
Bacterial Community Structure and Dynamic Changes in Different Functional Areas of a Piggery Wastewater Treatment System



Abstract
:1. Introduction
2. Materials and Methods
2.1. Description of Pig Farm and Sample Collection
2.2. Water Quality Detection
2.3. DNA Extraction, PCR Amplification and Illumina Sequencing
2.4. Data Analysis
3. Results and Discussion
3.1. Detection of Wastewater Quality
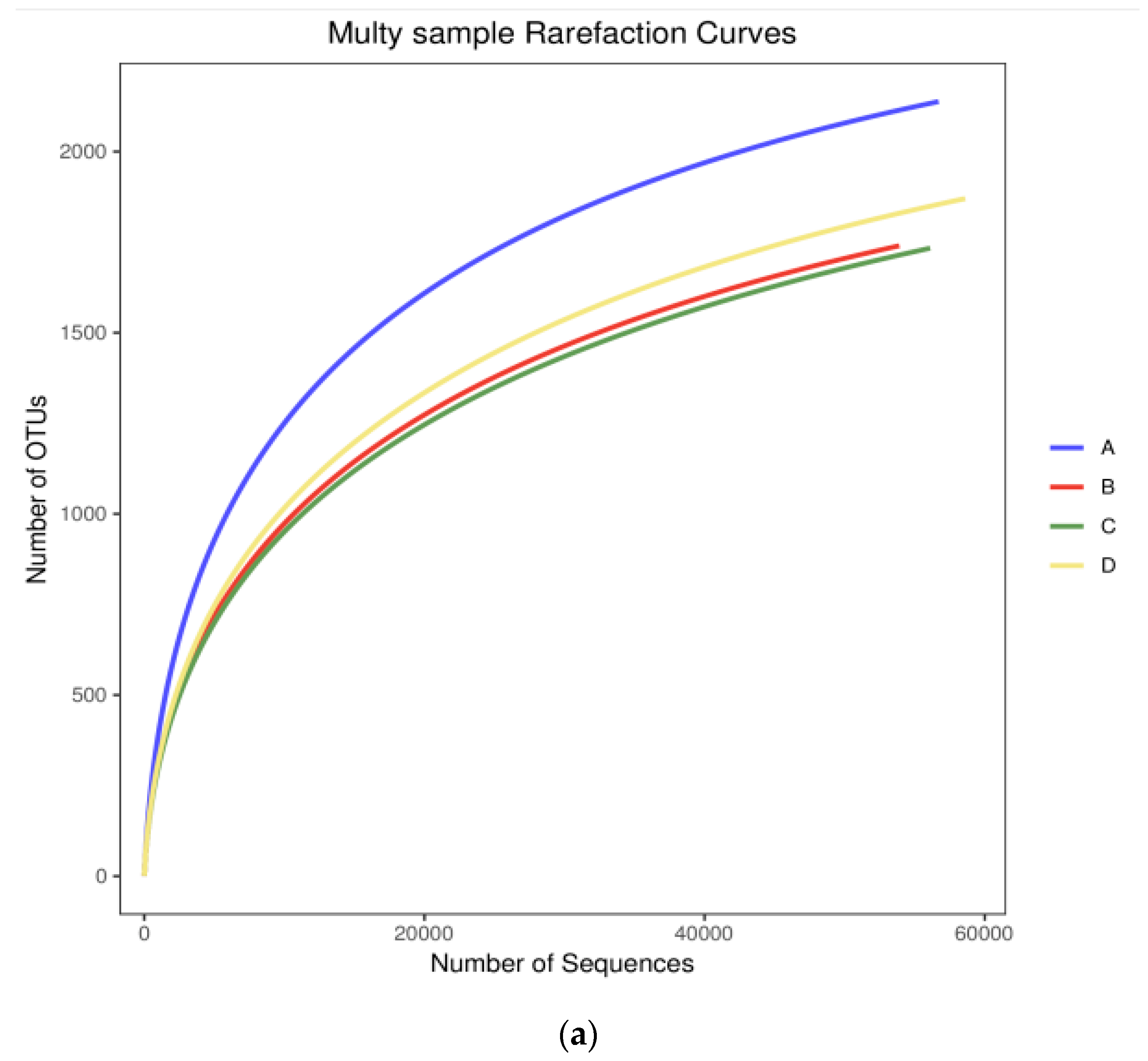
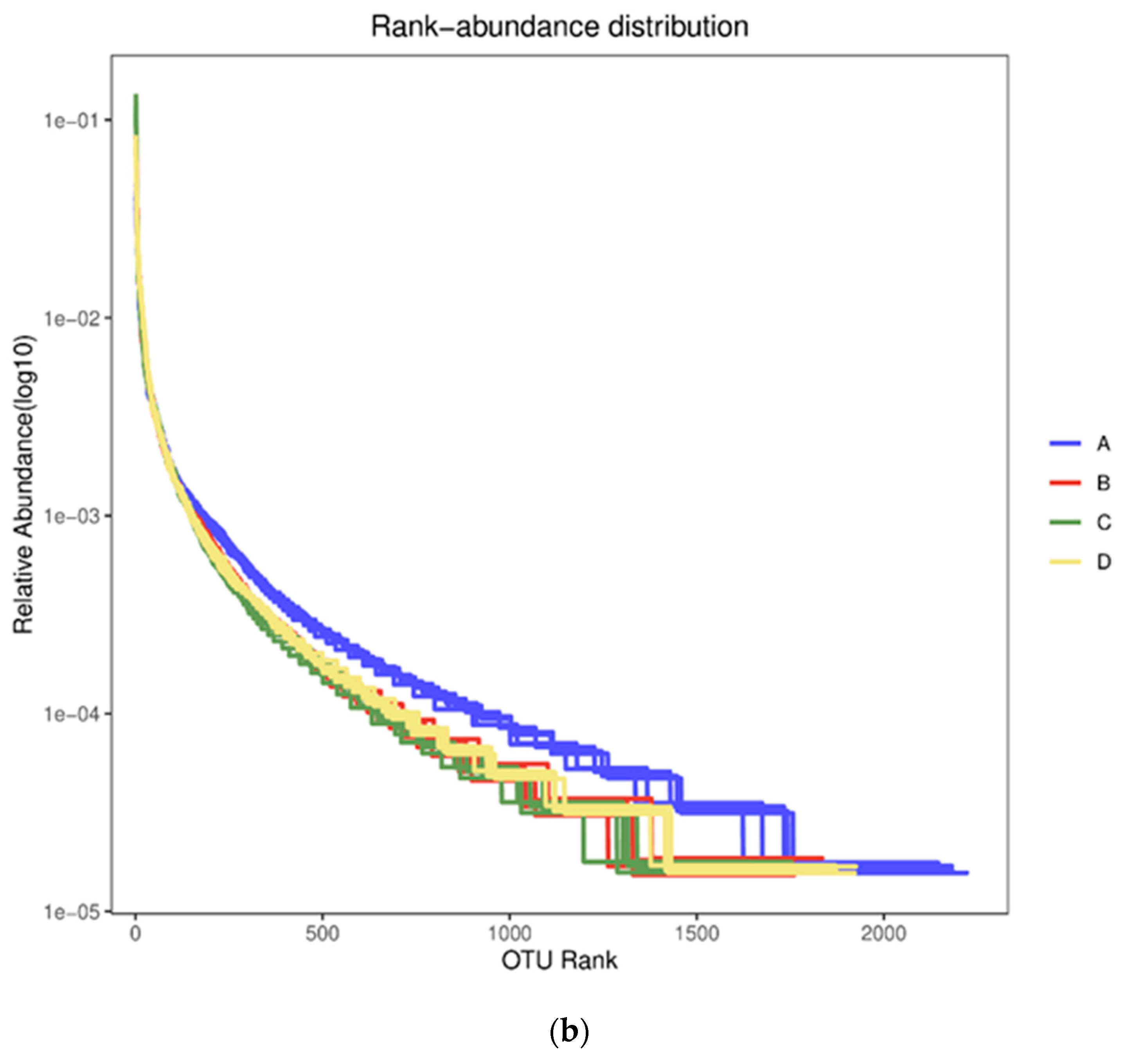
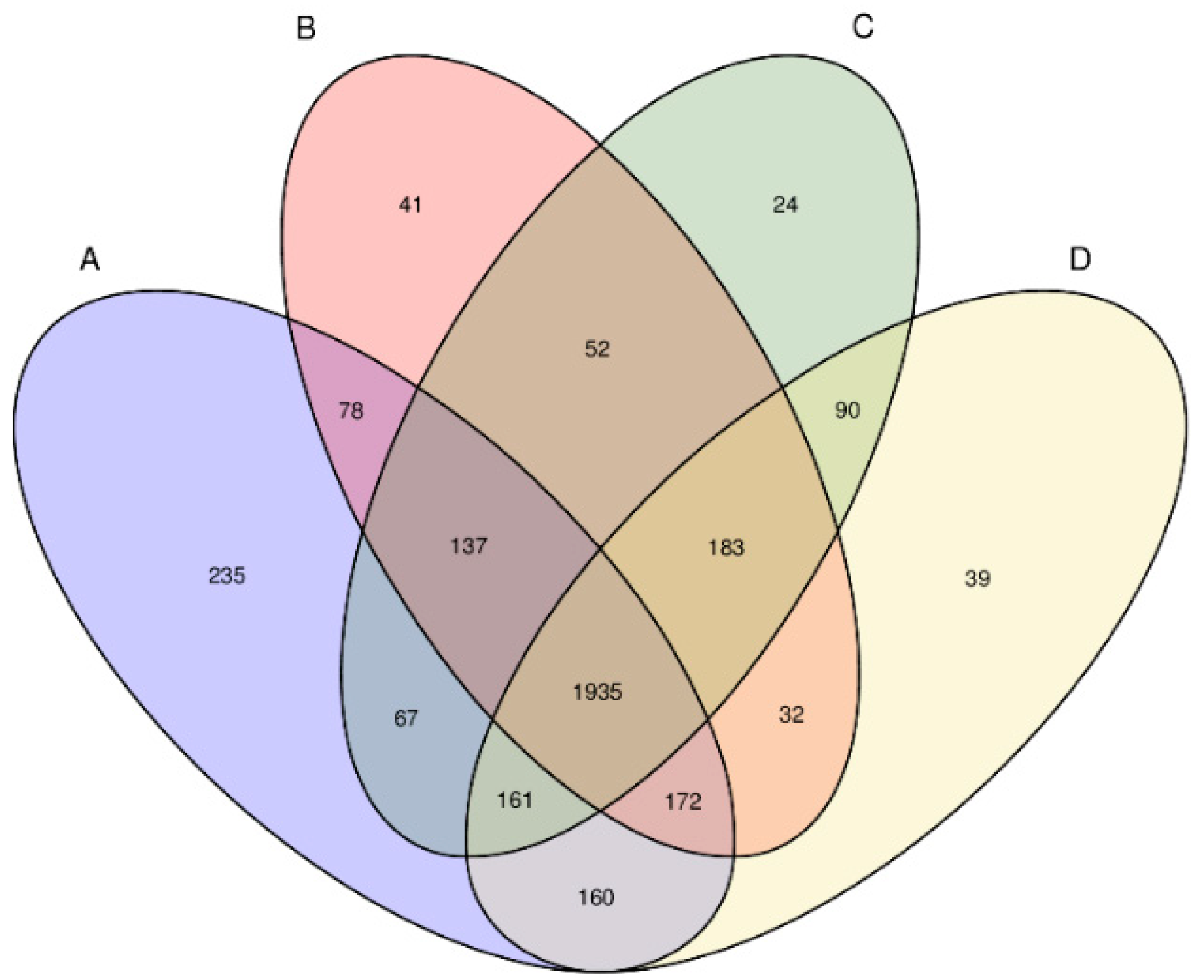
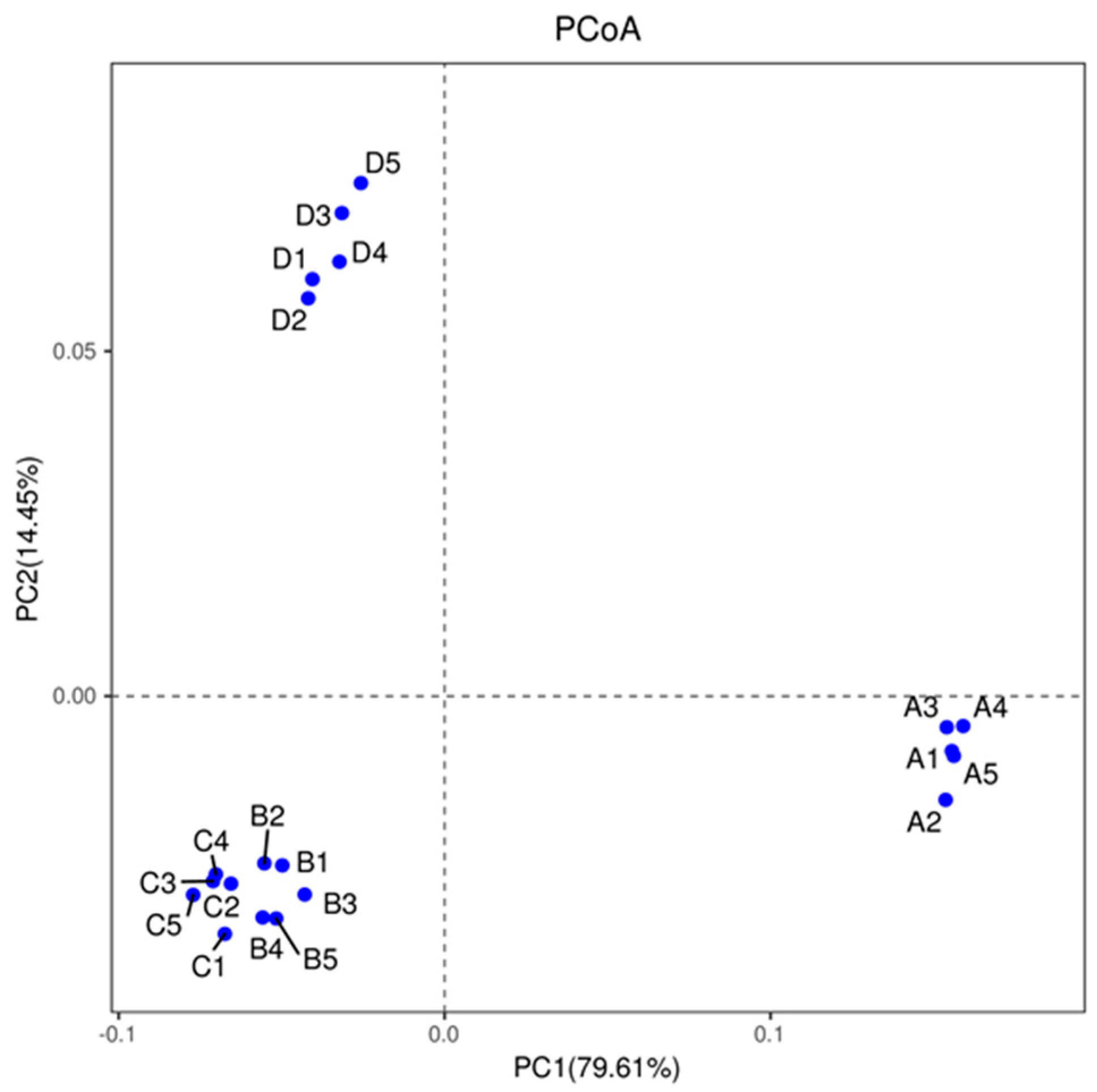
3.2. Diversity Analysis for Bacterial Communities
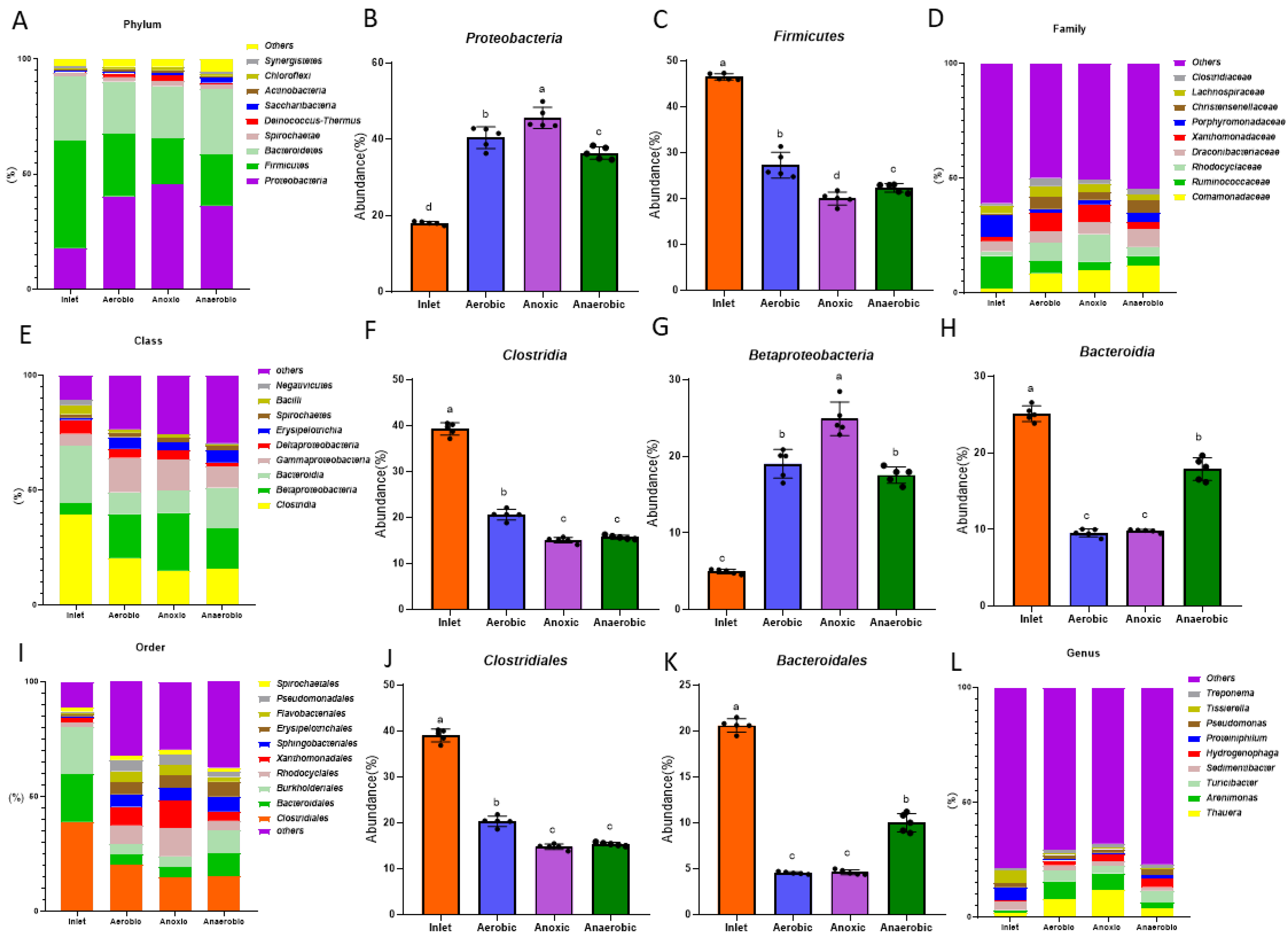
3.3. Bacterial Community Structure among the Biological Treatment Systems
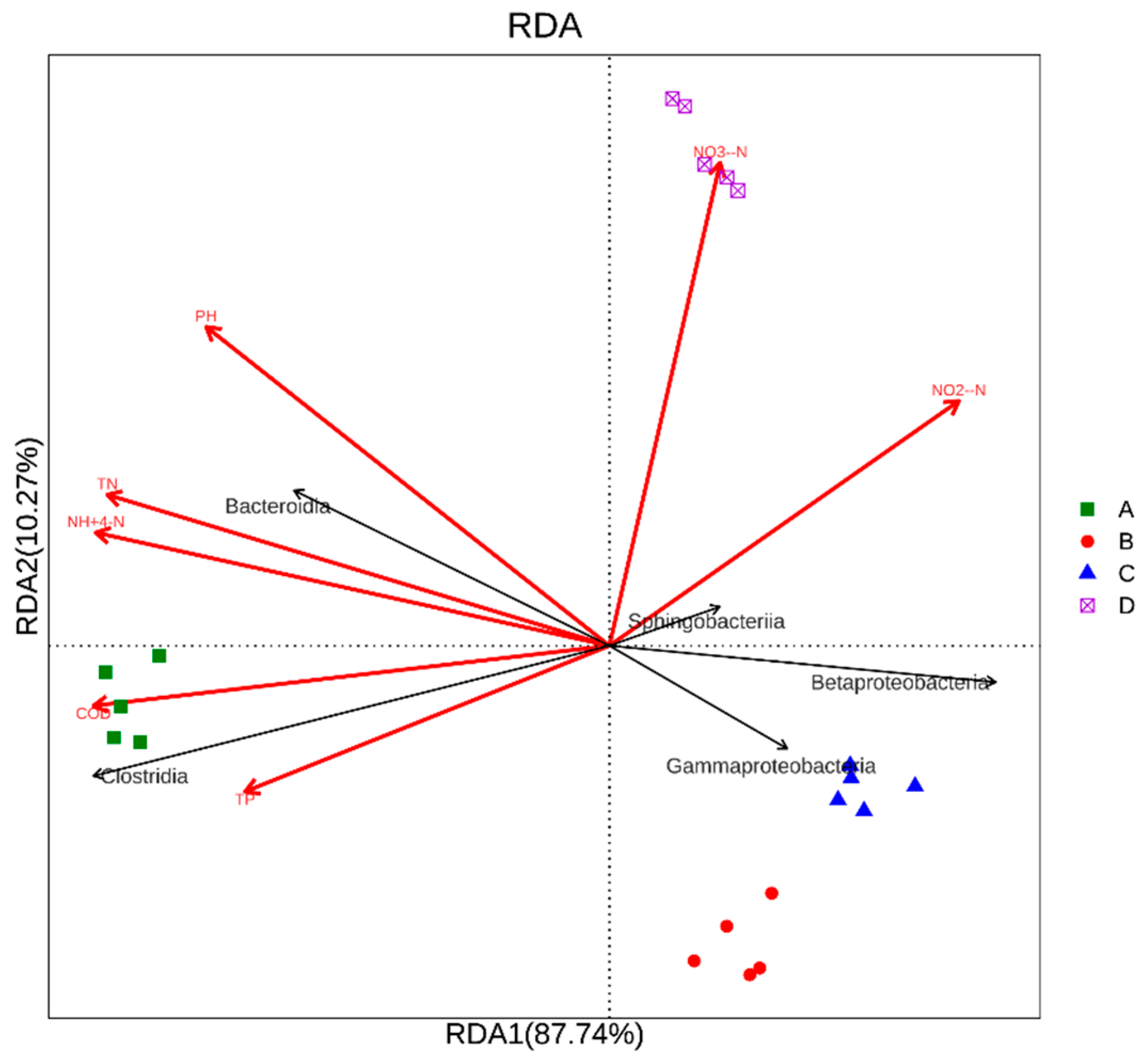
3.4. Environmental Factor Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.; Ling, J.; Chen, P.; Chen, J.; Dai, R.; Liao, J.; Yu, J.; Xu, Y. Pseudomonas mendocina LYX: A novel aerobic bacterium with advantage of removing nitrate high effectively by assimilation and dissimilation simultaneously. Front. Environ. Sci. Eng. 2021, 15, 1–10. [Google Scholar] [CrossRef]
- Gómez-Basurto, F.; Vital-Jácome, M.; Gómez-Acata, E.S.; Thalasso, F.; Luna-Guido, M.; Dendooven, L. Microbial community dynamics during aerobic granulation in a sequencing batch reactor (SBR). PeerJ 2019, 7, e7152. [Google Scholar] [CrossRef] [PubMed]
- Mainardis, M.; Buttazzoni, M.; Goi, D. Up-Flow Anaerobic Sludge Blanket (UASB) technology for energy recovery: A review on state-of-the-art and recent technological advances. Bioengineering 2020, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, Y.; Li, Y.; Liao, J.; Ling, J.; Li, J.; Xie, G. Effective removal of nitrate by denitrification re-enforced with a two-stage anoxic/oxic (A/O) process from a digested piggery wastewater with a low C/N ratio. J. Environ. Manag. 2019, 240, 19–26. [Google Scholar] [CrossRef]
- Niestępski, S.; Harnisz, M.; Ciesielski, S.; Korzeniewska, E.; Osińska, A. Environmental fate of Bacteroidetes, with particular emphasis on Bacteroides fragilis group bacteria and their specific antibiotic resistance genes, in activated sludge wastewater treatment plants. J. Hazard. Mater. 2020, 394, 122544. [Google Scholar] [CrossRef]
