
A Mouse Model Suggests That Heart Failure and Its Common Comorbidity Sleep Fragmentation Have No Synergistic Impacts on the Gut Microbiome

 , , ,
, , ,  , and
, and
Abstract
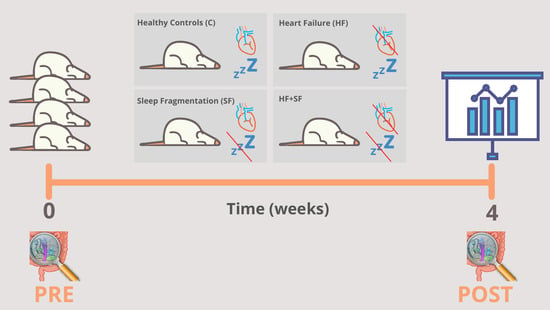
:
1. Introduction
2. Materials and Methods
2.1. Animal Models Experiments
2.2. DNA Extraction, Library Preparation and Sequencing
2.3. Microbiome Analysis
- “filterAndTrim(fnFs, filtFs, fnRs, filtRs, truncLen = c(285,240), maxN = 0, maxEE = c(10,10), truncQ = 1, rm.phix = TRUE, trimLeft = c(10,10), compress = TRUE, multithread = TRUE)”
- “prune_samples(sample_sums(object) ≥ 950, object)”
- “filter_taxa(object, function(x) sum(x > 0.001) > (0.05* length(x)), prune = TRUE)”
2.4. Statistical Analysis
3. Results
3.1. Experimental Modelling of HF and SF
- Heart failure (HF): This condition was induced by continuous infusion of isoproterenol, as previously described [18].
- Sleep fragmentation (SF): SF was induced daily by means of a previously validated device for mice (Lafayette Instruments, Lafayette, IN), based on automated intermittent tactile stimulation. Stimulation was applied in 2-min intervals during the murine sleep period (8 a.m.–8 p.m.), as described earlier [18].
- Combination of HF and SF (HF + SF) in which both conditions were induced in the same mice.
- Control: where no condition was induced. Before the start of the experiment and at the end of the 4-week experiment (HF, SF, HF + SF and control), fecal samples were obtained directly from stool expulsion and frozen at −80 °C until further analysis (Figure 1).
3.2. Characterization of the Microbiome
3.3. Alpha Diversity
3.4. Changes in Microbial Composition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Camps-Vilaró, A.; Delgado-Jiménez, J.F.; Farré, N.; Tizón-Marcos, H.; Álvarez-García, J.; Cinca, J.; Dégano, I.R.; Marrugat, J. Estimated Population Prevalence of Heart Failure with Reduced Ejection Fraction in Spain, According to DAPA-HF Study Criteria. J. Clin. Med. 2020, 9, 2089. [Google Scholar] [CrossRef] [PubMed]
- Farré, N.; Vela, E.; Clèries, M.; Bustins, M.; Cainzos-Achirica, M.; Enjuanes, C.; Moliner, P.; Ruiz, S.; Verdú-Rotellar, J.M.; Comín-Colet, J. Real world heart failure epidemiology and outcome: A population-based analysis of 88,195 patients. PLoS ONE 2017, 12, e0172745. [Google Scholar] [CrossRef]
- Cowie, M.R.; Gallagher, A.M. Sleep Disordered Breathing and Heart Failure: What Does the Future Hold? JACC Heart Fail. 2017, 5, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Javaheri, S.; Brown, L.K.; Khayat, R.N. Update on Apneas of Heart Failure with Reduced Ejection Fraction: Emphasis on the Physiology of Treatment: Part 2: Central Sleep Apnea. Chest 2020, 157, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nat. Cell Biol. 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Mashaqi, S.; Gozal, D. Obstructive Sleep Apnea and Systemic Hypertension: Gut Dysbiosis as the Mediator? J. Clin. Sleep Med. 2019, 15, 1517–1527. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, M.; Winkler, T.; Heinsen, F.-A.; Rühlemann, M.C.; Spehlmann, M.E.; Bajrovic, A.; Lieb, W.; Franke, A.; Ott, S.J.; Frey, N. Heart failure is associated with depletion of core intestinal microbiota. ESC Hear. Fail. 2017, 4, 282–290. [Google Scholar] [CrossRef]
