
Potential Association between Vaginal Microbiota and Cervical Carcinogenesis in Korean Women: A Cohort Study

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. HPV Assay and HPV Genotyping
2.3. DNA Extraction and High-Throughput Sequencing
2.4. Bioinformatic Analysis
2.5. Statistical Analysis
3. Results
3.1. Participants’ Characteristics
3.2. Vaginal Microbiota in Disease Progression
3.3. Comparative Analysis of the Vaginal Microbiota between the Groups
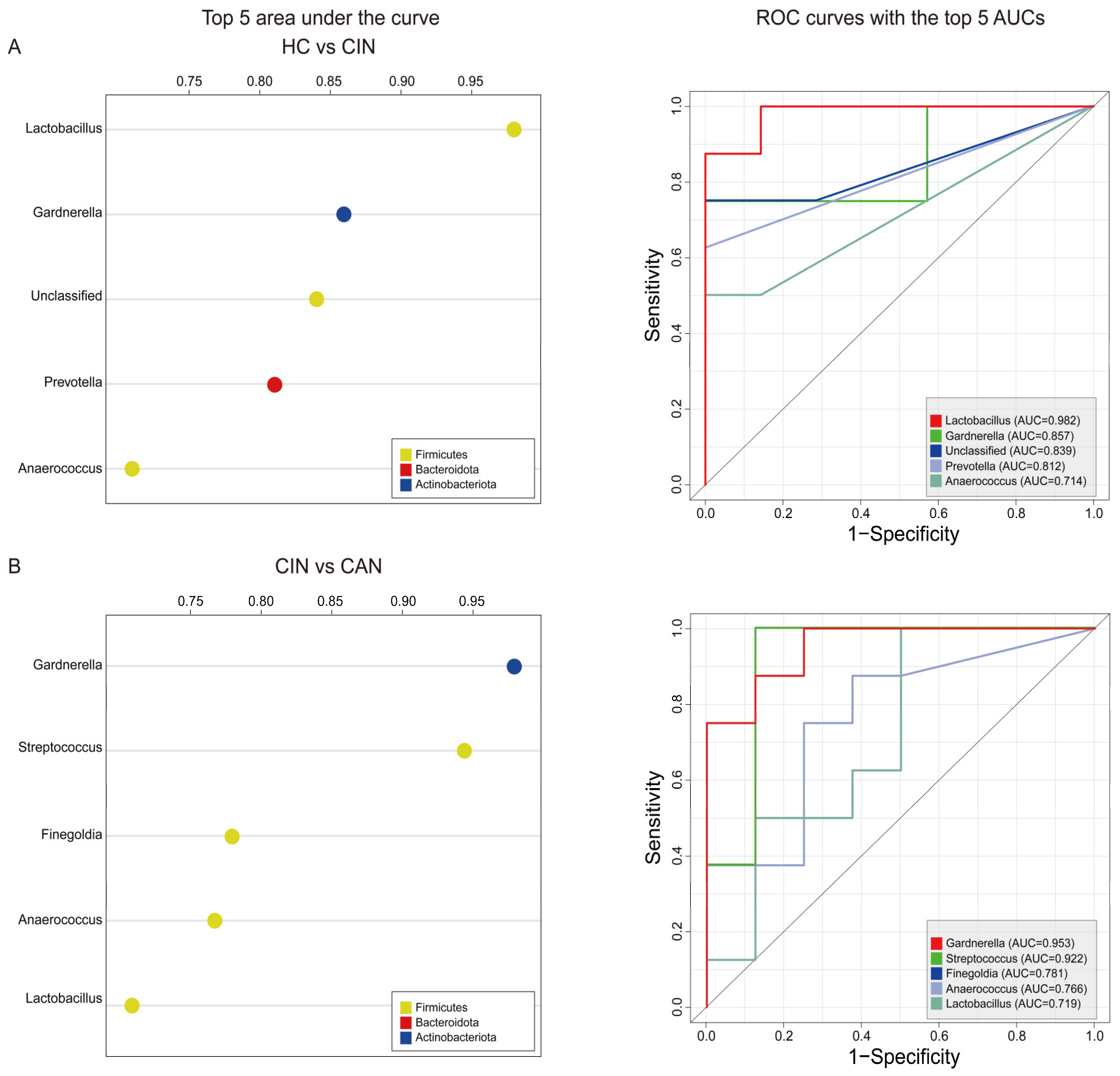
3.4. Predictive Ability of the Proposed Biomarkers
3.5. The Impact of HPV Infection across the Vaginal Microbiota
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muñoz, N. Human papillomavirus and cancer: The epidemiological evidence. J. Clin. Virol. 2000, 19, 1–5. [Google Scholar] [CrossRef]
- Castellsagué, X. Natural history and epidemiology of HPV infection and cervical cancer. Gynecol. Oncol. 2008, 110, S4–S7. [Google Scholar] [CrossRef]
- Clifford, G.M.; Smith, J.S.; Aguado, T.; Franceschi, S. Comparison of HPV type distribution in high-grade cervical lesions and cervical cancer: A meta-analysis. Br. J. Cancer 2003, 89, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Kyrgiou, M.; Mitra, A.; Moscicki, A.-B. Does the vaginal microbiota play a role in the development of cervical cancer? Transl. Res. 2017, 179, 168–182. [Google Scholar] [CrossRef] [Green Version]
- White, B.A.; Creedon, D.J.; Nelson, K.E.; Wilson, B.A. The vaginal microbiome in health and disease. Trends Endocrinol. Metab. 2011, 22, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Torcia, M. Interplay among vaginal microbiome, immune response and sexually transmitted viral infections. Int. J. Mol. Sci. 2019, 20, 266. [Google Scholar] [CrossRef] [Green Version]
- Campisciano, G.; Zanotta, N.; Licastro, D.; de Seta, F.; Comar, M. In Vivo microbiome and associated immune markers: New insights into the pathogenesis of vaginal dysbiosis. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.A.; Roach, P.J.; Montero, M.; Baroja-Fernández, E.; Muñoz, F.J.; Eydallin, G.; Viale, A.M.; Pozueta-Romero, J. Regulation of glycogen metabolism in yeast and bacteria. FEMS Microbiol. Rev. 2010, 34, 952–985. [Google Scholar] [CrossRef] [Green Version]
- Zacharof, M.; Lovitt, R.W. Bacteriocins produced by lactic acid bacteria a review article. Apcbee Procedia 2012, 2, 50–56. [Google Scholar] [CrossRef] [Green Version]
- O’Hanlon, D.E.; Cone, R.; Moench, T.R. Vaginal pH measured In Vivo: Lactobacilli determine pH and lactic acid concentration. BMC Microbiol. 2019, 19, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.A.; Beasley, D.E.; Dunn, R.R.; Archie, E.A. Lactobacilli dominance and vaginal pH: Why is the human vaginal microbiome unique? Front. Microbiol. 2016, 7, 1936. [Google Scholar] [CrossRef]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.K.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef] [Green Version]
