
Nicotine Increases Macrophage Survival through α7nAChR/NF-κB Pathway in Mycobacterium avium paratuberculosis Infection



Abstract
:1. Introduction
2. Materials and Methods
2.1. Monocyte-Derived Macrophages Preparation and Cell Culture
2.2. Measurement of Gene Expression in Treated Macrophages
2.3. Measurement of Cathelicidin Protein Expression in Treated Macrophages
2.4. Preparation of Macrophages Nuclear Protein Extract
2.5. Measurement of NF-κB p100/p52 Transcription Factor Activation in Treated Macrophages
2.6. Statistical Analysis
3. Results
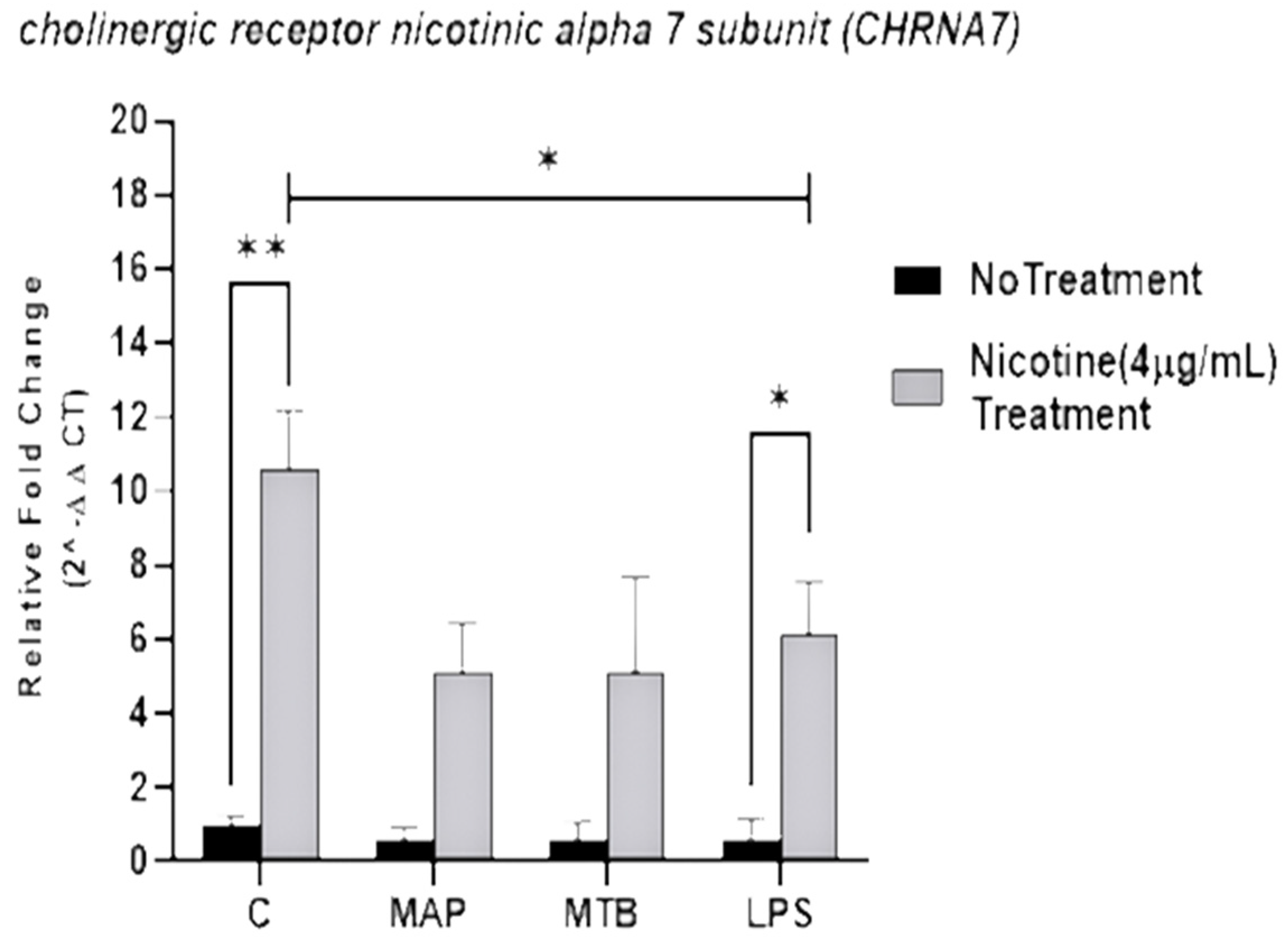
3.1. Nicotine Significantly Upregulates Nicotinic Acetylcholine Receptor (α7nAChR) Expression in MAP-Infected Macrophages
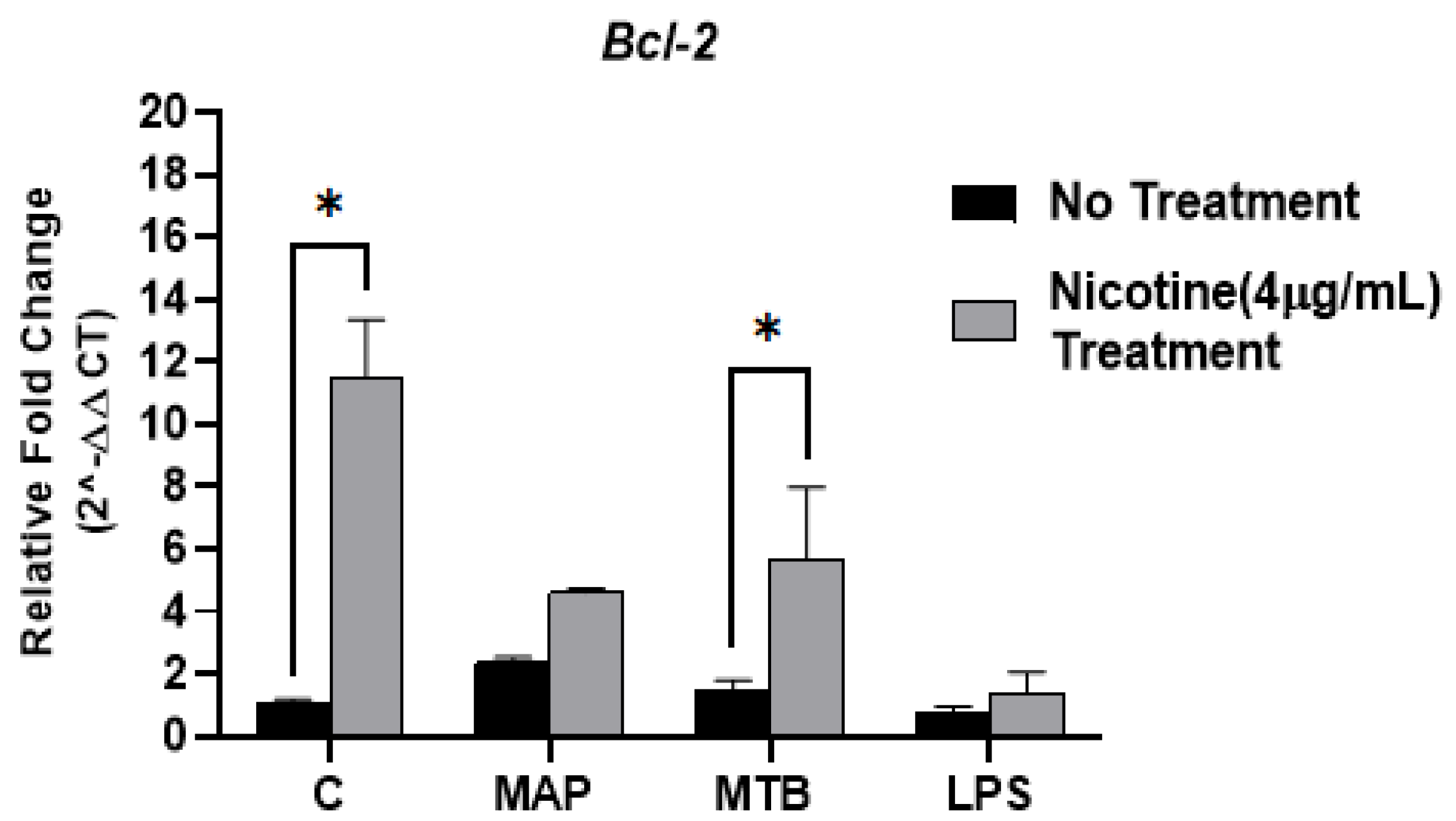
3.2. Nicotine Induces Modulation of Anti-Apoptotic Gene (Bcl2) in MAP-Infected Macrophages
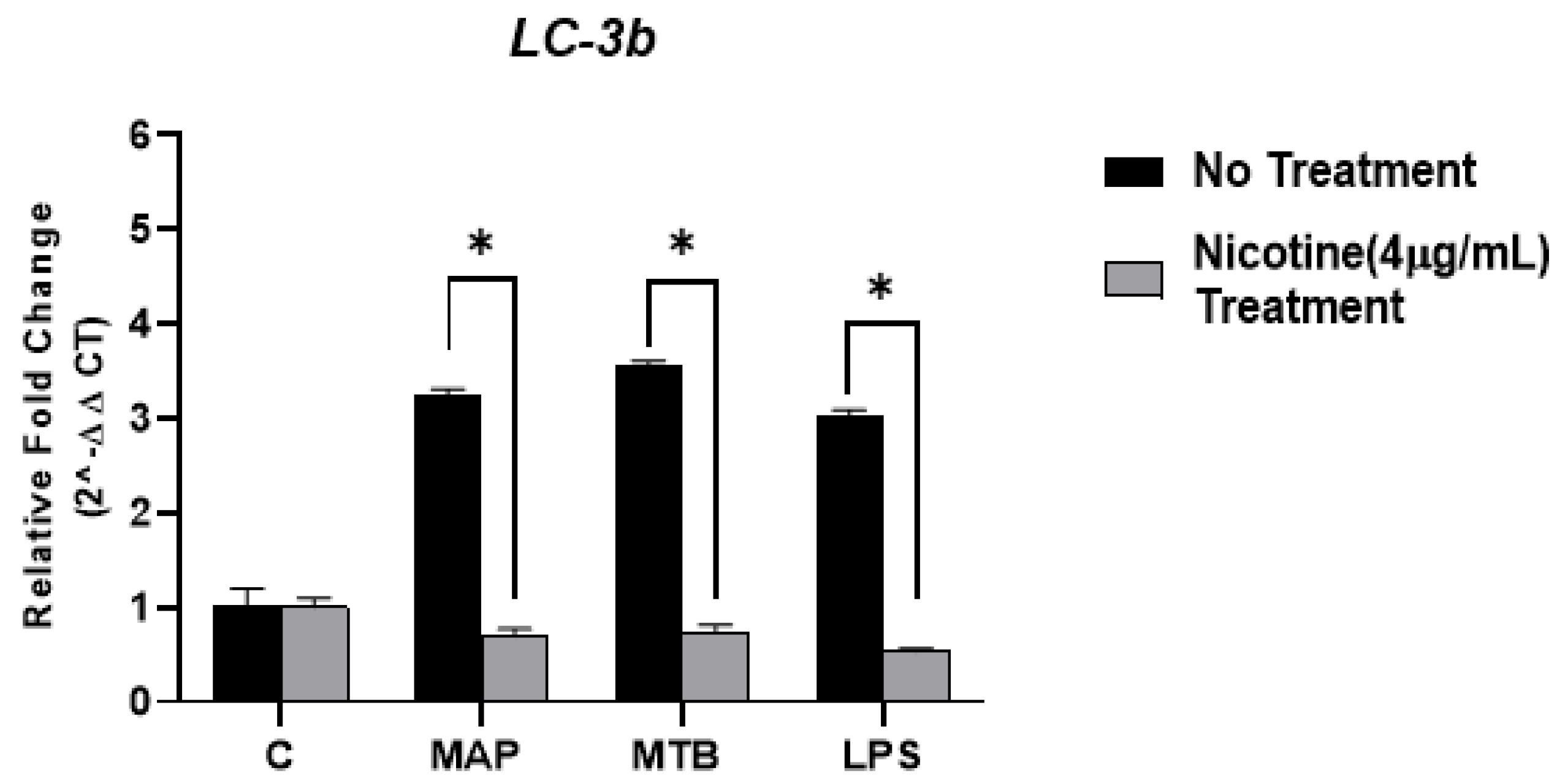
3.3. Nicotine Decreases Autophagy Marker Expression (LC-3b) While MAP Significantly Increases It
3.4. Nicotine Upregulates the Expression of REL-B and NF-ĸB p100 Subunit in MAP-Infected Macrophages
3.5. Nicotine Increases p52 Protein and NF-ĸB (Survival Pathway) Activity in MAP-Infected Macrophages
3.6. Nicotine Synergistically Inhibits Cathelicidin Production in MAP-Infected Macrophages