- Mayerhofer, C.C.; Kummen, M.; Holm, K.; Broch, K.; Awoyemi, A.; Vestad, B.; Storm-Larsen, C.; Seljeflot, I.; Ueland, T.; Bohov, P.; et al. Low fibre intake is associated with gut microbiota alterations in chronic heart failure. ESC Hear. Fail. 2020, 7, 456–466. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Heaney, L.M.; Bhandari, S.S.; Jones, D.J.L.; Ng, L.L. Trimethylamine N-oxide and prognosis in acute heart failure. Heart 2016, 102, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Genth-Zotz, S.; von Haehling, S.; Bolger, A.P.; Kalra, P.R.; Wensel, R.; Coats, A.J.S.; Anker, S.D. Pathophysiologic quantities of endotox-in-induced tumor necrosis factor-alpha release in whole blood from patients with chronic heart failure. Am. J. Cardiol. 2002, 90, 1226–1230. [Google Scholar] [CrossRef]
- Sandek, A.; Bauditz, J.; Swidsinski, A.; Buhner, S.; Weber-Eibel, J.; von Haehling, S.; Schroedl, W.; Karhausen, T.; Doehner, W.; Rauchhaus, M.; et al. Altered Intestinal Function in Patients with Chronic Heart Failure. J. Am. Coll. Cardiol. 2007, 50, 1561–1569. [Google Scholar] [CrossRef] [Green Version]
- Ko, C.-Y.; Liu, Q.-Q.; Su, H.-Z.; Zhang, H.-P.; Fan, J.-M.; Yang, J.-H.; Hu, A.-K.; Liu, Y.-Q.; Chou, D.; Zeng, Y.-M. Gut microbiota in obstructive sleep apnea–hypopnea syndrome: Disease-related dysbiosis and metabolic comorbidities. Clin. Sci. 2019, 133, 905–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Indias, I.; Torres, M.; Montserrat, J.M.; Sanchez-Alcoholado, L.; Cardona, F.; Tinahones, F.J.; Gozal, D.; Poroyko, V.A.; Navajas, D.; Queipo-Ortuño, M.I.; et al. Intermittent hypoxia alters gut microbiota diversity in a mouse model of sleep apnoea. Eur. Respir. J. 2015, 45, 1055–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, A.; Melnik, A.V.; Xue, J.; Poulsen, O.; Meehan, M.J.; Humphrey, G.; Jiang, L.; Ackermann, G.; McDonald, D.; Zhou, D.; et al. Intermittent Hypoxia and Hypercapnia, a Hallmark of Obstructive Sleep Apnea, Alters the Gut Microbiome and Metabolome. mSystems 2018, 3, e00018–e00020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poroyko, V.A.; Carreras, A.; Khalyfa, A.; Khalyfa, A.A.; Leone, V.; Peris, E.; Almendros, I.; Gileles-Hillel, A.; Qiao, Z.; Hubert, N.; et al. Chronic Sleep Disruption Alters Gut Microbiota, Induces Systemic and Adipose Tissue Inflammation and Insulin Resistance in Mice. Sci. Rep. 2016, 6, 35405. [Google Scholar] [CrossRef] [Green Version]
- Farré, N.; Farré, R.; Gozal, D. Sleep Apnea Morbidity: A Consequence of Microbial-Immune Cross-Talk? Chest 2018, 154, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Aguilera, I.; Benito, B.; Tajes, M.; Farré, R.; Gozal, D.; Almendros, I.; Farré, N. Chronic Sleep Fragmentation Mimicking Sleep Apnea Does Not Worsen Left-Ventricular Function in Healthy and Heart Failure Mice. Front. Neurol. 2020, 10, 1364. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Nearing, J.T.; Douglas, G.M.; Comeau, A.M.; Langille, M.G. Denoising the Denoisers: An independent evaluation of microbiome sequence error-correction approaches. PeerJ 2018, 6, e5364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Schliep, K.P. Phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [Green Version]
- Erik, S.W. Using DECIPHER v2.0 to Analyze Big Biological Sequence Data in R. R J. 2016, 8, 352–359. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [Green Version]
- Gloor, G.B.; Reid, G. Compositional analysis: A valid approach to analyze microbiome high-throughput sequencing data. Can. J. Microbiol. 2016, 62, 692–703. [Google Scholar] [CrossRef]
