
Predicting the Role of the Human Gut Microbiome in Constipation Using Machine-Learning Methods: A Meta-Analysis

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
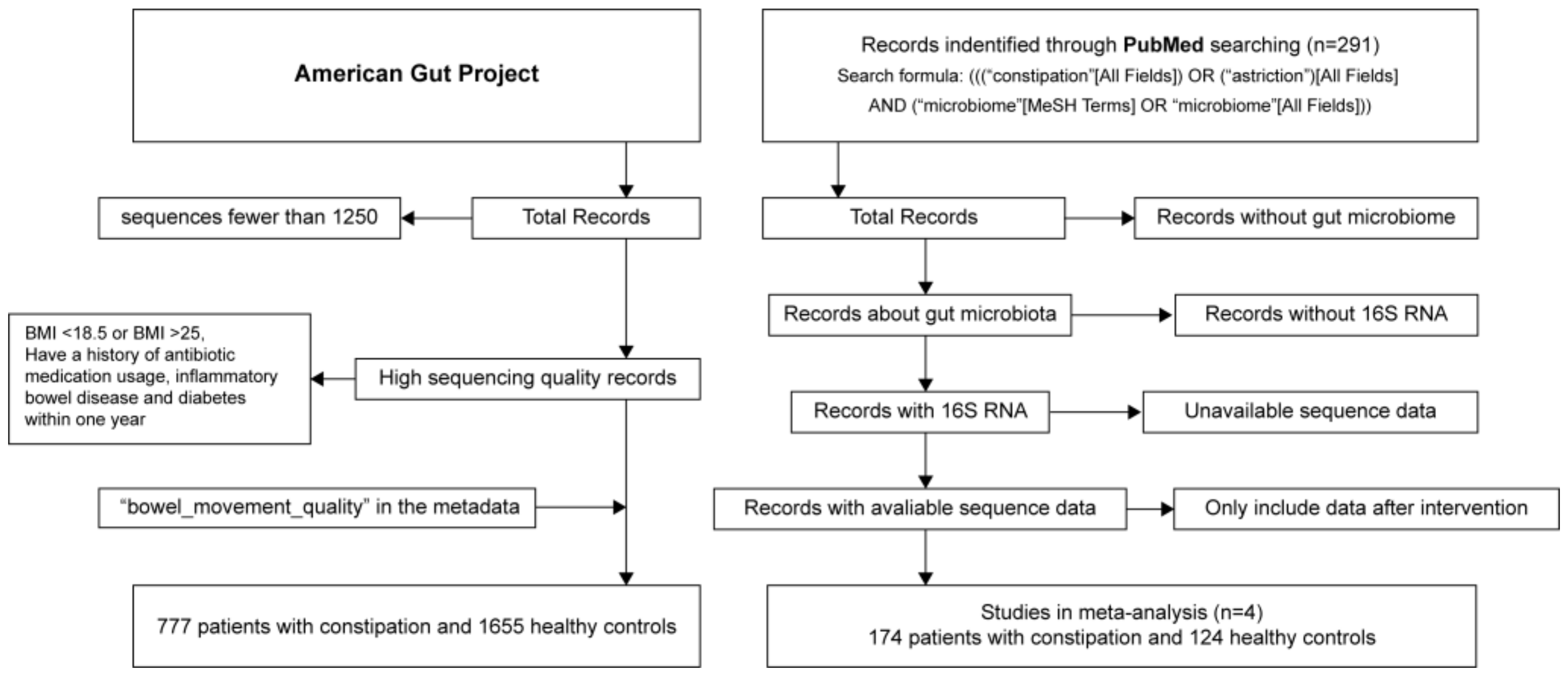
2.1. Data Inclusion Criteria and Data Collection
2.2. Bioinformatic Processing
2.3. Gut Microbiome Diversity Analysis
2.4. Taxonomic Analyses
2.5. Microbial Co-Abundance Network Construction
2.6. Data Normalization
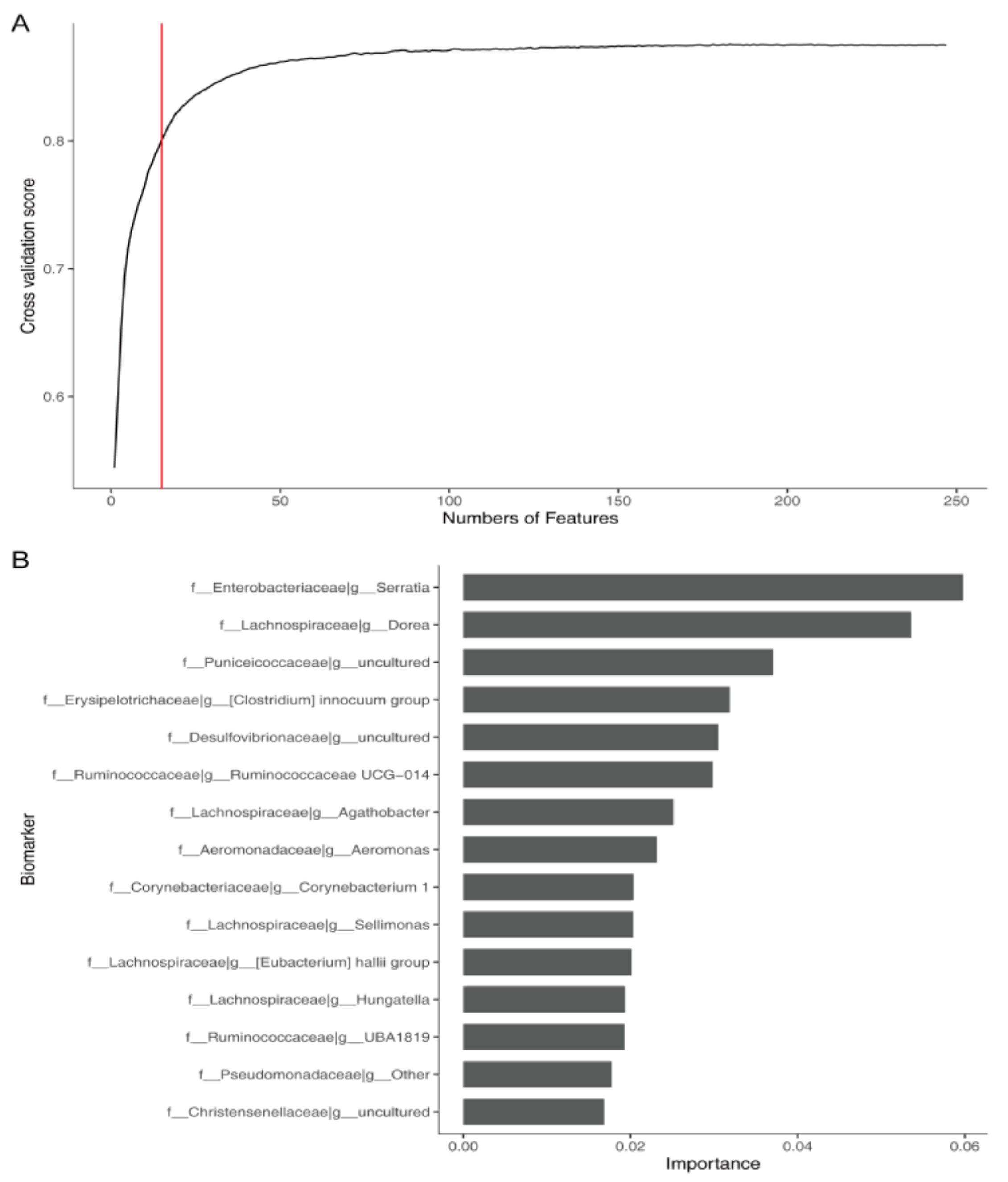
2.7. Feature Selection
2.8. Model Construction and Grid Search
3. Results
3.1. Characteristics of Study Population
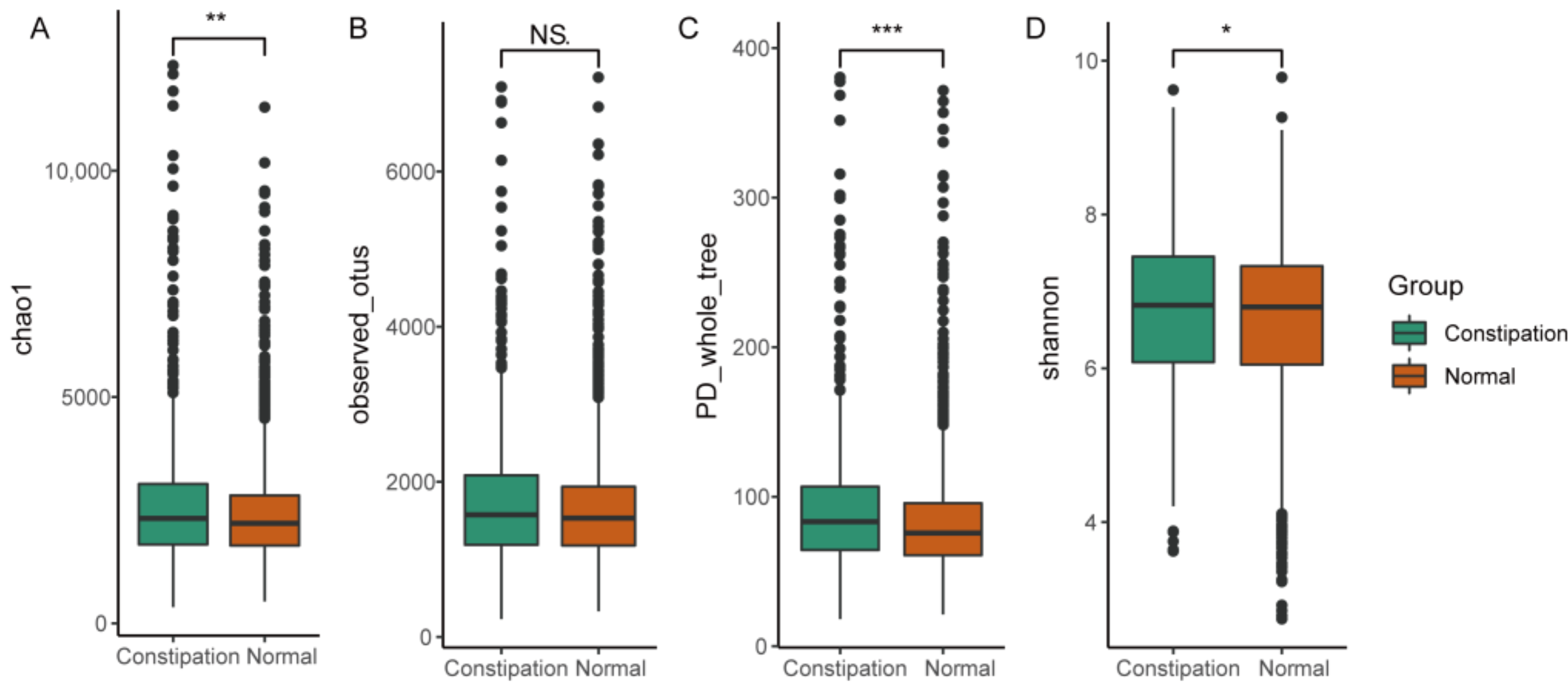
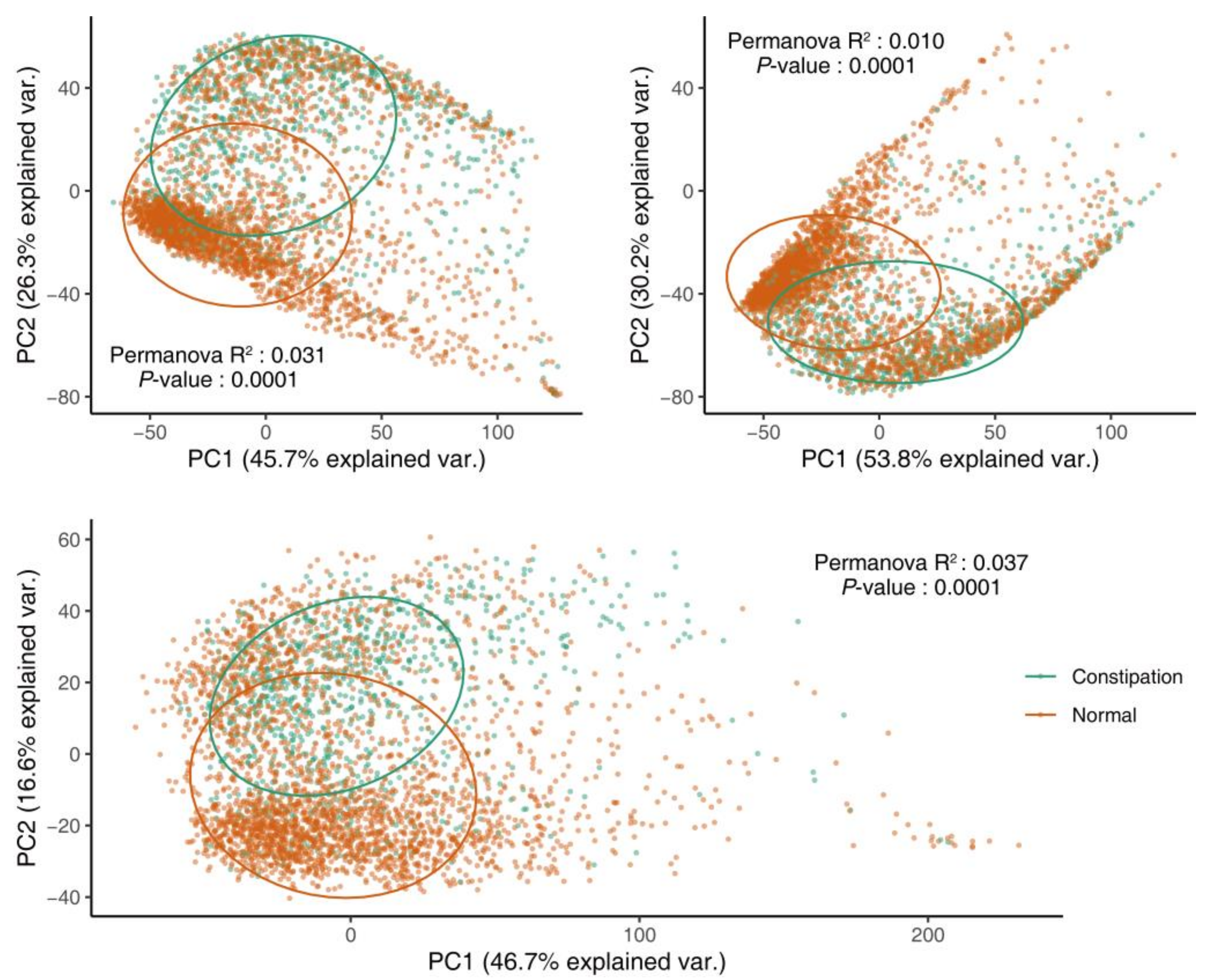
3.2. Gut Microbial Diversity in Patients with Constipation
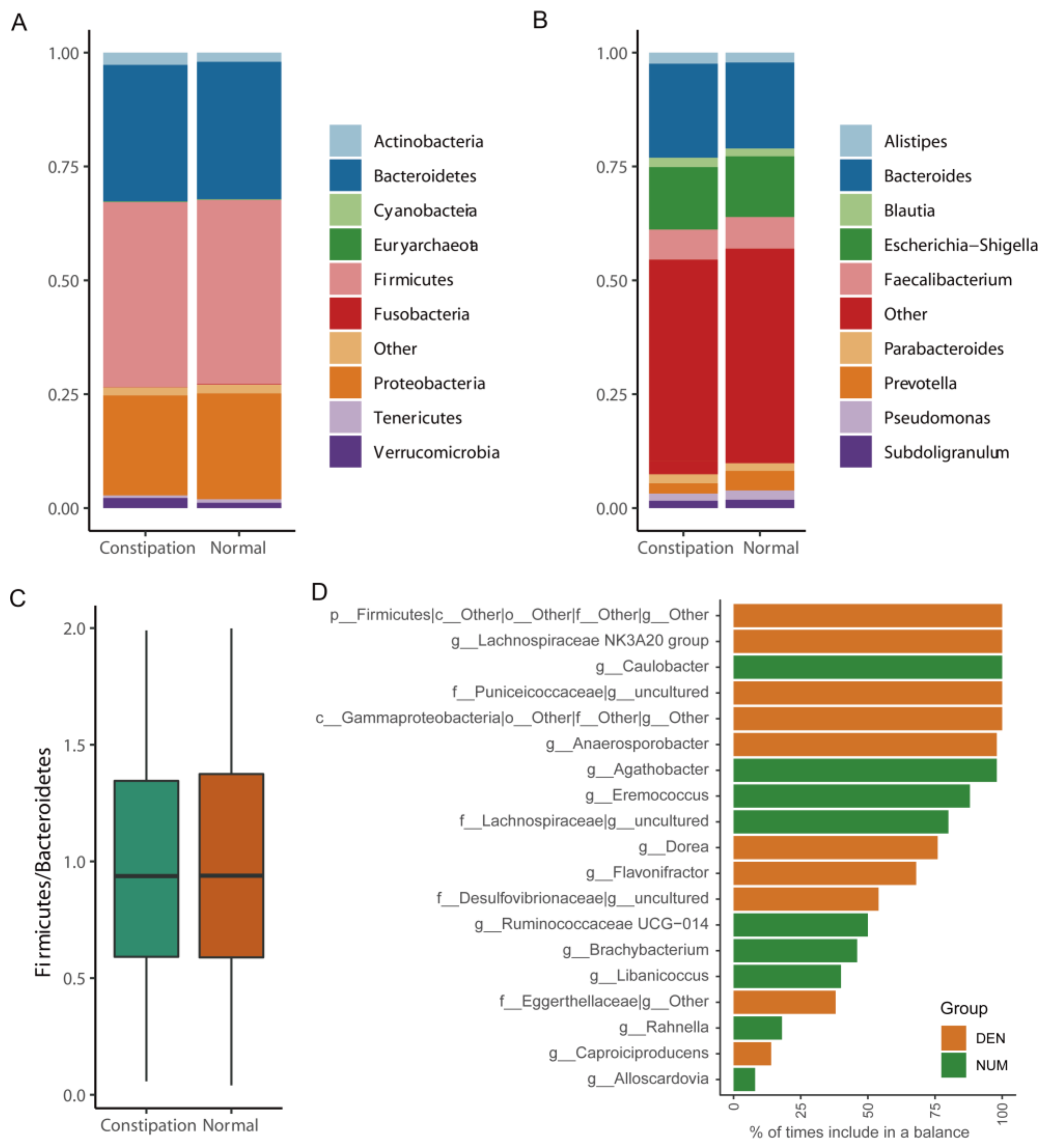
3.3. Phylogenetic Profiles of the Gut Microbiome of Patients with Constipation
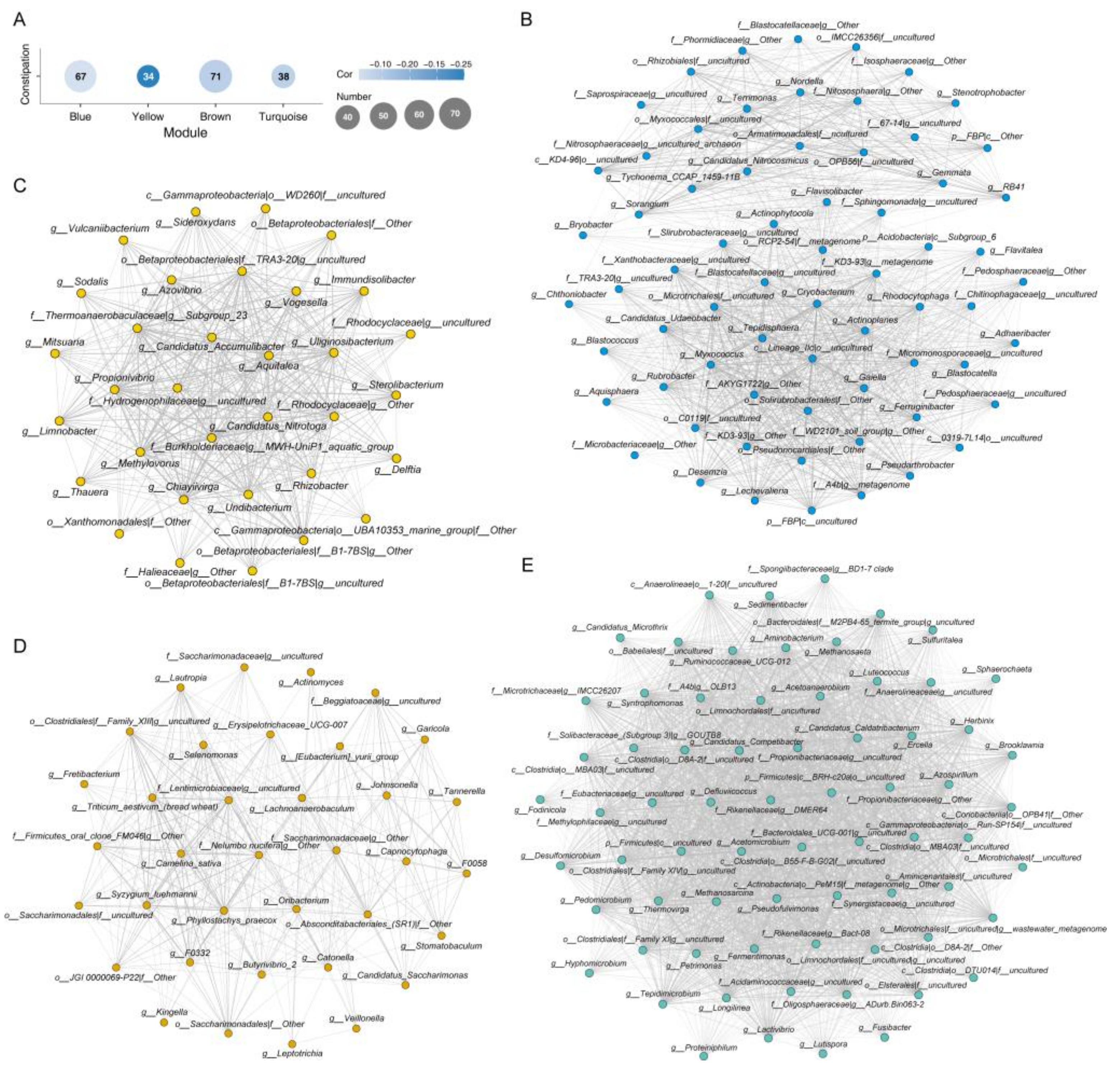