- Haahr, T.; Zacho, J.; Bräuner, M.; Shathmigha, K.; Skov Jensen, J.; Humaidan, P. Reproductive outcome of patients un-dergoing In Vitro fertilisation treatment and diagnosed with bacterial vaginosis or abnormal vaginal microbiota: A sys-tematic PRISMA review and meta-analysis. BJOG Int. J. Obs. Gynaecol. 2019, 126, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Di Pietro, M.; Filardo, S.; Porpora, M.G.; Recine, N.; Latino, M.A.; Sessa, R. HPV/Chlamydia trachomatis co-infection: Metagenomic analysis of cervical microbiota in asymptomatic women. New Microbiol. 2018, 41, 34–41. [Google Scholar]
- Stone, L. Vaginal microbiota and infectious infertility. Nat. Rev. Urol. 2018, 15, 136. [Google Scholar] [CrossRef]
- Liu, M.-B.; Xu, S.-R.; He, Y.; Deng, G.-H.; Sheng, H.-F.; Huang, X.-M.; Ouyang, C.-Y.; Zhou, H.-W. Diverse vaginal mi-crobiomes in reproductive-age women with vulvovaginal candidiasis. PLoS ONE 2013, 8, e79812. [Google Scholar]
- Lewis, F.M.T.; Bernstein, K.T.; Aral, S.O. Vaginal microbiome and its relationship to behavior, sexual health, and sexually transmitted diseases. Obs. Gynecol. 2017, 129, 643–654. [Google Scholar] [CrossRef]
- Hočevar, K.; Maver, A.M.; Vidmar Šimic, M.; Hodžić, A.; Haslberger, A.; Premru-Sršen, T.; Peterlin, B. Vaginal micro-biome signature is associated with spontaneous preterm delivery. Front. Med. 2019, 6, 201. [Google Scholar] [CrossRef]
- Fettweis, J.M.; Serrano, M.G.; Brooks, J.L.; Edwards, D.J.; Girerd, P.H.; Parikh, H.I.; Huang, B.; Arodz, T.J.; Edupuganti, L.; Glascock, A.L.; et al. The vaginal microbiome and preterm birth. Nat. Med. 2019, 25, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Macintyre, D.A.; Lee, Y.S.; Smith, A.; Marchesi, J.R.; Lehne, B.; Bhatia, R.; Lyons, D.; Paraskevaidis, E.; Li, J.V.; et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 2015, 5, 16865. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Ar-umugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The vegan package. Community Ecol. Package 2007, 10, 719. [Google Scholar]
- Warnes, M.G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W. Package “gplots”: Various R Programming Tools for Plotting Data. 2011. Available online: http://cran.r-project.org/web/packages/gplots/index.html (accessed on 2 December 2020).
- Robin, X.A.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Wei, T.; Simko, V.; Levy, M.; Xie, Y.; Jin, Y.; Zemla, J. Package “corrplot”. Statistician 2017, 56, e24. [Google Scholar]
