
Prokaryotic Diversity of the Composting Thermophilic Phase: The Case of Ground Coffee Compost

 , ,
, ,  ,
, 

Abstract
:1. Introduction
2. Materials and Methods
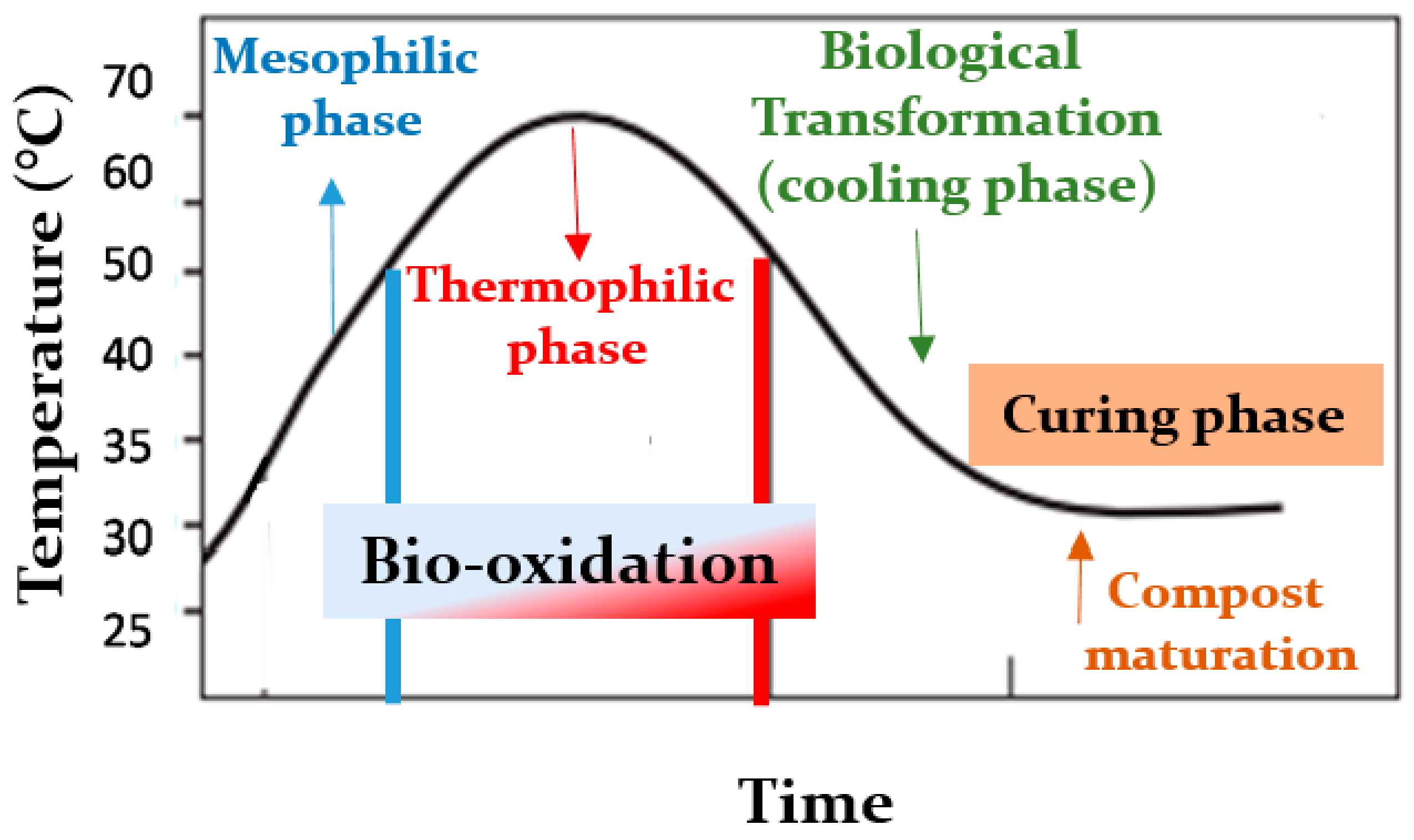
2.1. Sampling of the Compost
2.2. NMR Spectroscopy of the Compost
2.3. Isolation and Characterization of the Thermophilic Strains: A Culture-Dependent Approach
2.3.1. Isolation of the Thermophilic Strains
2.3.2. Determination Optimal Growth Conditions and Phenotypic Characterization
2.3.3. Molecular Characterization and Phylogenetic Analysis
2.4. Metagenomic Analysis: DNA Extraction from the Compost Samples
2.4.1. Amplification of 16S rRNA Genes and Illumina Sequencing
2.4.2. Post-Run Analysis
2.4.3. Analysis of the Prokaryotic Community
2.4.4. Predictive Functional Profiling
3. Results
3.1. NMR Spectroscopy of the Compost
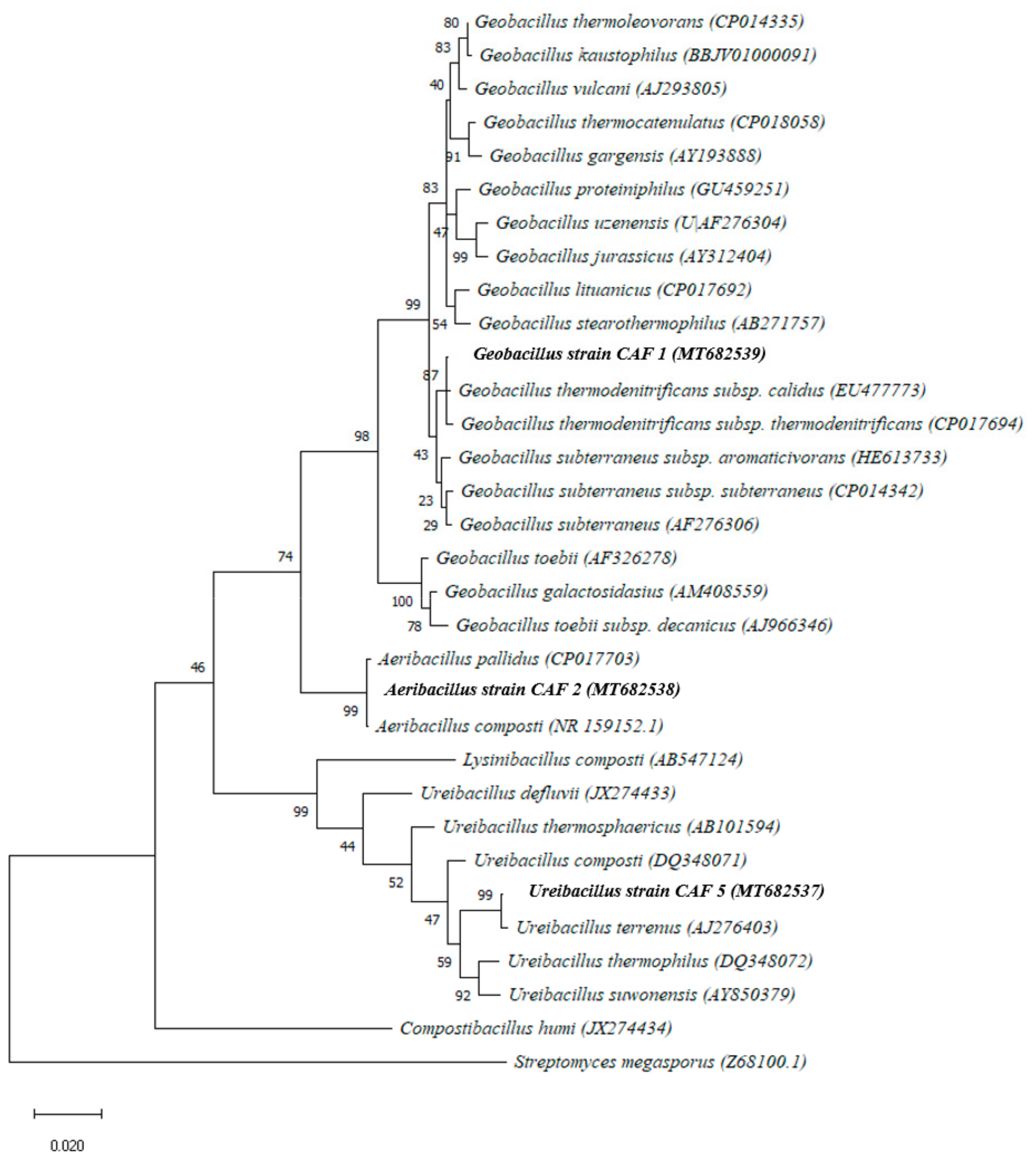
3.2. Isolation and the Phenotypic and Phylogenetic Characterization of Isolates Using a Culture-Dependent Approach
3.3. Metagenomic Analysis: 16S rRNA Gene Amplicon Sequencing
3.3.1. Bacteria
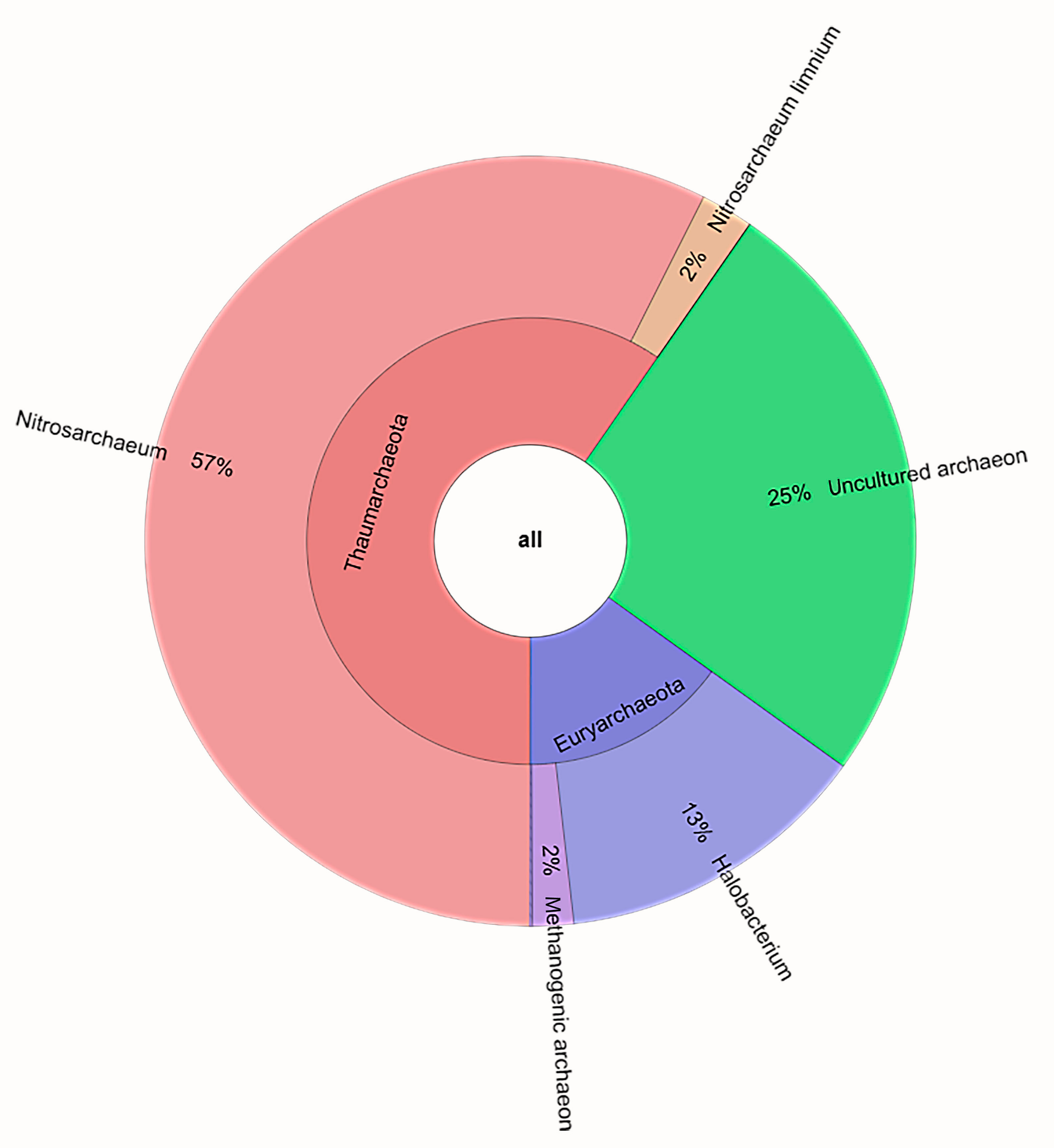
3.3.2. Archaea
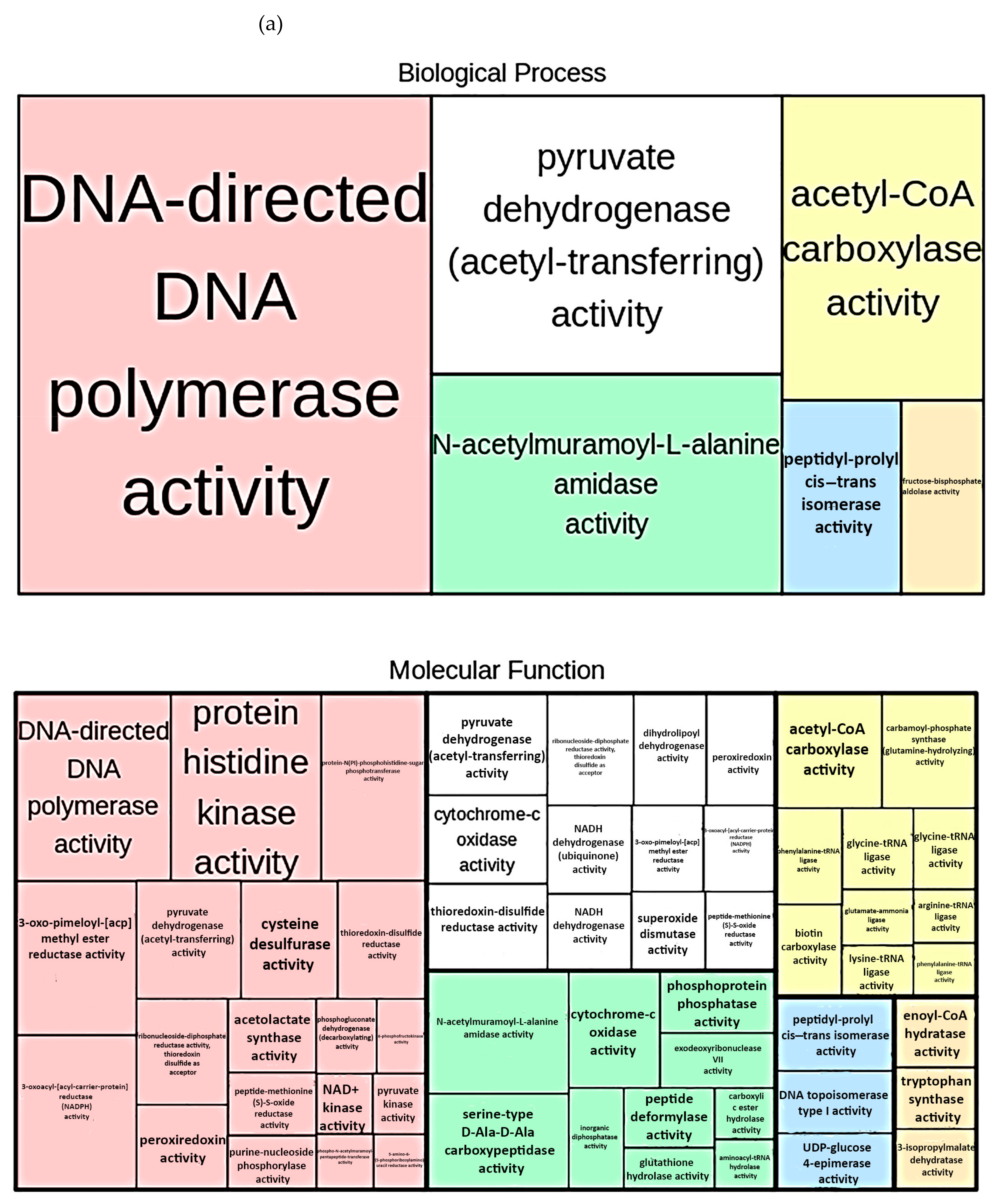
3.4. Functional Profiling
4. Discussion
4.1. Isolation and the Phenotypic and Phylogenetic Characterization of Isolates Using a Culture-Dependent Approach
4.2. Metagenomic Analysis: 16S rRNA Gene Amplicon Sequencing
4.3. Functional Profiling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amore, A.; Pepe, O.; Ventorino, V.; Birolo, L.; Giangrande, C.; Faraco, V. Industrial waste based compost as a source of novel cellulolytic strains and enzymes. FEMS Microbiol. Lett. 2013, 339, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; de Haro Marti, M.; Moore, A.; Falen, C. The Composting Process: Dairy Compost Production and Use in Idaho; CIS 1179; University of Idaho: Moscow, ID, USA, 2011. [Google Scholar]
- Yanan, Y.; Jie, G.; Xiaojuan, W.; Yajun, Z.; Wei, Z.; Rong, C.; Xiaochang, W. Effects of rhamnolipid and Tween-80 on cellulase activities and metabolic functions of the bacterial community during chicken manure composting. Bioresour. Technol. 2019, 288, 121507. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chu, C.; Li, X.; Wang, W.; Ren, N. Succession of bacterial community function in cow manure composing. Bioresour. Technol. 2018, 267, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Mao, H.; Wang, Z.; Tian, Y. Succession of organics metabolic function of bacterial community in swine manure composting. J. Hazard. Mater. 2018, 360, 471–480. [Google Scholar] [CrossRef]