- Qin, H.; Ji, B.; Zhang, S.; Kong, Z. Study on the bacterial and archaeal community structure and diversity of activated sludge from three wastewater treatment plants. Mar. Pollut. Bull. 2018, 135, 801–807. [Google Scholar] [CrossRef]
- Guo, J.; Ni, B.-J.; Han, X.; Chen, X.; Bond, P.; Peng, Y.; Yuan, Z. Unraveling microbial structure and diversity of activated sludge in a full-scale simultaneous nitrogen and phosphorus removal plant using metagenomic sequencing. Enzym. Microb. Technol. 2017, 102, 16–25. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Y.; Zhang, X.; Jiang, X.; Wu, S.; Shen, J.; Sun, X.; Li, J.; Lu, L.; Wang, L. Aerobic granulation strategy for bioaugmentation of a sequencing batch reactor (SBR) treating high strength pyridine wastewater. J. Hazard. Mater. 2015, 295, 153–160. [Google Scholar] [CrossRef]
- Gu, Y.; Wei, Y.; Xiang, Q.; Zhao, K.; Yu, X.; Zhang, X.; Li, C.; Chen, Q.; Xiao, H.; Zhang, X. C:N ratio shaped both taxonomic and functional structure of microbial communities in livestock and poultry breeding wastewater treatment reactor. Sci. Total. Environ. 2019, 651, 625–633. [Google Scholar] [CrossRef]
- Yin, Z.; Bi, X.; Xu, C. Ammonia-Oxidizing Archaea (AOA) play with Ammonia-Oxidizing Bacteria (AOB) in nitrogen removal from wastewater. Archaea 2018, 2018, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Zhang, X.-X.; Miao, Y.; Zhao, Y.; Ye, L.; Li, B.; Zhang, T. Fate of antibiotic resistance genes and their associations with bacterial community in livestock breeding wastewater and its receiving river water. Water Res. 2017, 124, 259–268. [Google Scholar] [CrossRef]
- Cai, L.; Tian, R.-M.; Zhou, G.; Tong, H.; Wong, Y.H.; Zhang, W.; Chui, A.P.Y.; Xie, J.Y.; Qiu, J.-W.; Ang, P.O.; et al. Exploring coral microbiome assemblages in the South China Sea. Sci. Rep. 2018, 8, 2428. [Google Scholar] [CrossRef]
- Islam, W.; Noman, A.; Naveed, H.; Huang, Z.; Chen, H.Y.H. Role of environmental factors in shaping the soil microbiome. Environ. Sci. Pollut. Res. 2020, 27, 41225–41247. [Google Scholar] [CrossRef]
