
The Shiga Toxin Receptor Globotriaosylceramide as Therapeutic Target in Shiga Toxin E. coli Mediated HUS

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Immunofluorescence Staining and Imaging of Bound Shiga Toxin Subunit B and CD-31
2.4. Analysis of Shiga Toxin Subunit B Binding by Flow Cytometry
2.5. Protein Synthesis by Radiolabeled 3H-Leucine Incorporation Assay
2.6. Cellular Gb3 and Ceramide Levels by Liquid Chromatography-Mass Spectrometry
2.7. Statistical Analysis
3. Results
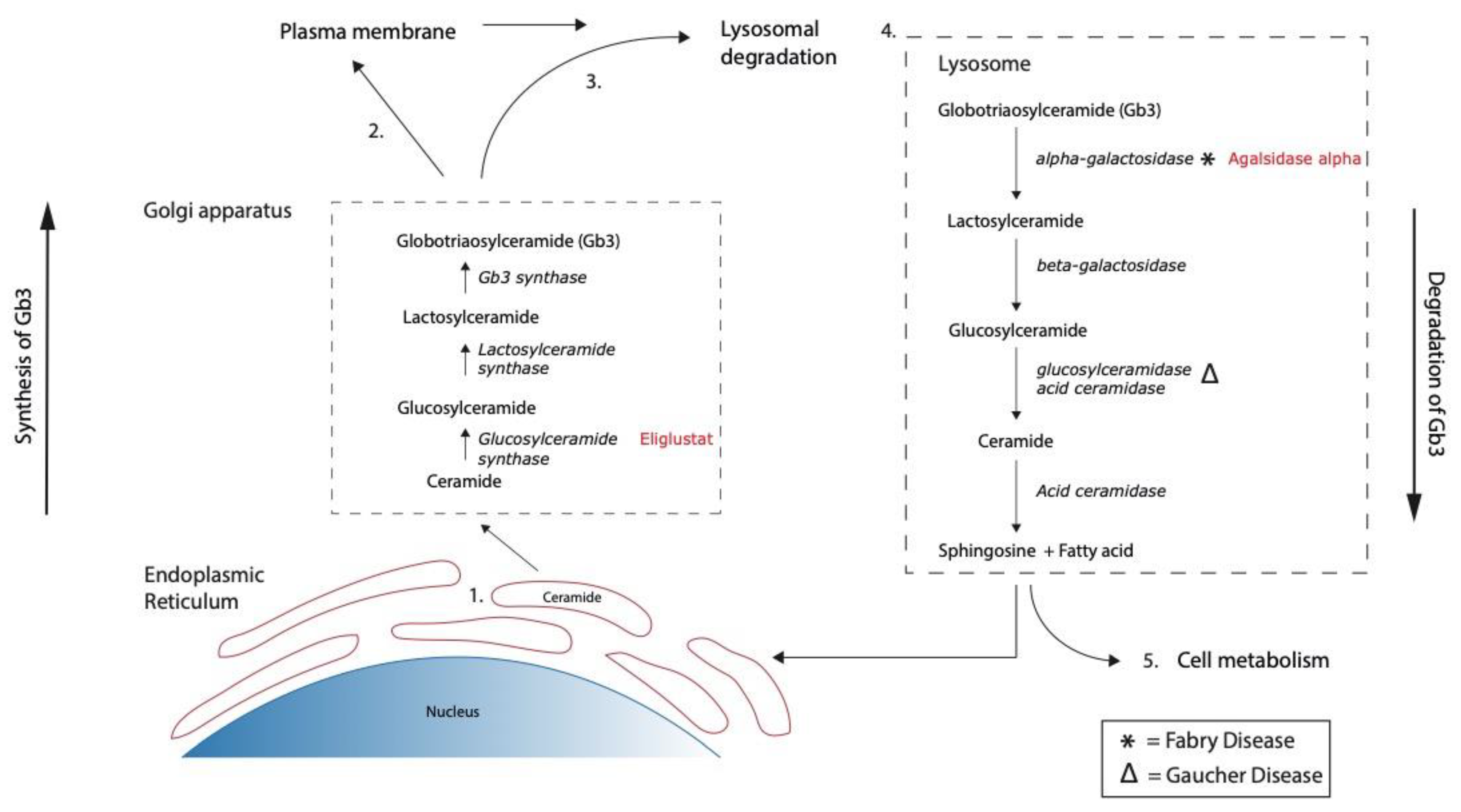
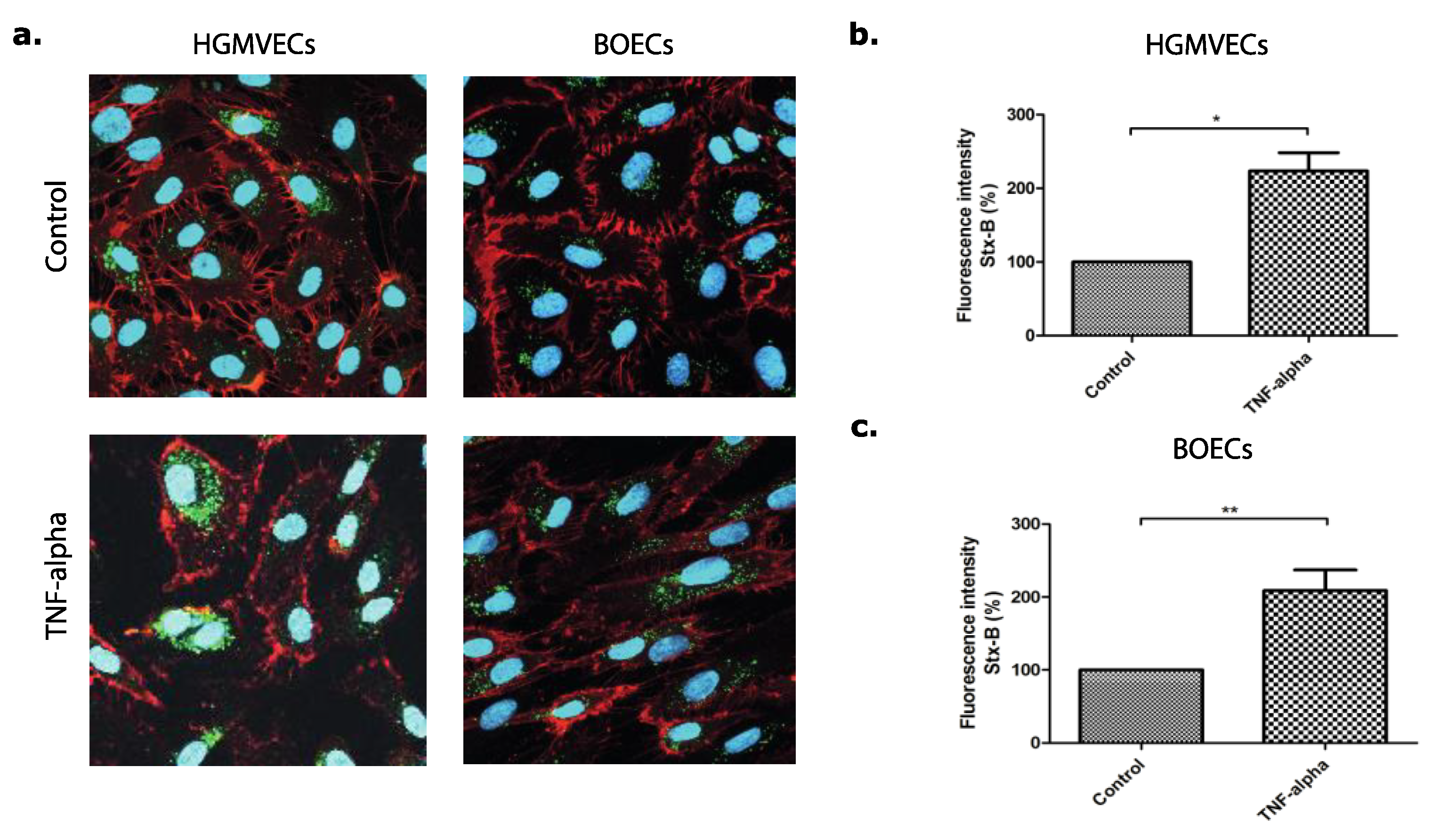
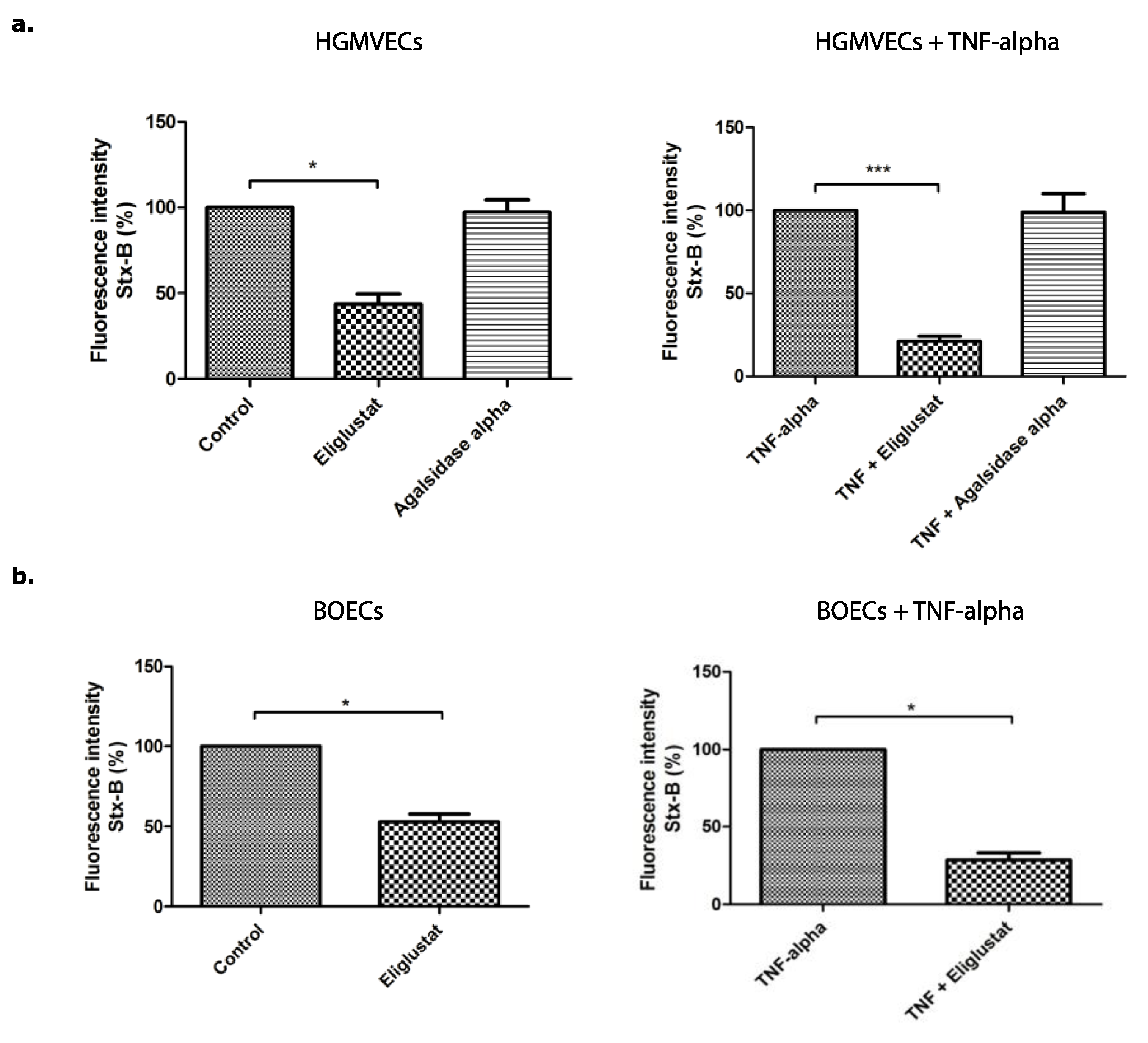
3.1. The Effect of Eliglustat and Agalsidase Alpha on Shiga Toxin Binding to the Endothelial Cell Surface
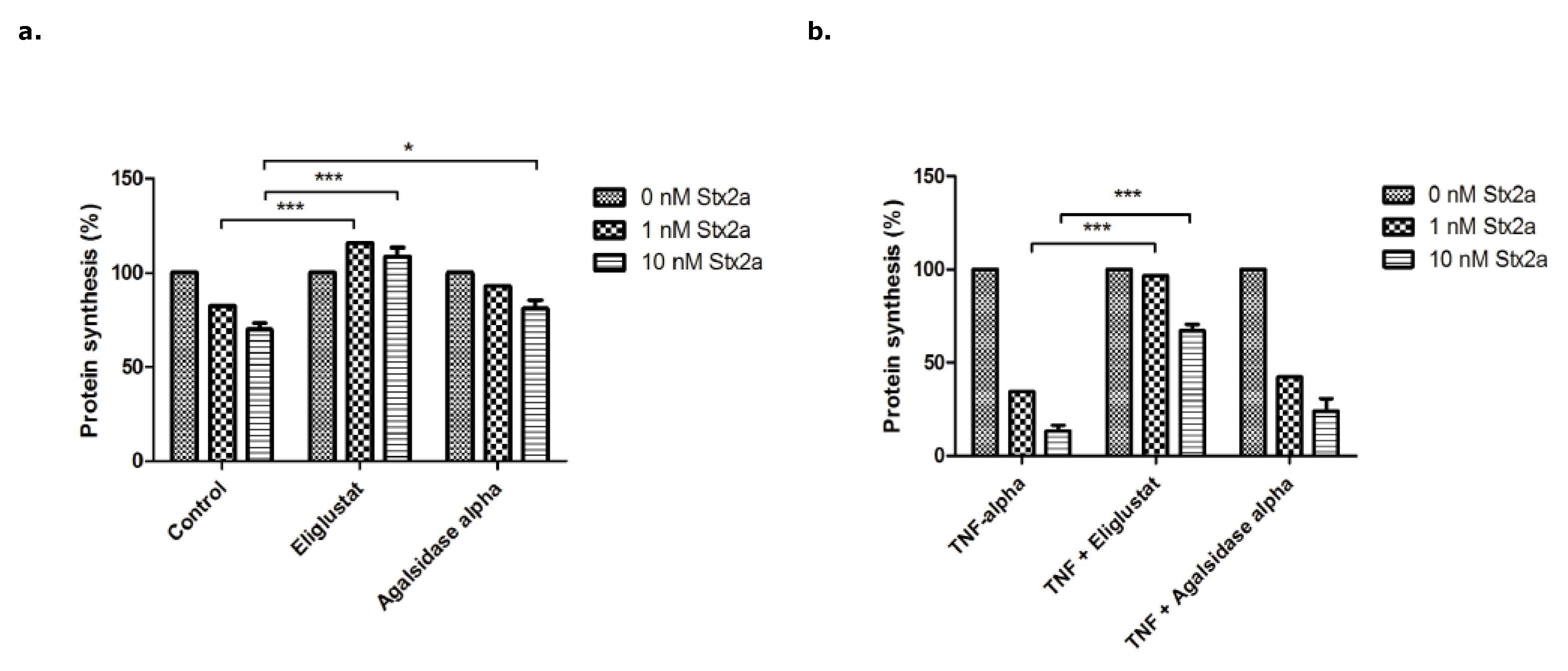
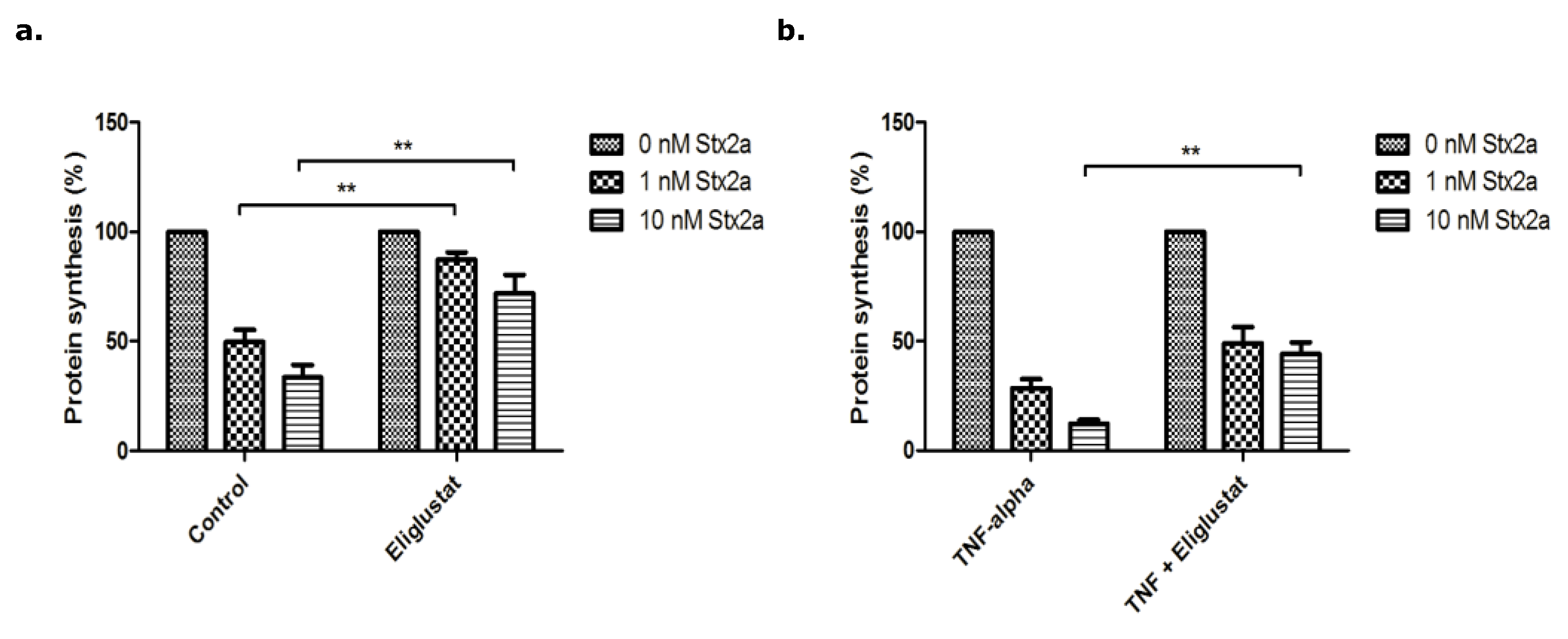
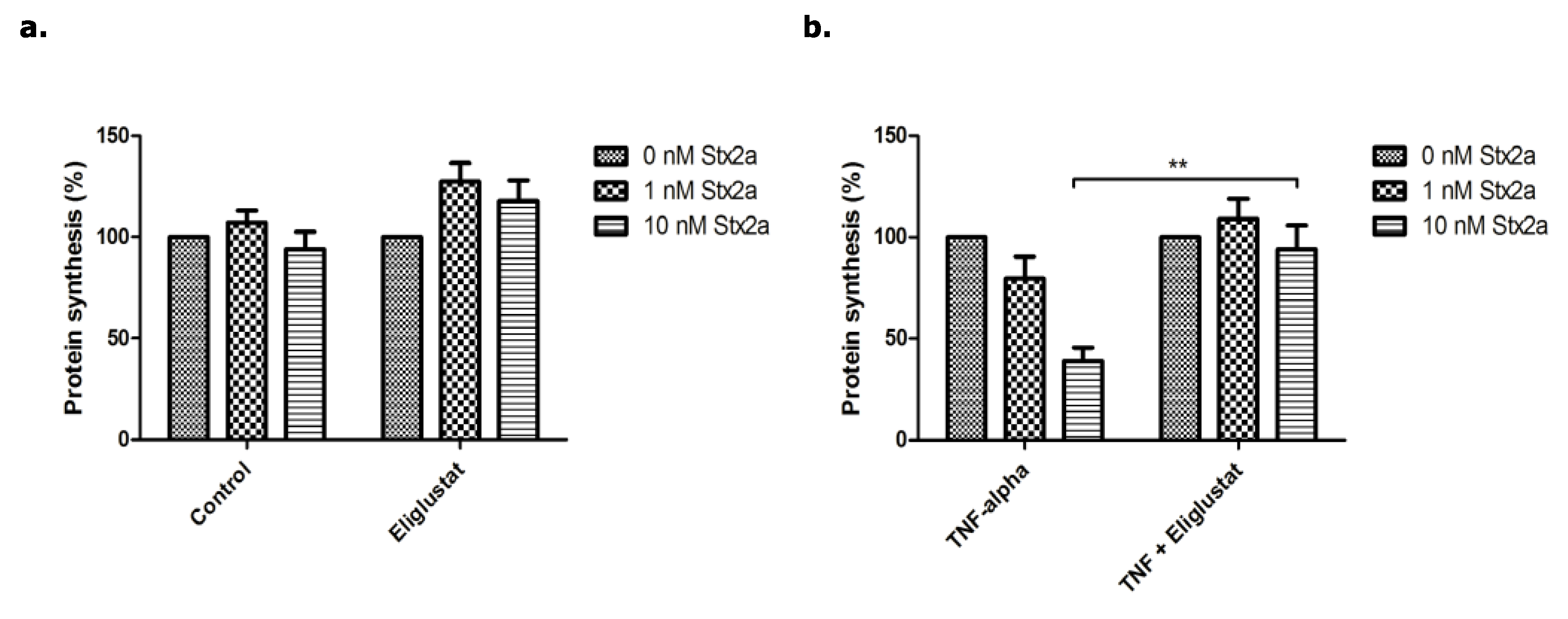
3.2. The Effect of Eliglustat and Agalsidase Alpha on Protein Synthesis Inhibition by Stx2a in Endothelial Cells
3.3. The Effect of Eliglustat and Agalsidase Alpha on Cellular Gb3 and Ceramide Levels
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- Noris, M.; Mescia, F.; Remuzzi, G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat. Rev. Nephrol. 2012, 8, 622–633. [Google Scholar] [CrossRef]
- Spinale, J.M.; Ruebner, R.L.; Copelovitch, L.; Kaplan, B.S. Long-term outcomes of Shiga toxin hemolytic uremic syndrome. Pediatr. Nephrol. 2013, 28, 2097–2105. [Google Scholar] [CrossRef]
- Garg, A.X.; Suri, R.S.; Barrowman, N.; Rehman, F.; Matsell, D.; Rosas-Arellano, M.P.; Salvadori, M.; Haynes, R.B.; Clark, W.F. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: A systematic review, meta-analysis, and meta-regression. JAMA 2003, 290, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Rosales, A.; Hofer, J.; Zimmerhackl, L.B.; Jungraithmayr, T.C.; Riedl, M.; Giner, T.; Strasak, A.; Orth-Höller, D.; Würzner, R.; Karch, H.; et al. Need for long-term follow-up in enterohemorrhagic Escherichia coli-associated hemolytic uremic syndrome due to late-emerging sequelae. Clin. Infect. Dis. 2012, 54, 1413–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ståhl, A.L.; Arvidsson, I.; Johansson, K.E.; Chromek, M.; Rebetz, J.; Loos, S.; Kristoffersson, A.C.; Békássy, Z.D.; Mörgelin, M.; Karpman, D. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. 2015, 11, e1004619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villysson, A.; Tontanahal, A.; Karpman, D. Microvesicle Involvement in Shiga Toxin-Associated Infection. Toxins 2017, 9, 376. [Google Scholar] [CrossRef] [Green Version]
- Johansson, K.; Willysson, A.; Kristoffersson, A.C.; Tontanahal, A.; Gillet, D.; Ståhl, A.L.; Karpman, D. Shiga Toxin-Bearing Microvesicles Exert a Cytotoxic Effect on Recipient Cells Only When the Cells Express the Toxin Receptor. Front. Cell. Infect. Microbiol. 2020, 10, 212. [Google Scholar] [CrossRef]
- Brigotti, M.; He, X.; Carnicelli, D.; Arfilli, V.; Porcellini, E.; Galassi, E.; Tazzari, P.L.; Ricci, F.; Patfield, S.A.; Testa, S.; et al. Particulate Shiga Toxin 2 in Blood is Associated to the Development of Hemolytic Uremic Syndrome in Children. Thromb. Haemost. 2020, 120, 107–120. [Google Scholar] [CrossRef]
- Waddell, T.; Cohen, A.; Lingwood, C.A. Induction of verotoxin sensitivity in receptor-deficient cell lines using the receptor glycolipid globotriosylceramide. Proc. Natl. Acad. Sci. USA 1990, 87, 7898–7901. [Google Scholar] [CrossRef] [Green Version]