- Moreno, J.; López, M.J.; Vargas-García, M.C.; Suárez-Estrella, F. Recent advances in microbial aspects of compost production and use. Acta Horticult. 2013, 1013, 443–457. [Google Scholar] [CrossRef]
- López-González, J.A.; Suárez-Estrella, F.; Vargas-García, M.C.; López, M.J.; Jurado, M.M.; Moreno, J. Dynamics of bacterial microbiota during lignocellusic waste composting: Studies upon its structure, functionality and biodiversity. Bioresour. Technol. 2015, 175, 406–416. [Google Scholar] [CrossRef]
- Jurado, M.M.; Camelo-Castillo, A.J.; Suárez-Estrella, F.; López, M.J.; López-González, J.A.; Estrella-González, M.J.; Siles-Castellano, A.B.; Moreno, J. Integral approach using bacterial microbiome to stabilize municipal solid waste. J. Environ. Manag. 2020, 265, 110528. [Google Scholar] [CrossRef]
- de Gannes, V.D.; Eudoxie, G.; Hickey, W.J. Prokaryotic successions and diversity in composts as revealed by 454-pyrosequencing. Bioresour. Technol. 2013, 133, 573–580. [Google Scholar] [CrossRef]
- Hultman, J.; Vasara, T.; Partanen, P.; Kurola, J.; Kontro, M.H.; Paulin, L.; Auvinen, P.; Romantschuk, M. Determination of fungal succession during municipal solid waste composting using a cloning-based analysis. J. Appl. Microbiol. 2010, 108, 472–487. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Pletschke, B.I. Thermophilic Bacilli and their Enzymes in Composting. In Composting for Sustainable Agriculture; Maheshwari, D.K., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 103–124. [Google Scholar] [CrossRef]
- Poli, A.; Romano, I.; Caliendo, G.; Nicolaus, G.; Orlando, P.; Falco, A.; Lama, L.; Gambacorta, A.; Nicolaus, B. Geobacillus toebii subsp. decanicus subsp. nov., a hydrocarbon-degrading, heavy metal resistant bacterium from hot compost. J. Gen. Appl. Microbiol. 2006, 52, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, A.; Laezza, G.; Gul-Guven, R.; Orlando, P.; Nicolaus, B. Geobacillus galactosidasius sp. nov., a new thermophilic galactosidase-producing bacterium isolated from compost. Syst. Appl. Microbiol. 