3.4. Microbial Co-Abundance Network Modules and Constipation Associations
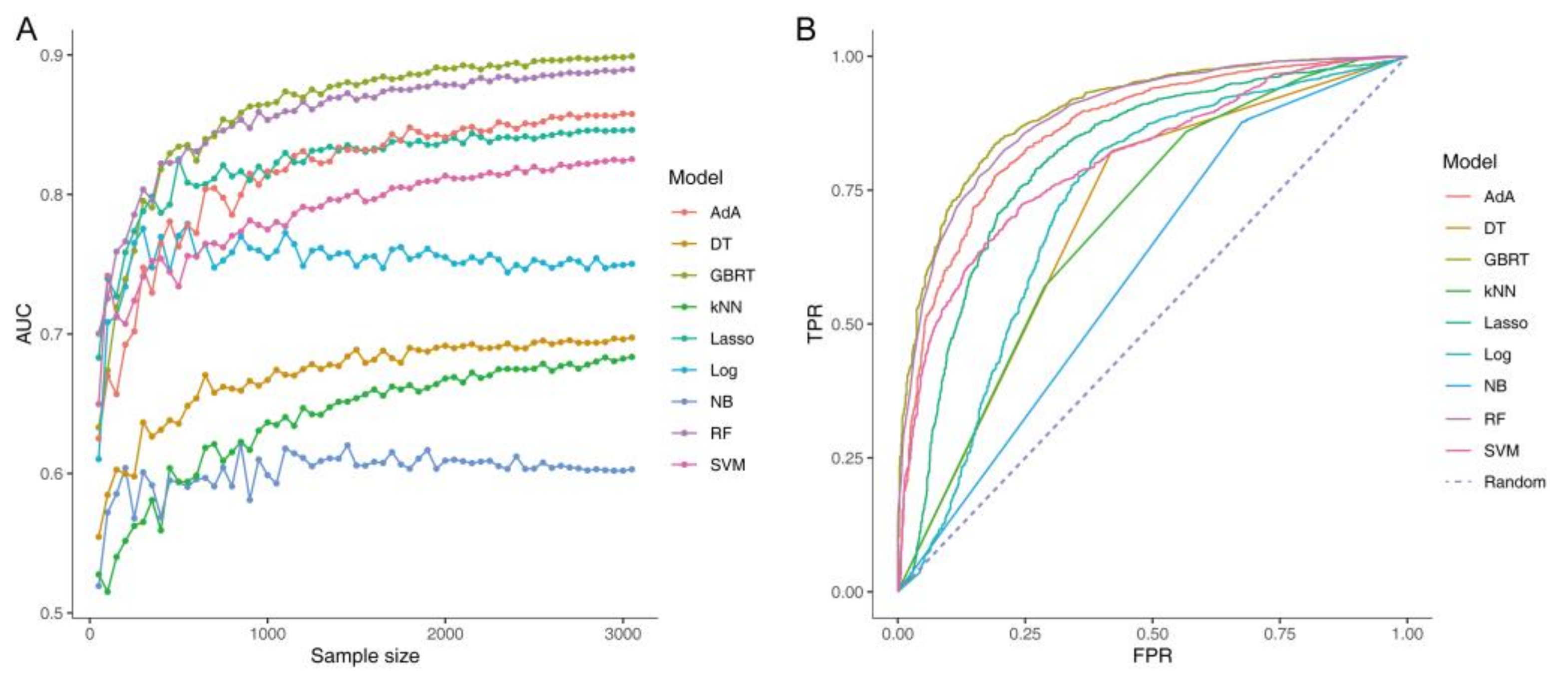
3.5. Detection of Constipation Based on the Gut Microbiome
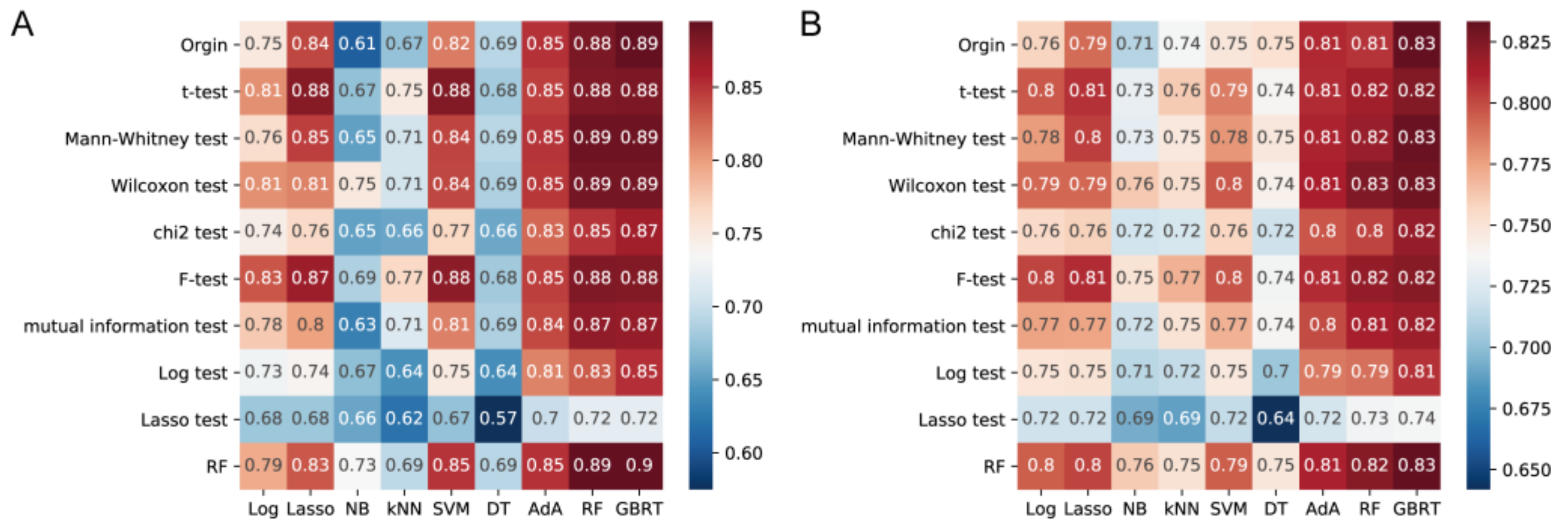
3.6. Validation and Tuning the Parameters of Classifier Models for Constipation
3.7. Identification of Microbial Markers for Constipation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Suares, N.C.; Ford, A.C. Prevalence of, and risk factors for, chronic idiopathic constipation in the community: Systematic review and meta-analysis. Am. J. Gastroenterol. 2011, 106, 1582–1591. [Google Scholar] [CrossRef]
- Rao, S.S.C.; Rattanakovit, K.; Patcharatrakul, T. Diagnosis and management of chronic constipation in adults. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 295–305. [Google Scholar] [CrossRef]