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valdez-Miramontes, C.E.; Martínez, L.A.T.; Torres-Juárez, F.; Carlos, A.R.; Marin-Luévano, S.P.; de Haro-Acosta, J.P.; Enciso-Moreno, J.A.; Rivas-Santiago, B. Nicotine modulates molecules of the innate immune response in epithelial cells and macrophages during infection with M. tuberculosis. Clin. Exp. Immunol. 2020, 199, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.A.O.; Rhodes, J.; Ingram, J.R. Mechanisms of disease: Nicotine—A review of its actions in the context of gastrointestinal disease. Nature Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 536–544. [Google Scholar] [CrossRef]
- AlQasrawi, D.; Qasem, A.; Naser, S.A. Divergent Effect of Cigarette Smoke on Innate Immunity in Inflammatory Bowel Disease: A Nicotine-Infection Interaction. Int. J. Mol. Sci. 2020, 21, 5801. [Google Scholar] [CrossRef] [PubMed]
- Naser, S.A.; Sagramsingh, S.R.; Naser, A.S.; Thanigachalam, S. Mycobacterium avium subspecies paratuberculosis causes Crohn’s disease in some inflammatory bowel disease patients. World J. Gastroenterol. WJG 2014, 20, 7403. [Google Scholar] [CrossRef] [PubMed]
- Rashid, T.; Wilson, C.; Ebringer, A. The link between ankylosing spondylitis, Crohn’s disease, Klebsiella, and starch consumption. Clin. Dev. Immunol. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Nazareth, N.; Magro, F.; Machado, E.; Ribeiro, T.G.; Martinho, A.; Rodrigues, P.; Alves, R.; Macedo, G.N.; Gracio, D.; Coelho, R.; et al. Prevalence of Mycobacteriumavium subsp. paratuberculosis and Escherichia coli in blood samples from patients with inflammatory bowel disease. Med Microbiol. Immunol. 