2011, 34, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Finore, I.; Gioiello, A.; Leone, L.; Orlando, P.; Romano, I.; Nicolaus, B.; Poli, A. Aeribacillus composti sp. nov., a thermophilic bacillus isolated from olive mill pomace compost. Int. J. System. Evolut. Microbiol. 2017, 67, 4830–4835. [Google Scholar] [CrossRef]
- Cozzolino, V.; Di Meo, V.; Monda, H.; Spaccini, R.; Piccolo, A. The molecular characteristics of compost affect plant growth, arbuscular mycorrhizal fungi, and soil microbial community composition. Biol. Fertil. Soils 2016, 52, 15–29. [Google Scholar] [CrossRef]
- Romano, I.; Poli, A.; Lama, L.; Gambacorta, A.; Nicolaus, B. Geobacillus thermoleovorans subsp. stromboliensis subsp. nov., isolated from the geothermal volcanic environment. J. Gen. Appl. Microbiol. 2005, 51, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Finore, I.; Vigneron, A.; Vincent, W.F.; Leone, L.; Di Donato, P.; Schiano Moriello, A.; Nicolaus, B.; Poli, A. Novel psychrophiles and exopolymers from permafrost thaw lake sediments. Microorganisms 2020, 8, 1282. [Google Scholar] [CrossRef]
- Finore, I.; Poli, A.; Di Donato, P.; Lama, L.; Trincone, A.; Fagnano, M.; Mori, M.; Nicolaus, B.; Tramice, A. The hemicellulose extract from Cynara cardunculus: A source of value-added biomolecules produced by xylanolytic thermozymes. Green Chem. 2016, 18, 2460–2472. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G + C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Palinska, K.A.; Vogt, J.C.; Surosz, W. Biodiversity analysis of the unique geothermal microbial ecosystem of the Blue Lagoon (Iceland) using next-generation sequencing (NGS). Hydrobiologia 2018, 811, 93–102. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 28. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, 17. [Google Scholar] [CrossRef] [Green Version]
- Grigatti, M.; Giorgioni, M.E.; Ciavatta, C. Compost-based growing media: Influence on growth and nutrient use of bedding plants. Biores. Technol. 2007, 98, 3526–3534. [Google Scholar] [CrossRef]
- Varma, V.S.; Dhamodharan, K.; Kalamdhad, A.S. Characterization of bacterial community structure during in-vessel composting of agricultural waste by 16S rRNA sequencing. 3 Biotech. 2018, 8, 018–1319. [Google Scholar] [CrossRef]