- Camilleri, M.; Ford, A.C.; Mawe, G.M.; Dinning, P.G.; Rao, S.S.; Chey, W.D.; Simrén, M.; Lembo, A.; Young-Fadok, T.M.; Chang, L. Chronic constipation. Nat. Rev. Dis. Primers 2017, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Armour, C.R.; Nayfach, S.; Pollard, K.S.; Sharpton, T.J. A Metagenomic Meta-analysis Reveals Functional Signatures of Health and Disease in the Human Gut Microbiome. mSystems 2019, 4, e00332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Tran, D.Q.; Rhoads, J.M. Probiotics in Disease Prevention and Treatment. J. Clin. Pharm. 2018, 58 (Suppl. 10), S164–S179. [Google Scholar] [CrossRef] [PubMed]
- Bharucha, A.E.; Pemberton, J.H.; Locke, G.R. American Gastroenterological Association technical review on constipation. Gastroenterology 2013, 144, 218–238. [Google Scholar] [CrossRef] [Green Version]
- Noergaard, M.; Traerup Andersen, J.; Jimenez-Solem, E.; Bring Christensen, M. Long term treatment with stimulant laxatives–clinical evidence for effectiveness and safety? Scand. J. Gastroenterol. 2019, 54, 27–34. [Google Scholar] [CrossRef]