- Ma, Y.; Ding, S.; Liu, G.; Fang, J.; Yan, W.; Duraipandiyan, V.; Al-Dhabi, N.A.; Esmail, G.A.; Jiang, H. Egg protein transferrin-derived peptides IRW and IQW regulate citrobacter rodentium-induced, inflammation-related microbial and metabolomic profiles. Front. Microbiol. 2019, 10, 643. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Ma, Y.; Liu, G.; Yan, W.; Jiang, H.; Fang, J. Lactobacillus brevis alleviates DSS-induced colitis by reprograming intestinal microbiota and influencing serum metabolome in murine model. Front. Physiol. 2019, 10, 1152. [Google Scholar] [CrossRef]
- Gao, P.; Xu, W.; Sontag, P.; Li, X.; Xue, G.; Liu, T.; Sun, W. Correlating microbial community compositions with environmental factors in activated sludge from four full-scale municipal wastewater treatment plants in Shanghai, China. Appl. Microbiol. Biotechnol. 2016, 100, 4663–4673. [Google Scholar] [CrossRef]
- Yan, W.; Wang, N.; Wei, D.; Liang, C.; Chen, X.; Liu, L.; Shi, J. Bacterial community compositions and nitrogen metabolism function in a cattle farm wastewater treatment plant revealed by Illumina high-throughput sequencing. Environ. Sci. Pollut. Res. 2021, 28, 40895–40907. [Google Scholar] [CrossRef]
- Zhang, L.; Shen, Z.; Fang, W.; Gao, G. Composition of bacterial communities in municipal wastewater treatment plant. Sci. Total. Environ. 2019, 689, 1181–1191. [Google Scholar] [CrossRef]
- Ospina-Betancourth, C.; Acharya, K.; Allen, B.; Head, I.M.; Sanabria, J.; Curtis, T.P. Valorization of pulp and paper industry wastewater using sludge enriched with nitrogen-fixing bacteria. Water Environ. Res. 2021, 93, 1734–1747. [Google Scholar] [CrossRef]
- APHA. Standard Methods for the Examination of Water and Wastewater, 23rd ed.; American Public Health Association: Washington, DC, USA, 2017. [Google Scholar]
- Wang, Q.; Liang, J.; Zhang, S.; Yoza, B.A.; Li, Q.X.; Zhan, Y.; Ye, H.; Zhao, P.; Chen, C. Characteristics of bacterial populations in an industrial scale petrochemical wastewater treatment plant: Composition, function and their association with environmental factors. Environ. Res. 2020, 189, 109939. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, L.; Xiang, F.; Zhao, L.; Qiao, Z. Activated Sludge microbial community and treatment performance of wastewater treatment plants in industrial and municipal zones. Int. J. Environ. Res. Public Health 2020, 17, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Meshref, M.N.; Dhar, B.R. Optimization of thermal hydrolysis process for enhancing anaerobic digestion in a wastewater treatment plant with existing primary sludge fermentation. Bioresour. Technol. 2021, 321, 124498. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Peng, Y.; Wang, B.; Li, X.; Zhang, Q. A novel SNPR process for advanced nitrogen and phosphorus removal from mainstream wastewater based on anammox, endogenous partial-denitrification and denitrifying dephosphatation. Water Res. 2020, 170, 115363. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhou, J.; He, X.; He, L.; Lin, Z.; Shi, S.; Zhou, J. Simultaneous nitrogen and phosphorus removal from simulated digested piggery wastewater in a single-stage biofilm process coupling anammox and intracellular carbon metabolism. Bioresour. Technol. 2021, 333, 125152. [Google Scholar] [CrossRef]
- Zhao, W.; Peng, Y.; Wang, M.; Huang, Y.; Li, X. Nutrient removal and microbial community structure variation in the two-sludge system treating low carbon/nitrogen domestic wastewater. Bioresour. Technol. 2019, 294, 122161. [Google Scholar] [CrossRef]
- Ma, X.; Wu, C.; Huang, J.; Zhou, R.; Shi, B. Microbial community of tannery wastewater involved in nitrification revealed by illumina MiSeq sequencing. J. Microbiol. Biotechnol. 2018, 28, 1168–1177. [Google Scholar] [CrossRef] [Green Version]
- Aziz, A.; Basheer, F.; Sengar, A.; Irfanullah; Khan, S.U.; Farooqi, I.H. Biological wastewater treatment (anaerobic-aerobic) technologies for safe discharge of treated slaughterhouse and meat processing wastewater. Sci. Total. Environ. 2019, 686, 681–708. [Google Scholar] [CrossRef]
- Kumar, H.; Na Jang, Y.; Kim, K.; Park, J.; Jung, M.W.; Park, J.-E. Compositional and functional characteristics of swine slurry microbes through 16S rRNA metagenomic sequencing approach. Anim. 2020, 10, 1372. [Google Scholar] [CrossRef]