- Margaryan, A.; Shahinyan, G.; Hovhannisyan, P.; Panosyan, H.; Birkeland, N.K.; Trchounian, A. Geobacillus and Anoxybacillus spp. from Terrestrial Geothermal Springs Worldwide: Diversity and Biotechnological Applications. In Extremophiles in Eurasian Ecosystems: Ecology, Diversity, and Applications; Microorganisms for Sustainability; Egamberdieva, D., Birkeland, N.K., Panosyan, H., Li, W.J., Eds.; Springer: Singapore, 2018; Volume 8. [Google Scholar] [CrossRef]
- Tuomela, M.; Vikman, M.; Hatakka, A.; Itävaara, M. Biodegradation of lignin in a compost environment: A review. Biores. Technol. 2000, 72, 169–183. [Google Scholar] [CrossRef]
- Di Piazza, S.; Houbraken, J.; Meijer, M.; Cecchi, G.; Kraak, B.; Rosa, E.; Zotti, M. Thermotolerant and thermophilic mycobiota in different steps of Compost Maturation. Microorganisms 2020, 8, 880. [Google Scholar] [CrossRef]
- Poli, A.; Finore, I.; Romano, I.; Gioiello, A.; Lama, L.; Nicolaus, B. Microbial Diversity in Extreme Marine Habitats and Their Biomolecules. Microorganisms 2017, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Antunes, L.P.; Martins, L.F.; Pereira, R.V.; Thomas, A.M.; Barbosa, D.; Lemos, L.N.; Silva, G.M.; Moura, L.M.; Epamino, G.W.; Digiampietri, L.A.; et al. Microbial community structure and dynamics in thermophilic composting viewed through metagenomics and metatranscriptomics. Sci. Rep. 2016, 6, 38915. [Google Scholar] [CrossRef]
- Kurola, J.M.; Arnold, M.; Kontro, M.H.; Talves, M.; Romantschuk, M. Wood ash for application in municipal biowaste composting. Bioresour. Technol. 2011, 102, 5214–5220. [Google Scholar] [CrossRef]
- Tran, Q.N.M.; Mimoto, H.; Koyama, M.; Nakasaki, K. Lactic acid bacteria modulate organic acid production during early stages of food waste composting. Sci. Total Environ. 2019, 687, 341–347. [Google Scholar] [CrossRef]
- Partanen, P.; Hultman, J.; Paulin, L.; Auvinen, P.; Romantschuk, M. Bacterial diversity at different stages of the composting process. BMC Microbiol. 2010, 10, 1471–2180. [Google Scholar] [CrossRef] [Green Version]