- Gupta, K.; Stapleton, A.E.; Hooton, T.M.; Roberts, P.L.; Fennell, C.L.; Stamm, W.E. Inverse Association of H2O2-producing Lactobacilli and vaginal Escherichia coli colonization in women with recurrent urinary tract infections. J. Infect. Dis. 1998, 178, 446–450. [Google Scholar] [CrossRef] [Green Version]
- Sewankambo, N.; Gray, R.H.; Wawer, M.J.; Paxton, L.; McNairn, D.; Wabwire-Mangen, F.; Serwadda, D.; Li, C.; Kiwanuka, N.; Hillier, S.L.; et al. HIV-1 infection associated with abnormal vaginal flora morphology and bacterial vaginosis. Lancet 1997, 350, 546–550. [Google Scholar] [CrossRef]
- Wiesenfeld, H.C.; Hillier, S.L.; Krohn, M.A.; Landers, D.V.; Sweet, R.L. Bacterial vaginosis is a strong predictor of Neisseria gonorrhoeae and Chlamydia trachomatis infection. Clin. Infect. Dis. 2003, 36, 663–668. [Google Scholar] [CrossRef] [Green Version]
- Van de Wijgert, J.; Mason, P.R.; Gwanzura, L.; Mbizvo, M.T.; Chirenje, Z.M.; Iliff, V.; Shiboski, S.; Padian, N.S. Intravaginal practices, vaginal flora disturbances, and acquisition of sexually transmitted diseases in zimbabwean women. J. Infect. Dis. 2000, 181, 587–594. [Google Scholar] [CrossRef]
- Onywera, H.; Williamson, A.-L.; Mbulawa, Z.Z.; Coetzee, D.; Meiring, T.L. The cervical microbiota in reproductive-age South African women with and without human papillomavirus infection. Papillomavirus Res. 2019, 7, 154–163. [Google Scholar] [CrossRef]
- Karpiński, T.M. Role of oral microbiota in cancer development. Microorganisms 2019, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of gut microbiota in inflammatory bowel disease (IBD): Cause or consequence? IBD treatment targeting the gut microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [Green Version]
- Nørreslet, L.B.; Agner, T.; Clausen, M.-L. The skin microbiome in inflammatory skin diseases. Curr. Derm. Rep. 2020, 9, 141–151. [Google Scholar] [CrossRef]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 di-abetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, K.; Mulak, A. Brain-gut-microbiota axis in alzheimer’s disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Rajpoot, M.; Sharma, A.K.; Gupta, G.K. Understanding the microbiome: Emerging biomarkers for exploiting the microbiota for personalized medicine against cancer. Semin. Cancer Biol. 2018, 52, 1–8. [Google Scholar] [CrossRef]