- Ergonul, Z.; Hughes, A.K.; Kohan, D.E. Induction of apoptosis of human brain microvascular endothelial cells by shiga toxin 1. J. Infect. Dis. 2003, 187, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Betz, J.; Dorn, I.; Kouzel, I.U.; Bauwens, A.; Meisen, I.; Kemper, B.; Bielaszewska, M.; Mormann, M.; Weymann, L.; Sibrowski, W.; et al. Shiga toxin of enterohaemorrhagic Escherichia coli directly injures developing human erythrocytes. Cell. Microbiol. 2016, 18, 1339–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legros, N.; Pohlentz, G.; Steil, D.; Müthing, J. Shiga toxin-glycosphingolipid interaction: Status quo of research with focus on primary human brain and kidney endothelial cells. Int. J. Med. Microbiol. 2018, 308, 1073–1084. [Google Scholar] [CrossRef]
- Dettmar, A.K.; Binder, E.; Greiner, F.R.; Liebau, M.C.; Kurschat, C.E.; Jungraithmayr, T.C.; Saleem, M.A.; Schmitt, C.P.; Feifel, E.; Orth-Höller, D.; et al. Protection of human podocytes from shiga toxin 2-induced phosphorylation of mitogen-activated protein kinases and apoptosis by human serum amyloid P component. Infect. Immun. 2014, 82, 1872–1879. [Google Scholar] [CrossRef] [Green Version]
- van Setten, P.A.; van Hinsbergh, V.W.; van der Velden, T.J.; van de Kar, N.C.; Vermeer, M.; Mahan, J.D.; Assmann, K.J.; van den Heuvel, L.P.; Monnens, L.A. Effects of TNF alpha on verocytotoxin cytotoxicity in purified human glomerular microvascular endothelial cells. Kidney Int. 1997, 51, 1245–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locatelli, M.; Buelli, S.; Pezzotta, A.; Corna, D.; Perico, L.; Tomasoni, S.; Rottoli, D.; Rizzo, P.; Conti, D.; Thurman, J.M.; et al. Shiga toxin promotes podocyte injury in experimental hemolytic uremic syndrome via activation of the alternative pathway of complement. J. Am. Soc. Nephrol. 2014, 25, 1786–1798. [Google Scholar] [CrossRef] [Green Version]