2015, 204, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Benfer, B.A. Source Book of Substance Abuse and Addiction. J. Psychosoc. Nurs. Ment. Health Serv. 1997, 35, 44–45. [Google Scholar] [CrossRef]
- Kazuto, M.; Klein, T.W.; Friedman, H.; Yamamoto, Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J. Immunol. 2001, 167, 6518–6524. [Google Scholar]
- Karlin, A. Emerging structure of the nicotinic acetylcholine receptors. Nature Rev. Neurosci. 2002, 3, 102–114. [Google Scholar] [CrossRef]
- Báez-Pagán, C.A.; Delgado-Vélez, M.; Lasalde-Dominicci, J.A. Activation of the macrophage α7 nicotinic acetylcholine receptor and control of inflammation. J. Neuroimmune Pharmacol. 2015, 10, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Dania, A.; Abdelli, L.S.; Naser, S.A. Mystery Solved: Why Smoke Extract Worsens Disease in Smokers with Crohn’s Disease and Not Ulcerative Colitis? Gut MAP! Microorganisms 2020, 8, 666. [Google Scholar]
- Qasem, A.; Naser, S.A. TNFα inhibitors exacerbate Mycobacterium paratuberculosis infection in tissue culture: A rationale for poor response of patients with Crohn’s disease to current approved therapy. BMJ Open Gastroenterol. 