- Ohkusa, T.; Koido, S.; Nishikawa, Y.; Sato, N. Gut microbiota and chronic constipation: A review and update. Front. Med. 2019, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Liu, W.; Alkhouri, R.; Baker, R.D.; Bard, J.E.; Quigley, E.M.; Baker, S.S. Structural changes in the gut microbiome of constipated patients. Physiol. Genomics 2014, 46, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Parthasarathy, G.; Chen, J.; Chen, X.; Chia, N.; Oconnor, H.M.; Wolf, P.G.; Gaskins, H.R.; Bharucha, A.E. Relationship Between Microbiota of the Colonic Mucosa vs Feces and Symptoms, Colonic Transit, and Methane Production in Female Patients With Chronic Constipation. Gastroenterology 2016, 150, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.M. A role for the gut microbiota in IBS. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 497–505. [Google Scholar] [CrossRef]
- Blatchford, P.; Stoklosinski, H.; Eady, S.L.; Wallace, A.J.; Butts, C.A.; Gearry, R.B.; Gibson, G.R.; Ansell, J. Consumption of kiwifruit capsules increases Faecalibacterium prausnitzii abundance in functionally constipated individuals: A randomised controlled human trial. J. Nutr. Sci. 2017, 6, e52. [Google Scholar] [CrossRef] [Green Version]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef] [Green Version]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-Generation Machine Learning for Biological Networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meij, T.G.J.; De Groot, E.F.; Eck, A.; Budding, A.E.; Kneepkens, C.M.F.; Benninga, M.A.; Van Bodegraven, A.A.; Savelkoul, P.H.M. Characterization of Microbiota in Children with Chronic Functional Constipation. PLoS ONE 2016, 11, e0164731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American