- Lingwood, C. Verotoxin Receptor-Based Pathology and Therapies. Front. Cell. Infect. Microbiol. 2020, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, J.; Walsh, M.T.; Hammad, S.M.; Hussain, M.M. Sphingolipids and Lipoproteins in Health and Metabolic Disorders. Trends Endocrinol. Metab. 2017, 28, 506–518. [Google Scholar] [CrossRef]
- Sandhoff, K.; Kolter, T. Biosynthesis and degradation of mammalian glycosphingolipids. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 847–861. [Google Scholar] [CrossRef]
- Maruyama, H.; Miyata, K.; Mikame, M.; Taguchi, A.; Guili, C.; Shimura, M.; Murayama, K.; Inoue, T.; Yamamoto, S.; Sugimura, K.; et al. Effectiveness of plasma lyso-Gb3 as a biomarker for selecting high-risk patients with Fabry disease from multispecialty clinics for genetic analysis. Genet. Med. 2019, 21, 44–52. [Google Scholar] [CrossRef]
- Hennermann, J.B.; Arash-Kaps, L.; Fekete, G.; Schaaf, A.; Busch, A.; Frischmuth, T. Pharmacokinetics, pharmacodynamics, and safety of moss-aGalactosidase A in patients with Fabry disease. J. Inherit. Metab. Dis. 2019, 42, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Bennett, L.L.; Turcotte, K. Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease. Drug Des. Devel. Ther. 2015, 9, 4639–4647. [Google Scholar] [CrossRef] [Green Version]
- Mistry, P.K.; Lukina, E.; Ben Turkia, H.; Shankar, S.P.; Baris, H.; Ghosn, M.; Mehta, A.; Packman, S.; Pastores, G.; Petakov, M.; et al. Outcomes after 18 months of eliglustat therapy in treatment-naïve adults with Gaucher disease type 1: The phase 3 ENGAGE trial. Am. J. Hematol. 2017, 92, 1170–1176. [Google Scholar] [CrossRef]