- Luo, Y.; Yao, J.; Wang, X.; Zheng, M.; Guo, D.; Chen, Y. Efficient municipal wastewater treatment by oxidation ditch process at low temperature: Bacterial community structure in activated sludge. Sci. Total. Environ. 2020, 703, 135031. [Google Scholar] [CrossRef]
- Xu, S.; Yao, J.; Ainiwaer, M.; Hong, Y.; Zhang, Y. Analysis of bacterial community structure of activated sludge from wastewater treatment plants in winter. BioMed Res. Int. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Mao, Y.; Wang, Z.; Zhang, T. Diversity and functions of bacterial community in drinking water biofilms revealed by high-throughput sequencing. Sci. Rep. 2015, 5, 10044. [Google Scholar] [CrossRef]
- Morin, L.; Goubet, A.; Madigou, C.; Pernelle, J.-J.; Palmier, K.; Labadie, K.; Lemainque, A.; Michot, O.; Astoul, L.; Barbier, P.; et al. Colonization kinetics and implantation follow-up of the sewage microbiome in an urban wastewater treatment plant. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.-X.; Lu, X.; Liu, B.; Li, Y.; Long, C.; Li, A. Abundance and diversity of bacterial nitrifiers and denitrifiers and their functional genes in tannery wastewater treatment plants revealed by high-throughput sequencing. PLoS ONE 2014, 9, e113603. [Google Scholar] [CrossRef]
- Wang, P.; Wu, D.; You, X.; Su, Y.; Xie, B. Antibiotic and metal resistance genes are closely linked with nitrogen-processing functions in municipal solid waste landfills. J. Hazard. Mater. 2021, 403, 123689. [Google Scholar] [CrossRef]
- Joshi, D.R.; Zhang, Y.; Tian, Z.; Gao, Y.; Yang, M. Performance and microbial community composition in a long-term sequential anaerobic-aerobic bioreactor operation treating coking wastewater. Appl. Microbiol. Biotechnol. 2016, 100, 8191–8202. [Google Scholar] [CrossRef]
- Xin, X.; Yang, H.; Guan, L.; Liu, S.; Liu, J. Responses of nitrogen and phosphorus removal performance and microbial community to Fe3O4@SiO2 nanoparticles in a sequencing batch reactor. Appl. Biochem. Biotechnol. 2021, 193, 544–559. [Google Scholar] [CrossRef]
- Tang, J.; Bu, Y.; Zhang, X.-X.; Huang, K.; He, X.; Ye, L.; Shan, Z.; Ren, H. Metagenomic analysis of bacterial community composition and antibiotic resistance genes in a wastewater treatment plant and its receiving surface water. Ecotoxicol. Environ. Saf. 2016, 132, 260–269. [Google Scholar] [CrossRef]