- Sundberg, C.; Yu, D.; Franke-Whittle, I.; Kauppi, S.; Smårs, S.; Insam, H.; Romantschuk, M.; Jönsson, H. Effects of pH and microbial composition on odour in food waste composting. Waste Manag. 2013, 33, 204–211. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Bonilla, G.A.; Lopes, C.M.; Durrer, A.; Alves, P.R.L.; Passaglia, N.; Cardoso, E. Effect of phosphate-solubilizing bacteria on phosphorus dynamics and the bacterial community during composting of sugarcane industry waste. Syst. Appl. Microbiol. 2017, 40, 308–313. [Google Scholar] [CrossRef]
- Neher, D.A.; Weicht, T.R.; Bates, S.T.; Leff, J.W.; Fierer, N. Changes in bacterial and fungal communities across compost recipes, preparation methods, and composting times. PLoS ONE 2013, 8, e79512. [Google Scholar] [CrossRef] [Green Version]
- Reis, V.M.; Teixeira, K.R. Nitrogen fixing bacteria in the family Acetobacteraceae and their role in agriculture. J. Basic Microbiol. 2015, 55, 931–949. [Google Scholar] [CrossRef]
- Yu, H.; Zeng, G.; Huang, H.; Xi, X.; Wang, R.; Huang, D.; Huang, G.; Li, J. Microbial community succession and lignocellulose degradation during agricultural waste composting. Biodegradation 2007, 18, 793–802. [Google Scholar] [CrossRef]
- Steger, K.; Jarvis, A.; Vasara, T.; Romantschuk, M.; Sundh, I. Effects of differing temperature management on development of Actinobacteria populations during composting. Res. Microbiol. 2007, 158, 617–624. [Google Scholar] [CrossRef]
- Schäfer, J.; Klug, K.; van Kampen, V.; Jäckel, U. Quantification of Saccharopolyspora rectivirgula in composting plants: Assessment of the relevance of S. rectivirgula. Ann. Occup. Hyg. 2013, 57, 875–883. [Google Scholar]
- Veillette, M.; Bonifait, L.; Mbareche, H.; Marchand, G.; Duchaine, C. Preferential aerosolization of Actinobacteria during handling of composting organic matter. J. Aer. Sci. 2018, 116, 83–91. [Google Scholar] [CrossRef]
- Thummes, K.; Schäfer, J.; Kämpfer, P.; Jäckel, U. Thermophilic methanogenic archaea in compost material: Occurrence, persistence and possible mechanisms for their distribution to other environments. Syst. Appl. Microbiol. 2007, 30, 634–643. [Google Scholar] [CrossRef]
- de Gannes, V.; Eudoxie, G.; Dyer, D.H.; William, J. Hickey. Diversity and abundance of ammonia oxidizing archaea in tropical compost systems. Front. Microbiol. 2012, 3, 244. [Google Scholar] [CrossRef] [Green Version]