- Shayman, J.A. Eliglustat tartrate, a prototypic glucosylceramide synthase inhibitor. Expert. Rev. Endocrinol. Metab. 2013, 8, 491–504. [Google Scholar] [CrossRef]
- Garimano, N.; Amaral, M.M.; Ibarra, C. Endocytosis, Cytotoxicity, and Translocation of Shiga Toxin-2 Are Stimulated by Infection of Human Intestinal (HCT-8) Monolayers With an Hypervirulent. Front. Cell. Infect. Microbiol. 2019, 9, 396. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, D.S.; Fischer Sigel, L.K.; Balestracci, A.; Ibarra, C.; Amaral, M.M.; Silberstein, C. Eliglustat prevents Shiga toxin 2 cytotoxic effects in human renal tubular epithelial cells. Pediatr. Res. 2021. [Google Scholar] [CrossRef]
- Martin-Ramirez, J.; Hofman, M.; van den Biggelaar, M.; Hebbel, R.P.; Voorberg, J. Establishment of outgrowth endothelial cells from peripheral blood. Nat. Protoc. 2012, 7, 1709–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feitz, W.J.C.; van de Kar, N.C.A.J.; Cheong, I.; van der Velden, T.J.A.M.; Ortiz-Sandoval, C.G.; Orth-Höller, D.; van den Heuvel, L.P.J.W.; Licht, C. Primary Human Derived Blood Outgrowth Endothelial Cells: An Appropriate In Vitro Model to Study Shiga Toxin Mediated Damage of Endothelial Cells. Toxins 2020, 12, 483. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groener, J.E.; Poorthuis, B.J.; Kuiper, S.; Helmond, M.T.; Hollak, C.E.; Aerts, J.M. HPLC for simultaneous quantification of total ceramide, glucosylceramide, and ceramide trihexoside concentrations in plasma. Clin. Chem. 2007, 53, 742–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, H.; Mirzaian, M.; Dekker, N.; Joao Ferraz, M.; Lugtenburg, J.; Codée, J.D.; van der Marel, G.A.; Overkleeft, H.S.; Linthorst, G.E.; Groener, J.E.; et al. Quantification of globotriaosylsphingosine in plasma and urine of fabry patients by stable isotope ultraperformance liquid chromatography-tandem mass spectrometry. Clin. Chem. 2013, 59, 547–556. [Google Scholar] [CrossRef] [Green Version]