2018, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qasem, A.; Elkamel, E.; Naser, S.A. Anti-MAP Triple Therapy Supports Immunomodulatory Therapeutic Response in Crohn’s Disease through Downregulation of NF-κB Activation in the Absence of MAP Detection. Biomedicines 2020, 8, 513. [Google Scholar] [CrossRef]
- Radek, K.A.; Elias, P.M.; Taupenot, L.; Mahata, S.K.; O’Connor, D.T.; Gallo, R.L. Neuroendocrine nicotinic receptor activation increases susceptibility to bacterial infections by suppressing antimicrobial peptide production. Cell Host Microbe 2010, 7, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhou, X.-D.; Kolosov, V.P.; Perelman, J.M. Nicotine reduces TNF-α expression through a α7 nAChR/MyD88/NF-ĸB pathway in HBE16 airway epithelial cells. Cell. Physiol. Biochem. 2011, 27, 605–612. [Google Scholar] [CrossRef]
- Gwilt, C.R.; Donnelly, L.E.; Rogers, D.F. The non-neuronal cholinergic system in the airways: An unappreciated regulatory role in pulmonary inflammation? Pharmacol. Ther. 2007, 115, 208–222. [Google Scholar] [CrossRef]
- Chong, I.-W.; Lin, S.-R.; Hwang, J.-J.; Huang, M.-S.; Wang, T.-H.; Hung, J.-Y.; Paulauskis, J.D. Expression and regulation of the macrophage inflammatory protein-1alpha gene by nicotine in rat alveolar macrophages. Eur. Cytokine Netw. 2002, 13, 242–249. [Google Scholar]