- Brochier-Armanet, C.; Boussau, B.; Gribaldo, S.; Forterre, P. Mesophilic crenarchaeota: Proposal for a third archaeal phylum, the Thaumarchaeota. Nat. Rev. Microbiol. 2008, 6, 245–252. [Google Scholar] [CrossRef]
- Tolar, B.B.; Boye, K.; Bobb, C.; Maher, K.; Bargar, J.R.; Francis, C.A. Stability of floodplain subsurface microbial communities through seasonal hydrological and geochemical cycles. Front. Earth Sci. 2020, 8. [Google Scholar] [CrossRef]
- Jung, M.Y.; Islam, M.A.; Gwak, J.H.; Kim, J.G.; Rhee, S.K. Nitrosarchaeum koreense gen. nov., sp. nov., an aerobic and mesophilic, ammonia-oxidizing archaeon member of the phylum Thaumarchaeota isolated from agricultural soil. Int. J. Syst. Evol. Microbiol. 2018, 68, 3084–3095. [Google Scholar] [CrossRef]
- Cardarelli, E.L.; Bargar, J.R.; Francis, C.A. Diverse Thaumarchaeota dominate subsurface ammonia-oxidizing communities in semi-arid floodplains in the Western United States. Microb. Ecol. 2020, 80, 778–792. [Google Scholar] [CrossRef]
- Billings, S.A.; Ziegler, S. Altered patterns of soil carbon substrate usage and heterotrophic respiration in a pine forest with elevated CO2 and N fertilization. Glob. Change Biol. 2008, 14, 1025–1036. [Google Scholar] [CrossRef]
- Rashid, M.I.; Mujawar, L.H.; Shahzad, T.; Almeelbi, T.; Ismail, I.M.; Oves, M. Bacteria and fungi can contribute to nutrients bioavailability and aggregate formation in degraded soils. Microbiol. Res. 2016, 183, 26–41. [Google Scholar] [CrossRef]
- Vimal, S.R.; Singh, J.S.; Arora, N.K.; Singh, S. Soil-Plant-Microbe interactions in stressed agriculture management: A Review. Pedosphere 2017, 27, 177–192. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon Species (ppm Intervals) | Aliphatic C (0–45) | O-Substituted Alkyl C (45–60) | O-Alkyl C (60–110) | Aromatic C (110–145) | O-Aryl C (145–160) | Carboxyl C (160–190) | HB/HI a | A/OA b |
|---|---|---|---|---|---|---|---|---|
| Relative % | 33.5 | 15.2 | 23.3 | 14.8 | 3.2 | 10.0 | 2.7 | 0.7 |
| Reads | Bacteria OTUs | Archaea OTUs | |||||||
|---|---|---|---|---|---|---|---|---|---|
| DNA Replicate Name’s Samples | Total Number | Clipping- Trimming- Filtering | Good Quality (%) | Reads Missed (%) | Final Reads Number | Number | Shannon Index | Number | Shannon Index |