- van de Kar, N.C.; Monnens, L.A.; Karmali, M.A.; van Hinsbergh, V.W. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: Implications for the pathogenesis of the hemolytic uremic syndrome. Blood 1992, 80, 2755–2764. [Google Scholar] [CrossRef] [Green Version]
- Stricklett, P.K.; Hughes, A.K.; Ergonul, Z.; Kohan, D.E. Molecular basis for up-regulation by inflammatory cytokines of Shiga toxin 1 cytotoxicity and globotriaosylceramide expression. J. Infect. Dis. 2002, 186, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.S.; Busch, A.; Day, T.S.; Meng, X.L.; Yu, C.I.; Dabrowska-Schlepp, P.; Fode, B.; Niederkrüger, H.; Forni, S.; Chen, S.; et al. Mannose receptor-mediated delivery of moss-made α-galactosidase A efficiently corrects enzyme deficiency in Fabry mice. J. Inherit. Metab. Dis. 2016, 39, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silberstein, C.; Copeland, D.P.; Chiang, W.-L.; Repetto, H.A.; Ibarra, C. A Glucosylceramide Synthase Inhibitor Prevents the Cytotoxic Effects of Shiga Toxin-2 on Human Renal Tubular Epithelial Cells. J. Epithel. Biol. Pharmacol. 2008, 1, 71–75. [Google Scholar] [CrossRef]
- Silberstein, C.; Lucero, M.S.; Zotta, E.; Copeland, D.P.; Lingyun, L.; Repetto, H.A.; Ibarra, C. A glucosylceramide synthase inhibitor protects rats against the cytotoxic effects of shiga toxin 2. Pediatr. Res. 2011, 69, 390–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, A.; Arend, L.J.; Lee, L.; Lingwood, C.; Brady, R.O.; Shayman, J.A. Glycosphingolipid depletion in fabry disease lymphoblasts with potent inhibitors of