- Révész, F.; Farkas, M.; Kriszt, B.; Szoboszlay, S.; Benedek, T.; Táncsics, A. Effect of oxygen limitation on the enrichment of bacteria degrading either benzene or toluene and the iden-tification of Malikia spinosa (Comamonadaceae) as prominent aerobic benzene-, toluene-, and ethylbenzene-degrading bacterium: Enrichment, isolation and whole-genome analysis. Environ. Sci. Pollut. Res. Int. 2020, 27, 31130–31142. [Google Scholar]
- Wang, Z.; Li, W.; Li, H.; Zheng, W.; Guo, F. Phylogenomics of rhodocyclales and its distribution in wastewater treatment systems. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Xin, X.; Liu, S.; Qin, J.; Ye, Z.; Liu, W.; Fang, S.; Yang, J. Performances of simultaneous enhanced removal of nitrogen and phosphorus via biological aerated filter with biochar as fillers under low dissolved oxygen for digested swine wastewater treatment. Bioprocess. Biosyst. Eng. 2021, 44, 1741–1753. [Google Scholar] [CrossRef]
- Li, C.; Zeng, W.; Li, N.; Guo, Y.; Peng, Y. Population structure and morphotype analysis of Candidatus Accumulibacter using fluorescence in situ hybridization-staining-flow cytometry. Appl. Environ. Microbiol. 2019, 85, e02943-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIlroy, S.J.; Onetto, C.A.; McIlroy, B.; Herbst, F.A.; Dueholm, M.S.; Kirkegaard, R.H.; Fernando, E.; Karst, S.M.; Nierychlo, M.; Kristensen, J.M.; et al. Genomic and in situ analyses reveal the micropruina spp. as abundant fermentative glycogen accu-mulating organisms in enhanced biological phosphorus removal systems. Front. Microbiol. 2018, 9, 1004. [Google Scholar] [CrossRef] [PubMed]
- Semedo, M.; Wittorf, L.; Hallin, S.; Song, B. Differential expression of clade I and II N2O reductase genes in denitrifying Thauera linaloolentis 47LolT under different nitrogen conditions. FEMS Microbiol. Lett. 2021, 367. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhang, Y.; Zhang, Z.; Dong, J.; Fu, J.; Xu, X.; Zhu, L. Enhancement of PPCPs removal by shaped microbial community of aerobic granular sludge under condition of low C/N ratio influent. J. Hazard. Mater. 2020, 394, 122583. [Google Scholar] [CrossRef]