- Palarea-Albaladejo, J.; Martín-Fernández, J.A. zCompositions—R package for multivariate imputation of left-censored data under a compositional approach. Chemom. Intell. Lab. Syst. 2015, 143, 85–96. [Google Scholar] [CrossRef]
- Reynolds, A.P.; Richards, G.; De La Iglesia, B.; Rayward-Smith, V.J. Clustering Rules: A Comparison of Partitioning and Hierarchical Clustering Algorithms. J. Math. Model. Algorithms 2006, 5, 475–504. [Google Scholar] [CrossRef]
- Bates, D.M.; DebRoy, S. Linear mixed models and penalized least squares. J. Multivar. Anal. 2004, 91, 1–17. [Google Scholar] [CrossRef]
- Fox, J.; Friendly, M.; Weisberg, S. Hypothesis Tests for Multivariate Linear Models Using the car Package. R J. 2013, 5, 39–52. [Google Scholar] [CrossRef] [Green Version]
- Hothorn, T.; Bretz, F.; Westfall, P. Simultaneous Inference in General Parametric Models. Biom. J. 2008, 50, 346–363. [Google Scholar] [CrossRef] [Green Version]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Bäckhed, F.; Blaser, M.J.; Bushman, F.D.; De Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Kim, S.-E.; Kim, A.-R.; Kang, S.; Park, M.-Y.; Sung, M.-K. Dietary fat intake and age modulate the composition of the gut microbiota and colonic inflammation in C57BL/6J mice. BMC Microbiol. 2019, 19, 1–11. [Google Scholar] [CrossRef]
- Langille, M.G.; Meehan, C.J.; Koenig, J.E.; Dhanani, A.S.; Rose, R.A.; Howlett, S.E.; Beiko, R.G. Microbial shifts in the aging mouse gut. Microbiome 2014, 2, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Mika, A.; Van Treuren, W.; González, A.; Herrera, J.J.; Knight, R.; Fleshner, M. Exercise Is More Effective at Altering Gut Microbial Composition and Producing Stable Changes in Lean Mass in Juvenile versus Adult Male F344 Rats. PLoS ONE 2015, 10, e0125889. [Google Scholar] [CrossRef] [PubMed]
- Yuzefpolskaya, M.; Bohn, B.; Nasiri, M.; Zuver, A.M.; Onat, D.D.; Royzman, E.A.; Nwokocha, J.; Mabasa, M.; Pinsino, A.; Brunjes, D.; et al. Gut microbiota, endotoxemia, inflammation, and oxidative stress in patients with heart failure, left ventricular assist device, and transplant. J. Hear. Lung Transplant. 2020, 39, 880–890. [Google Scholar] [CrossRef]
- Yang, J.-Y.; Lee, Y.-S.; Kim, Y.; Lee, S.-H.; Ryu, S.; Fukuda, S.; Hase, K.; Yang, C.S.; Lim, H.S.; Kim, M.S.; et al. Gut commensal Bacteroides acidifaciens prevents obesity and im-proves insulin sensitivity in mice. Mucosal Immunol. 2017, 10, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Marques, F.Z.; Nelson, E.; Chu, P.-Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef]
- Duong, A.T.H.; Reitz, C.J.; Louth, E.L.; Creighton, S.D.; Rasouli, M.; Zwaiman, A.; Kroetsch, J.T.; Bolz, S.-S.; Winters, B.D.; Bailey, C.D.C.; et al. The Clock Mechanism Influences Neurobiology and Adaptations to Heart Failure in Clock ∆19/∆19 Mice with Implications for Circadian Medicine. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chatham, J.C.; Young, M.E. Circadian Regulation of Cardiac Physiology: Rhythms That Keep the Heart Beating. Annu. Rev. Physiol. 2020, 82, 79–101. [Google Scholar] [CrossRef]