glucosylceramide synthase. Kidney Int. 2000, 57, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.C.; Sacerdoti, F.; Rivera, F.P.; Repetto, H.A.; Ibarra, C.; Amaral, M.M. Prevention of renal damage caused by Shiga toxin type 2: Action of Miglustat on human endothelial and epithelial cells. Toxicon 2015, 105, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Burrow, T.A.; Charrow, J.; Goker-Alpan, O.; Kaplan, P.; Kishnani, P.S.; Mistry, P.; Ruskin, J.; Weinreb, N. Recommendations for the use of eliglustat in the treatment of adults with Gaucher disease type 1 in the United States. Mol. Genet. Metab. 2016, 117, 95–103. [Google Scholar] [CrossRef]
- van der Veen, S.J.; Hollak, C.E.M.; van Kuilenburg, A.B.P.; Langeveld, M. Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 2020, 43, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Karpman, D.; Andreasson, A.; Thysell, H.; Kaplan, B.S.; Svanborg, C. Cytokines in childhood hemolytic uremic syndrome and thrombotic thrombocytopenic purpura. Pediatr. Nephrol. 1995, 9, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, J.; Gomez, T.; Doyle, M.P.; Wells, J.G.; Zhao, T.; Tauxe, R.V.; Griffin, P.M. Lessons from a large outbreak of Escherichia coli O157:H7 infections: Insights into the infectious dose and method of widespread contamination of hamburger patties. Epidemiol. Infect. 1999, 122, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.R.; Sperandio, V. Shiga toxin in enterohemorrhagic E.coli: Regulation and novel anti-virulence strategies. Front. Cell. Infect. Microbiol. 2012, 2, 81. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HGMVECs | ||||
| Gb3 (%) | LacCer (%) | GlcCer (%) | Cer (%) | |
| Control | 100 | 100 | 100 | 100 |
| TNF-alpha | 172 | 89 | 128 | 116 |
