | NC_003277.2 | NZ_CP063701.1 | CM001154.1 | |||
| Other Name | pSLT | pSENV | str. SG9 | |||
| Atmer Size | Description of Loci | Variation from pSLT | Variation from pSLT | pSLT Start | pSLT End | Gene Function |
| 9mer | Intergenic(PSLT039-PSLT038) | 1nt deletion | 1nt deletion | 27961 | 27969 | spvB-spvC;58nt upstream from spvC start |
| 10mer | Intergenic (PSLT041-PSLT042) | 1nt deletion | 2nt deletion | 32324 | 32333 | spvR-PSLT041: 10nt upstream from spvR start |
| 9mer | PSLT076 | present | absent | 62299 | 62307 | traY: conjugative transfer: oriT nicking |
| 8mer | PSLT088 | absent | present | 69093 | 69100 | traC: conjugative transfer: assembly |
| 8mer | PSLT102 | absent | absent | 82835 | 82842 | traS: conjugative transfer: surface exclusion |
| 8mer | PSLT111 | truncated | truncated | 93512 | 93519 | finO: conjugative transfer: regulation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guard, J.; Rivers, A.R.; Vaughn, J.N.; Rothrock, Jr., M.J.; Oladeinde, A.; Shah, D.H. AT Homopolymer Strings in Salmonella enterica Subspecies I Contribute to Speciation and Serovar Diversity. Microorganisms 2021, 9, 2075. https://doi.org/10.3390/microorganisms9102075
Guard J, Rivers AR, Vaughn JN, Rothrock, Jr. MJ, Oladeinde A, Shah DH. AT Homopolymer Strings in Salmonella enterica Subspecies I Contribute to Speciation and Serovar Diversity. Microorganisms. 2021; 9(10):2075. https://doi.org/10.3390/microorganisms9102075
Chicago/Turabian StyleGuard, Jean, Adam R. Rivers, Justin N. Vaughn, Michael J. Rothrock, Jr., Adelumola Oladeinde, and Devendra H. Shah. 2021. "AT Homopolymer Strings in Salmonella enterica Subspecies I Contribute to Speciation and Serovar Diversity" Microorganisms 9, no. 10: 2075. https://doi.org/10.3390/microorganisms9102075
APA StyleGuard, J., Rivers, A. R., Vaughn, J. N., Rothrock, Jr., M. J., Oladeinde, A., & Shah, D. H. (2021). AT Homopolymer Strings in Salmonella enterica Subspecies I Contribute to Speciation and Serovar Diversity. Microorganisms, 9(10), 2075. https://doi.org/10.3390/microorganisms9102075