- Temraz, S.; Nassar, F.; Nasr, R.; Charafeddine, M.; Mukherji, D.; Shamseddine, A.I. Gut microbiome: A promising biomarker for immunotherapy in colorectal cancer. Int. J. Mol. Sci. 2019, 20, 4155. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, Q.; Zhao, J.; Gong, L.; Zhang, Y.; Wang, X.; Yuan, Z. Altered diversity and composition of the gut mi-crobiome in patients with cervical cancer. AMB Express 2019, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Sankaranarayanan, R.; Nene, B.M.; Shastri, S.S.; Jayant, K.; Muwonge, R.; Malvi, S.G. Reply to S D Rathod’s Commentary on HPV screening for cervical cancer in rural India. Indian J. Med. Ethic 2011, 360, 1385–1394. [Google Scholar] [CrossRef] [Green Version]
- Bosch, F.X.; Manos, M.M.; Muñoz, N.; Sherman, M.; Jansen, A.M.; Peto, J.; Schiffman, M.H.; Moreno, V.; Kurman, R.J.; Shan, K.V.; et al. Prevalence of human papillomavirus in cervical cancer: A worldwide perspective. J. Natl. Cancer Inst. 1995, 87, 796–802. [Google Scholar] [CrossRef]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N.; et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Fredricks, D.; Fiedler, T.L.; Marrazzo, J.M. Molecular identification of bacteria associated with bacterial vaginosis. Engl. J. Med. 2005, 353, 1899–1911. [Google Scholar] [CrossRef] [Green Version]
- Filardo, S.; di Pietro, M.; Porpora, M.G.; Recine, N.; Farcomeni, A.; Latino, M.A.; Sessa, R. Diversity of Cervical microbiota in asymptomatic Chlamydia trachomatis genital infection: A pilot study. Front. Cell. Infect. Microbiol. 2017, 7, 321. [Google Scholar] [CrossRef]
- Freitas, A.C.; Bocking, A.; Hill, J.E.; Money, D.M. Increased richness and diversity of the vaginal microbiota and spon-taneous preterm birth. Microbiome 2018, 6, 117. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Dutilh, B.E.; Hall, N.; Peters, W.H.M.; Roelofs, R.; Boleij, A.; Tjalsma, H. Towards the human colorectal cancer microbiome. PLoS ONE 2011, 6, e20447. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2011, 22, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.; Fukuma, N.; Totsika, M.; Kenny, L.; Morrison, M.; Punyadeera, C. The performance of an oral microbiome bi-omarker panel in predicting oral cavity and oropharyngeal cancers. Front. Cell. Infect. Microbiol. 2018, 8, 267. [Google Scholar] [CrossRef] [Green Version]
- Patras, K.A.; Rösler, B.; Thoman, M.L.; Doran, K.S. Characterization of host immunity during persistent vaginal colo-nization by Group B Streptococcus. Mucosal Immunol. 2015, 8, 1339–1348. [Google Scholar] [CrossRef] [Green Version]
- Soares, G.C.M.T.; da Silva, B.A.; dos Santos, M.H.B.; da Costa, A.F.E.; dos Santos, A.L.S.; Morandi, V.; Nagao, P.E. Metallopeptidases produced by group B Streptococcus: Influence of proteolytic inhibitors on growth and on interaction with human cell lineages. Int. J. Mol. Med. 2008, 22, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Boris, S.; Suárez, J.E.; Vázquez, F.; Barbés, C. Adherence of human vaginal Lactobacilli to vaginal epithelial cells and interaction with uropathogens. Infect. Immun. 1998, 66, 1985–1989. [Google Scholar] [CrossRef] [Green Version]