| Eliglustat | 74 | 59 | 49 | 88 |
| Agalsidase alpha | 64 | 68 | 75 | 93 |
| TNF + Eliglustat | 61 | 56 | 51 | 110 |
| BOECs | ||||
| Gb3 (%) | LacCer (%) | GlcCer (%) | Cer (%) | |
| Control | 100 | 100 | 100 | 100 |
| TNF-alpha | 177 | 93 | 184 | 80 |
| Eliglustat | n.a | n.a | n.a. | n.a. |
| Agalsidase alpha | n.a | n.a. | n.a. | n.a. |
| TNF + Eliglustat | 79 | 65 | 84 | 91 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feitz, W.J.C.; Bouwmeester, R.; van der Velden, T.J.A.M.; Goorden, S.; Licht, C.; van den Heuvel, L.P.J.W.; van de Kar, N.C.A.J. The Shiga Toxin Receptor Globotriaosylceramide as Therapeutic Target in Shiga Toxin E. coli Mediated HUS. Microorganisms 2021, 9, 2157. https://doi.org/10.3390/microorganisms9102157
Feitz WJC, Bouwmeester R, van der Velden TJAM, Goorden S, Licht C, van den Heuvel LPJW, van de Kar NCAJ. The Shiga Toxin Receptor Globotriaosylceramide as Therapeutic Target in Shiga Toxin E. coli Mediated HUS. Microorganisms. 2021; 9(10):2157. https://doi.org/10.3390/microorganisms9102157
Chicago/Turabian StyleFeitz, Wouter J. C., Romy Bouwmeester, Thea J. A. M. van der Velden, Susan Goorden, Christoph Licht, Lambert P. J. W. van den Heuvel, and Nicole C. A. J. van de Kar. 2021. "The Shiga Toxin Receptor Globotriaosylceramide as Therapeutic Target in Shiga Toxin E. coli Mediated HUS" Microorganisms 9, no. 10: 2157. https://doi.org/10.3390/microorganisms9102157
APA StyleFeitz, W. J. C., Bouwmeester, R., van der Velden, T. J. A. M., Goorden, S., Licht, C., van den Heuvel, L. P. J. W., & van de Kar, N. C. A. J. (2021). The Shiga Toxin Receptor Globotriaosylceramide as Therapeutic Target in Shiga Toxin E. coli Mediated HUS. Microorganisms, 9(10), 2157. https://doi.org/10.3390/microorganisms9102157