Gut: An Open Platform for Citizen Science Microbiome Research. mSystems 2018, 3, e00031-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeber-Lubecka, N.; Kulecka, M.; Ambrozkiewicz, F.; Paziewska, A.; Goryca, K.; Karczmarski, J.; Rubel, T.; Wojtowicz, W.; Mlynarz, P.; Marczak, L. Limited prolonged effects of rifaximin treatment on irritable bowel syndrome-related differences in the fecal microbiome and metabolome. Gut Microbes 2016, 7, 397–413. [Google Scholar] [CrossRef] [Green Version]
- Ou, Y.; Chen, S.; Ren, F.; Zhang, M.; Ge, S.; Guo, H.; Zhang, H.; Zhao, L. Lactobacillus casei strain Shirota alleviates constipation in adults by increasing the pipecolinic acid level in the gut. Front. Microbiol. 2019, 10, 324. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Ou, Y.; Zhao, L.; Li, Y.; Qiao, Z.; Hao, Y.; Ren, F. Differential effects of Lactobacillus casei strain Shirota on patients with constipation regarding stool consistency in China. J. Neurogastroenterol. Motil. 2019, 25, 148. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Zhu, Q.; Qu, X.; Qin, H. Microbial treatment in chronic constipation. Sci. China Life Sci. 2018, 61, 744–752. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, L.; Zhai, Q.; Zhao, J.; Zhang, H.; Lee, Y.-K.; Lu, W.; Li, M.; Chen, W. Chinese gut microbiota and its associations with staple food type, ethnicity, and urbanization. Npj Biofilms Microbiomes 2021, 7, 71. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The vegan package. Community Ecol. Package 2007, 10, 719. [Google Scholar]
- Revelle, W.R. Psych: Procedures for Personality and Psychological Research. 2017. Available online: https://www.scholars.northwestern.edu/en/publications/psych-procedures-for-personality-and-psychological-research (accessed on 18 September 2021).