- Wagner, W.; Ciszewski, W.M.; Kania, L.-K.D. D-lactate enhance DNA repair and modulate the resistance of cervical carcinoma cells to anticancer drugs via histone deacetylase inhibition and hydroxycarboxylic acid receptor 1 activation. Cell Commun. Signal. 2015, 13, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Aroutcheva, A.A.; Simoes, J.A.; Behbakht, K.; Faro, S. Gardnerella vaginalisIsolated from Patients with Bacterial Vaginosis and from Patients with Healthy Vaginal Ecosystems. Clin. Infect. Dis. 2001, 33, 1022–1027. [Google Scholar] [CrossRef] [Green Version]
- Usyk, M.; Zolnik, C.P.; E Castle, P.; Porras, C.; Herrero, R.; Gradissimo, A.; Gonzalez, P.; Safaeian, M.; Schiffman, M.; Burk, R.D.; et al. Cervicovaginal microbiome and natural history of HPV in a longitudinal study. PLoS Pathog. 2020, 16, e1008376. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | HC (n = 7) | CIN (n = 8) | CAN (n = 8) |
|---|---|---|---|
| Age (years) | 47.4 ± 5.38 | 43.4 ± 12.8 | 47 ± 10.2 |
| Menopause (n, %) | 2 (28.6) | 3 (37.5) | 2 (25.0) |
| Marriage (n, %) | 5 (71.4) | 6 (75.0) | 7 (87.5) |
| Parity (n) | 1.1 ± 1.0 | 1 ± 0.9 | 1.8 ± 0.8 |
| Smoker (n, %) | 0 (0.0) | 4 (50.0) | 3 (37.5) |
| Contraceptive use (n, %) | 0 (0.0) | 3 (37.5) | 2 (25.0) |
| HPV positive (n, %) | 0 (0.0) | 7 (87.5) | 8 (100.0) |
| HPV16/18 positive (n, %) | 0 (0.0) | 0 (0.0) | 5 (62.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, G.-U.; Jung, D.-R.; Lee, Y.H.; Jeon, S.Y.; Han, H.S.; Chong, G.O.; Shin, J.-H. Potential Association between Vaginal Microbiota and Cervical Carcinogenesis in Korean Women: A Cohort Study. Microorganisms 2021, 9, 294. https://doi.org/10.3390/microorganisms9020294
Kang G-U, Jung D-R, Lee YH, Jeon SY, Han HS, Chong GO, Shin J-H. Potential Association between Vaginal Microbiota and Cervical Carcinogenesis in Korean Women: A Cohort Study. Microorganisms. 2021; 9(2):294. https://doi.org/10.3390/microorganisms9020294
Chicago/Turabian StyleKang, Gi-Ung, Da-Ryung Jung, Yoon Hee Lee, Se Young Jeon, Hyung Soo Han, Gun Oh Chong, and Jae-Ho Shin. 2021. "Potential Association between Vaginal Microbiota and Cervical Carcinogenesis in Korean Women: A Cohort Study" Microorganisms 9, no. 2: 294. https://doi.org/10.3390/microorganisms9020294
APA StyleKang, G.-U., Jung, D.-R., Lee, Y. H., Jeon, S. Y., Han, H. S., Chong, G. O., & Shin, J.-H. (2021). Potential Association between Vaginal Microbiota and Cervical Carcinogenesis in Korean Women: A Cohort Study. Microorganisms, 9(2), 294. https://doi.org/10.3390/microorganisms9020294