| caffA | 95,404 | 71,098 | 74.52 | 25.47 | 65,837 | 1439 | 7.12 | 99 | 1.996603 |
| caffB | 126,649 | 91,468 | 72.22 | 27.77 | 85,614 | 2183 | 7.46 | 164 | 1.969011 |
| caffE | 190,366 | 142,988 | 75.11 | 24.88 | 133,122 | 3265 | 7.95 | 253 | 2.184408 |
| Phylum/Class | Genus | Proportion of Total Reads Number (%) | ||||
|---|---|---|---|---|---|---|
| caffA | caffB | caffE | Average | St.Dev. | ||
| Actinobacteria | Frigoribacterium | 0.00 | 0.00 | 0.03 | 0.01 | ±0.02 |
| Sanguibacter | 0.07 | 0.00 | 0.00 | 0.02 | ±0.04 | |
| Pseudonocardia | 0.28 | 0.37 | 0.58 | 0.41 | ±0.16 | |
| Saccharomonospora | 0.14 | 0.27 | 0.74 | 0.38 | ±0.31 | |
| Saccharopolyspora | 0.35 | 0.64 | 4.38 | 1.79 | ±2.25 | |
| Streptomyces | 0.07 | 0.00 | 0.06 | 0.04 | ±0.04 | |
| Nocardiopsi | 0.00 | 0.05 | 0.03 | 0.03 | ±0.02 | |
| Thermomonospora | 0.00 | 0.05 | 0.00 | 0.02 | ±0.03 | |
| Bacteroidetes | Persicitalea | 0.00 | 0.05 | 0.03 | 0.03 | ±0.02 |
| Parapedobacter | 0.00 | 0.14 | 0.00 | 0.05 | ±0.08 | |
| Firmicutes | Aeribacillus | 0.00 | 0.05 | 0.09 | 0.05 | ±0.05 |
| Bacillus | 7.71 | 5.54 | 4.26 | 5.84 | ±1.75 | |
| Oceanobacillus | 0.21 | 0.78 | 0.06 | 0.35 | ±0.38 | |
| Ornithinibacillus | 0.28 | 0.14 | 0.34 | 0.25 | ±0.10 | |
| Terribacillus | 0.07 | 0.00 | 0.00 | 0.02 | ±0.04 | |
| Ureibacillus | 0.97 | 0.87 | 1.01 | 0.95 | ±0.07 | |
| Ammoniibacillus | 0.14 | 0.05 | 0.03 | 0.07 | ±0.06 | |
| Brevibacillu | 0.21 | 0.00 | 0.06 | 0.09 | ±0.11 | |
| Paenibacillus | 0.14 | 0.23 | 0.28 | 0.21 | ±0.07 | |
| Thermobacillus | 0.00 | 0.00 | 0.15 | 0.05 | ±0.09 | |
| Bhargavae | 0.00 | 0.14 | 0.00 | 0.05 | ±0.08 | |
| Lysinibacillus | 1.74 | 1.33 | 0.98 | 1.35 | ±0.38 | |
| Rummeliibacillus | 0.00 | 0.00 | 0.06 | 0.02 | ±0.04 | |
| Lactobacillus | 2.43 | 3.34 | 6.09 | 3.96 | ±1.91 | |
| Pediococcus | 0.07 | 0.05 | 0.28 | 0.13 | ±0.13 | |
| Leuconostoc | 0.00 | 0.00 | 0.03 | 0.01 | ±0.02 | |
| Weissella | 2.15 | 3.34 | 6.89 | 4.13 | ±2.46 | |
| Lactococcus | 0.00 | 0.00 | 0.03 | 0.01 | ±0.02 | |
| Tepidimicrobium | 0.00 | 0.00 | 0.15 | 0.05 | ±0.09 | |
| Alpha | Acetobacter | 17.16 | 21.48 | 8.33 | 15.66 | ±6.70 |
| Ameyamaea | 7.30 | 6.46 | 3.86 | 5.87 | ±1.79 | |
| Brevundimonas | 0.07 | 0.00 | 0.00 | 0.02 | ±0.04 | |
| Shinella | 0.00 | 0.00 | 0.03 | 0.01 | ±0.02 | |