- Gao, J.; Yan, K.-T.; Wang, J.-X.; Dou, J.; Wang, J.; Ren, M.; Ma, J.; Zhang, X.; Liu, Y. Gut microbial taxa as potential predictive biomarkers for acute coronary syndrome and post-STEMI cardiovascular events. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Ye, L.; Li, J.; Jin, L.; Wang, W.; Li, S.; Bao, M.; Wu, S.; Li, L.; Geng, B.; et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lataro, R.M.; Imori, P.F.M.; Santos, E.S.; Silva, L.E.V.; Duarte, R.T.D.; Silva, C.A.A.; Falcão, J.P.; Paton, J.F.R.; Salgado, H.C. Heart failure developed after myocardial infarction does not affect gut microbiota composition in the rat. Am. J. Physiol. Liver Physiol. 2019, 317, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Kain, V.; Van Der Pol, W.; Mariappan, N.; Ahmad, A.; Eipers, P.; Gibson, D.L.; Gladine, C.; Vigor, C.; Durand, T.; Morrow, C.; et al. Obesogenic diet in aging mice disrupts gut microbe composition and alters neutrophil:lymphocyte ratio, leading to inflamed milieu in acute heart failure. FASEB J. 2019, 33, 6456–6469. [Google Scholar] [CrossRef] [PubMed]
- Turek, F.; Gillette, M. Melatonin, sleep, and circadian rhythms: Rationale for development of specific melatonin agonists. Sleep Med. 2004, 5, 523–532. [Google Scholar] [CrossRef]
- Hong, F.; Pan, S.; Xu, P.; Xue, T.; Wang, J.; Guo, Y.; Jia, L.; Qiao, X.; Li, L.; Zhai, Y. Melatonin Orchestrates Lipid Homeostasis through the Hepatointestinal Circadian Clock and Microbiota during Constant Light Exposure. Cells 2020, 9, 489. [Google Scholar] [CrossRef] [Green Version]
- Lucking, E.F.; O’Connor, K.M.; Strain, C.R.; Fouhy, F.; Bastiaanssen, T.F.; Burns, D.P.; Golubeva, A.V.; Stanton, C.; Clarke, G.; Cryan, J.F.; et al. Chronic intermittent hypoxia disrupts cardiorespiratory homeostasis and gut microbiota composition in adult male guinea-pigs. EBioMedicine 2018, 38, 191–205. [Google Scholar] [CrossRef] [Green Version]
- Patke, A.; Murphy, P.J.; Onat, O.E.; Krieger, A.C.; Özçelik, T.; Campbell, S.S.; Young, M.W. Mutation of the Human Circadian Clock Gene CRY1 in Familial Delayed Sleep Phase Disorder. Cell 2017, 169, 203–215.e13. [Google Scholar] [CrossRef] [Green Version]
- Naghshin, J.; Rodriguez, R.H.; Davis, E.M.; Romano, L.C.; McGaffin, K.R.; O’Donnell, C.P. Chronic intermittent hypoxia exposure improves left ventricular contractility in transgenic mice with heart failure. J. Appl. Physiol. 2012, 113, 791–798. [Google Scholar] [CrossRef] [Green Version]
- Naghshin, J.; McGaffin, K.R.; Witham, W.G.; Mathier, M.A.; Romano, L.C.; Smith, S.H.; Janczewski, A.M.; Kirk, J.A.; Shroff, S.G.; O’Donnell, C.P. Chronic intermittent hypoxia increases left ventricular contractility in C57BL/6J mice. J. Appl. Physiol. 2009, 107, 787–793. [Google Scholar] [CrossRef]
- Farré, N.; Lupon, J.; Roig, E.; Gonzalez-Costello, J.; Vila, J.; Perez, S.; De Antonio, M.; Gonzalez, E.S.; Sánchez-Enrique, C.; Moliner, P.; et al. Clinical characteristics, one-year change in ejection fraction and long-term outcomes in patients with heart failure with mid-range ejection fraction: A multicentre prospective observational study in Catalonia (Spain). BMJ Open 2017, 7, e018719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mong, J.A.; Cusmano, D.M. Sex differences in sleep: Impact of biological sex and sex steroids. Philos. Trans. R. Soc. B: Biol. Sci. 2016, 371, 20150110. [Google Scholar] [CrossRef] [PubMed]
- Regitz-Zagrosek, V.; Seeland, U. Sex and gender differences in myocardial hypertrophy and heart failure. Wien. Med. Wochenschr. 2011, 161, 109–116. [Google Scholar] [CrossRef]