- Wu, X.; Wu, X.; Li, J.; Wu, Q.; Ma, Y.; Sui, W.; Zhao, L.; Zhang, X. Cross-feeding between members of Thauera spp. and Rhodococcus spp. drives quinoline-denitrifying deg-radation in a hypoxic bioreactor. mSphere 2020, 5, e00246-20. [Google Scholar] [CrossRef]
- Wang, Q.; He, J. Complete nitrogen removal via simultaneous nitrification and denitrification by a novel phosphate accumulating Thauera sp. strain SND5. Water Res. 2020, 185, 116300. [Google Scholar] [CrossRef]
- Tourova, T.; Sokolova, D.; Nazina, T.; Grouzdev, D.; Kurshev, E.; Laptev, A. Biodiversity of microorganisms colonizing the surface of polystyrene samples exposed to different aqueous environments. Sustainability 2020, 12, 3624. [Google Scholar] [CrossRef]
- Kang, X.-H.; Leng, Y.; O, M.M.; Zeng, X.-Y.; Li, S.-W. The seasonal changes of core bacterial community decide sewage purification in sub-plateau municipal sewage treatment plants. Bioprocess. Biosyst. Eng. 2020, 43, 1609–1617. [Google Scholar] [CrossRef]
- He, Y.; Li, K.-X.; Wang, J.-W.; Wang, W.; Fan, P.-C.; Chen, H.-H.; Wang, J.-J. Microbial community structure of waste water treatment plants in different seasons. Huan Jing Ke Xue 2021, 42, 1488–1495. [Google Scholar]
- Meerbergen, K.; Van Geel, M.; Waud, M.; Willems, K.A.; Dewil, R.; Van Impe, J.; Appels, L.; Lievens, B. Assessing the composition of microbial communities in textile wastewater treatment plants in comparison with municipal wastewater treatment plants. Microbiology 2016, 6, e00413. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Mhuantong, W.; Liu, S.-J.; Pisutpaisal, N.; Wongwilaiwalin, S.; Kanokratana, P.; Wang, A.-J.; Jiang, C.-Y.; Champreda, V.; Qiu, D.-R. Tropical and temperate wastewater treatment plants assemble different and diverse microbiomes. Appl. Microbiol. Biotechnol. 2021, 105, 853–867. [Google Scholar] [CrossRef]
- Hassa, J.; Maus, I.; Off, S.; Pühler, A.; Scherer, P.; Klocke, M.; Schlüter, A. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl. Microbiol. Biotechnol. 2018, 102, 5045–5063. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | pH | COD/mg/L | BOD5 | NH4+-N | TP | TN | NO3−-N | NO2−-N |