| Paracoccus | 0.07 | 0.00 | 0.00 | 0.02 | ±0.04 | |
| Ameyamaea | 0.00 | 0.41 | 0.06 | 0.16 | ±0.22 | |
| Gamma | Bordetella | 0.83 | 0.14 | 0.06 | 0.34 | ±0.43 |
| Lautropia | 0.00 | 0.05 | 0.00 | 0.02 | ±0.03 | |
| Orrella | 0.00 | 0.05 | 0.00 | 0.02 | ±0.03 | |
| Kingella | 0.00 | 0.05 | 0.00 | 0.02 | ±0.03 | |
| Franconibacter | 0.00 | 0.00 | 0.03 | 0.01 | ±0.02 | |
| Alcanivorax | 0.07 | 0.00 | 0.00 | 0.02 | ±0.04 | |
| Pseudomonas | 1.95 | 0.87 | 0.40 | 1.07 | ±0.79 | |
| Phylum/Class | Proportion of Total Reads Number (%) | |||||
|---|---|---|---|---|---|---|
| Genus | caffA | caffB | caffE | Average | St.Dev. | |
| Euryarchaeota | Halobacterium | 12.89 | 13.24 | 13.46 | 13.20 | ±0.28 |
| Halogeometricum | 0.00 | 0.00 | 0.12 | 0.04 | ±0.06 | |
| Methanogenic archaeon | 1.27 | 0.92 | 2.76 | 1.65 | ±0.97 | |
| Mathanomicrobiales archaeon | 0.00 | 0.00 | 0.12 | 0.04 | ±0.06 | |
| Thaumarchaeota | Nitrosarchaeum | 65.53 | 66.93 | 43.50 | 58.65 | ±13.14 |
| Nitrosarchaeum limnium | 2.53 | 2.53 | 1.73 | 2.26 | ±0.46 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papale, M.; Romano, I.; Finore, I.; Lo Giudice, A.; Piccolo, A.; Cangemi, S.; Di Meo, V.; Nicolaus, B.; Poli, A. Prokaryotic Diversity of the Composting Thermophilic Phase: The Case of Ground Coffee Compost. Microorganisms 2021, 9, 218. https://doi.org/10.3390/microorganisms9020218
Papale M, Romano I, Finore I, Lo Giudice A, Piccolo A, Cangemi S, Di Meo V, Nicolaus B, Poli A. Prokaryotic Diversity of the Composting Thermophilic Phase: The Case of Ground Coffee Compost. Microorganisms. 2021; 9(2):218. https://doi.org/10.3390/microorganisms9020218
Chicago/Turabian StylePapale, Maria, Ida Romano, Ilaria Finore, Angelina Lo Giudice, Alessandro Piccolo, Silvana Cangemi, Vincenzo Di Meo, Barbara Nicolaus, and Annarita Poli. 2021. "Prokaryotic Diversity of the Composting Thermophilic Phase: The Case of Ground Coffee Compost" Microorganisms 9, no. 2: 218. https://doi.org/10.3390/microorganisms9020218
APA StylePapale, M., Romano, I., Finore, I., Lo Giudice, A., Piccolo, A., Cangemi, S., Di Meo, V., Nicolaus, B., & Poli, A. (2021). Prokaryotic Diversity of the Composting Thermophilic Phase: The Case of Ground Coffee Compost. Microorganisms, 9(2), 218. https://doi.org/10.3390/microorganisms9020218