- Nguyen, T.L.A.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model. Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnett, M.; Fraser, A. Animal models of colitis: Lessons learned, and their relevance to the clinic. In Ulcerative Colitis—Treatments, Special Populations and the Future; InTech: Rijeka, Croatia, 2011. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linar Model—Fixed Effect | Phylum | Class | Order | Family | Genus | Species |
|---|---|---|---|---|---|---|
| (A)—Condition | 3 | 5 | 5 | 10 | 23 | 26 |
| (A)—Time | 4 | 9 | 10 | 19 | 41 | 47 |
| (B)—Condition | 1 | 2 | 4 | 14 | 30 | 32 |
| (B)—W.change | 1 | 1 | 1 | 3 | 9 | 9 |
| Condition | Change of Weight | |
|---|---|---|
| Bacteroides acidifaciens | 0.00015 | |
| Ileibacterium valens | 0.00113 | 0.00062 |
| Mucispirillum schaedleri | 0.00125 | 0.03626 |
| Olsenella spp. | 2.79 × 10–25 | |
| Bacteroides spp. | 0.00904 | |
| Odoribacter spp. | 0.03183 | |
| Muribaculum spp. | 0.01244 | |
| Prevotellaceae_UCG.001 spp. | 0.03238 | |
| Alistipes spp. | 3.44 × 10–5 | |
| O.Bacteroidales.UCS | 0.00117 | |
| Mucispirillum spp. | 0.00408 | |
| Lactococcus spp. | 0.00262 | |
| Defluviitaleaceae_UCG.011 spp. | 0.04673 | |
| Lachnoclostridium spp. | 0.00029 | |
| Lachnospiraceae_NK4A136_group spp. | 0.00637 | |
| F.Peptococcaceae.UCS | 1.57 × 10–6 | 0.00019 |
| Anaerotruncus spp. | 0.00799 | 0.02487 |
| Harryflintia spp. | 0.02105 | |
| Oscillibacter spp. | 0.01505 | |
| Ruminococcaceae_UCG.010 spp. | 0.04265 | |
| Ruminococcaceae_UCG.014 spp. | 8.72 × 10–6 | |
| Ruminococcus spp. | 3.73 × 10–6 | 0.00302 |
| F.Ruminococcaceae.UCS | 0.02286 | |
| Allobaculum spp. | 0.00087 | 0.01719 |
| Candidatus_Stoquefichus spp. | 0.04012 | |
| Dubosiella spp. | 0.00068 | |
| Faecalibaculum spp. | 0.03229 | |
| Bilophila spp. | 0.00968 | |
| F.Desulfovibrionaceae.UCS | 1.99 × 10–7 | |
| Oxalobacter spp. | 0.01909 | |
| Anaeroplasma spp. | 0.03002 | |
| O.Mollicutes_RF39.UCS | 0.03361 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khannous-Lleiffe, O.; Willis, J.R.; Saus, E.; Cabrera-Aguilera, I.; Almendros, I.; Farré, R.; Gozal, D.; Farré, N.; Gabaldón, T. A Mouse Model Suggests That Heart Failure and Its Common Comorbidity Sleep Fragmentation Have No Synergistic Impacts on the Gut Microbiome. Microorganisms 2021, 9, 641. https://doi.org/10.3390/microorganisms9030641
Khannous-Lleiffe O, Willis JR, Saus E, Cabrera-Aguilera I, Almendros I, Farré R, Gozal D, Farré N, Gabaldón T. A Mouse Model Suggests That Heart Failure and Its Common Comorbidity Sleep Fragmentation Have No Synergistic Impacts on the Gut Microbiome. Microorganisms. 2021; 9(3):641. https://doi.org/10.3390/microorganisms9030641
Chicago/Turabian StyleKhannous-Lleiffe, Olfat, Jesse R. Willis, Ester Saus, Ignacio Cabrera-Aguilera, Isaac Almendros, Ramon Farré, David Gozal, Nuria Farré, and Toni Gabaldón. 2021. "A Mouse Model Suggests That Heart Failure and Its Common Comorbidity Sleep Fragmentation Have No Synergistic Impacts on the Gut Microbiome" Microorganisms 9, no. 3: 641. https://doi.org/10.3390/microorganisms9030641
APA StyleKhannous-Lleiffe, O., Willis, J. R., Saus, E., Cabrera-Aguilera, I., Almendros, I., Farré, R., Gozal, D., Farré, N., & Gabaldón, T. (2021). A Mouse Model Suggests That Heart Failure and Its Common Comorbidity Sleep Fragmentation Have No Synergistic Impacts on the Gut Microbiome. Microorganisms, 9(3), 641. https://doi.org/10.3390/microorganisms9030641