|---|---|---|---|---|---|---|---|---|
| Inlet | 7.83 ± 0.03 | 3670 ± 9.06 | 968 ± 8.34 | 365 ± 4.41 | 75.2 ± 2.70 | 436 ± 9.52 | 18.9 ± 1.48 | 0.15 ± 0.01 |
| Anaerobic | 7.42 ± 0.03 | 970 ± 10.38 | 266 ± 14.31 | 200 ± 5.74 | 53.9 ± 6.62 | 284 ± 8.07 | 85.3 ± 1.45 | 26.4 ± 1.16 |
| Anoxic | 6.74 ± 0.03 | 927 ± 12.57 | 255 ± 15.64 | 105 ± 5.65 | 64.4 ± 1.03 | 177 ± 6.68 | 13.2 ± 0.65 | 33.8 ± 1.01 |
| Aerobic | 6.32 ± 0.03 | 984 ± 4.53 | 263 ± 5.65 | 130 ± 5.27 | 54.2 ± 1.67 | 190 ± 3.14 | 33.0 ± 1.79 | 1.48 ± 0.05 |
| Index | Inlet | Anaerobic | Anoxic | Aerobic |
|---|---|---|---|---|
| Sequences | 59833 ± 2774 a | 60799 ± 2530 a | 59419 ± 2998 a | 58550 ± 4079 a |
| Sobs | 2162 ± 49.9 a | 1887.6 ± 35.66 b | 1761 ± 71.6 c | 1777 ± 34.8 c |
| Chao1 | 2509 ± 49.5 a | 2275 ± 86.2 b | 2201 ± 103.1 b | 2177 ± 60.2 b |
| Ace | 2504 ± 52.03 a | 2308 ± 77.6 b | 2207 ± 109.4 bc | 2183 ± 45.2 c |
| Shannon | 5.87 ± 0.07 a | 5.37 ± 0.05 b | 5.15 ± 0.10 c | 5.24 ± 0.06 c |
| Simpson | 0.9 ± 0.1 d | 2.1 ± 0.1 b | 2.5 ± 0.4 a | 1.5 ± 0.1 c |
| Coverage | 99.2 ± 0.04 a | 99.2 ± 0.05 a | 99.2 ± 0.05 a | 99.2 ± 0.09 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, L.; Liu, N.; Liu, G.; Fang, J. Bacterial Community Structure and Dynamic Changes in Different Functional Areas of a Piggery Wastewater Treatment System. Microorganisms 2021, 9, 2134. https://doi.org/10.3390/microorganisms9102134
Shi L, Liu N, Liu G, Fang J. Bacterial Community Structure and Dynamic Changes in Different Functional Areas of a Piggery Wastewater Treatment System. Microorganisms. 2021; 9(10):2134. https://doi.org/10.3390/microorganisms9102134
Chicago/Turabian StyleShi, Lin, Naiyuan Liu, Gang Liu, and Jun Fang. 2021. "Bacterial Community Structure and Dynamic Changes in Different Functional Areas of a Piggery Wastewater Treatment System" Microorganisms 9, no. 10: 2134. https://doi.org/10.3390/microorganisms9102134
APA StyleShi, L., Liu, N., Liu, G., & Fang, J. (2021). Bacterial Community Structure and Dynamic Changes in Different Functional Areas of a Piggery Wastewater Treatment System. Microorganisms, 9(10), 2134. https://doi.org/10.3390/microorganisms9102134