- Rivera-Pinto, J.; Egozcue, J.J.; Pawlowsky-Glahn, V.; Paredes, R.; Noguera-Julian, M.; Calle, M.L. Balances: A new perspective for microbiome analysis. MSystems 2018, 3, e00053-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swami, A.; Jain, R. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2012, 12, 2825–2830. [Google Scholar]
- He, Y.; Wu, W.; Zheng, H.M.; Li, P.; McDonald, D.; Sheng, H.F.; Chen, M.X.; Chen, Z.H.; Ji, G.Y.; Zheng, Z.D.X.; et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 2018, 24, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Wassan, J.T.; Wang, H.; Browne, F.; Zheng, H. A Comprehensive Study on Predicting Functional Role of Metagenomes Using Machine Learning Methods. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 16, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Varshney, P.K.; Arora, M.K. Logistic regression for feature selection and soft classification of remote sensing data. IEEE Geosci. Remote Sens. Lett. 2006, 3, 491–494. [Google Scholar] [CrossRef]
- Liu, Y. A comparative study on feature selection methods for drug discovery. J. Chem. Inf. Comput. Sci. 2004, 44, 1823–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Bracken, M.B. Tree-based, two-stage risk factor analysis for spontaneous abortion. Am. J. Epidemiol. 1996, 144, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Hager, C.L.; Isham, N.; Schrom, K.P.; Chandra, J.; McCormick, T.; Miyagi, M.; Ghannoum, M.A. Effects of a novel probiotic combination on pathogenic bacterial-fungal polymicrobial biofilms. MBio 2019, 10, e00338-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Yang, M.; Jin, Y.; Li, Y.; Qian, W.; Xiong, H.; Song, J.; Hou, X. Involvement of shared mucosal-associated microbiota in the duodenum and rectum in diarrhea-predominant irritable bowel syndrome. J. Gastroenterol. Hepatol. 2018, 33, 1220–1226. [Google Scholar] [CrossRef]
- Ohara, T. Identification of the microbial diversity after fecal microbiota transplantation therapy for chronic intractable constipation using 16s rRNA amplicon sequencing. PLoS ONE 2019, 14, e0214085. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, C.; Cui, S.; Lee, Y.-K.; Wang, G.; Jianxin, Z.; Zhang, H.; Chen, W. Adhesive Bifidobacterium induced changes in cecal microbiome alleviated constipation in mice. Front. Microbiol. 2019, 10, 1721. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Lin, L.; Lin, Y.; Zhong, Y.; Zhang, S.; Liu, W.; Zou, B.; Liao, Q.; Xie, Z. Zengye decoction induces alterations to metabolically active gut microbiota in aged constipated rats. Biomed. Pharmacother. 2019, 109, 1361–1371. [Google Scholar] [CrossRef]
- Jin, M.; Li, D.; Ji, R.; Liu, W.; Xu, X.; Li, Y. Changes in intestinal microflora in digestive tract diseases during pregnancy. Arch. Gynecol. Obstet. 2020, 301, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Maharshak, N.; Ringel, Y.; Katibian, D.; Lundqvist, A.; Sartor, R.B.; Carroll, I.M.; Ringel-Kulka, T. Fecal and mucosa-associated intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Dig. Dis. Sci. 2018, 63, 1890–1899. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| before | after | |||||
|---|---|---|---|---|---|---|
| Train AUC | Test AUC | Validation AUC | Train AUC | Test AUC | Validation AUC | |
| F-Lasso | 86.8% | 84.5% | 49.9% | 86.9% | 84.8% | 50.6% |
| T-SVM | 88.1% | 83.5% | 52.1% | 88.4% | 84.5% | 54.3% |
| RF-RF | 89.4% | 89.7% | 52.6% | 90.3% | 90.6% | 49.4% |
| RF-GBRT | 89.5% | 89.9% | 49.9% | 90.8% | 91.1% | 55.5% |
| Chi2-GBRT | 86.5% | 86.8% | 62.7% | 87.3% | 87.5% | 70.7% |
| Log-GBRT | 85.2% | 85.4% | 65.1% | 85.9% | 86.2% | 70.8% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Wu, T.; Lu, W.; Yuan, W.; Pan, M.; Lee, Y.-K.; Zhao, J.; Zhang, H.; Chen, W.; Zhu, J.; et al. Predicting the Role of the Human Gut Microbiome in Constipation Using Machine-Learning Methods: A Meta-Analysis. Microorganisms 2021, 9, 2149. https://doi.org/10.3390/microorganisms9102149
Chen Y, Wu T, Lu W, Yuan W, Pan M, Lee Y-K, Zhao J, Zhang H, Chen W, Zhu J, et al. Predicting the Role of the Human Gut Microbiome in Constipation Using Machine-Learning Methods: A Meta-Analysis. Microorganisms. 2021; 9(10):2149. https://doi.org/10.3390/microorganisms9102149
Chicago/Turabian StyleChen, Yutao, Tong Wu, Wenwei Lu, Weiwei Yuan, Mingluo Pan, Yuan-Kun Lee, Jianxin Zhao, Hao Zhang, Wei Chen, Jinlin Zhu, and et al. 2021. "Predicting the Role of the Human Gut Microbiome in Constipation Using Machine-Learning Methods: A Meta-Analysis" Microorganisms 9, no. 10: 2149. https://doi.org/10.3390/microorganisms9102149
APA StyleChen, Y., Wu, T., Lu, W., Yuan, W., Pan, M., Lee, Y. -K., Zhao, J., Zhang, H., Chen, W., Zhu, J., & Wang, H. (2021). Predicting the Role of the Human Gut Microbiome in Constipation Using Machine-Learning Methods: A Meta-Analysis. Microorganisms, 9(10), 2149. https://doi.org/10.3390/microorganisms9102149