- Qasem, A.; Abdel-Aty, A.; Abu-Suwa, H.; Naser, A.S. Oxidative stress due to Mycobacterium avium subspecies paratuberculosis (MAP) infection upregulates selenium-dependent GPx activity. Gut Pathog. 2016, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Qasem, A.; Naser, A.E.; Naser, S.A. The alternate effects of anti-TNFα therapeutics and their role in mycobacterial granulomatous infection in Crohn’s disease. Expert Rev. Anti-Infect. Ther. 2017, 15, 637–643. [Google Scholar] [CrossRef]
- Qasem, A.; Ramesh, S.; Naser, S.A. Genetic polymorphisms in tumour necrosis factor receptors (TNFRSF1A/1B) illustrate differential treatment response to TNFα inhibitors in patients with Crohn’s disease. BMJ Open Gastroenterol. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Baik, Y.H.; Cho, J.H.; Kim, S.; Lee, K.-S.; Han, J.-S. Inhibition of lipopolysaccharide-induced nitric oxide synthesis by nicotine through S6K1-p42/44 MAPK pathway and STAT3 (Ser 727) phosphorylation in Raw 264.7 cells. Cytokine 2008, 44, 126–134. [Google Scholar] [CrossRef]
- Takahashi, H.K.; Iwagaki, H.; Hamano, R.; Kanke, T.; Liu, K.; Sadamori, H.; Yagi, T.; Yoshino, T.; Tanaka, N.; Nishibori, M. The immunosuppressive effects of nicotine during human mixed lymphocyte reaction. Eur. J. Pharmacol. 2007, 559, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Feldman, N.E.; Chmura, K.; Ovrutsky, A.R.; Su, W.-L.; Griffin, L.; Pyeon, D.; McGibney, M.T.; Strand, M.J.; Numata, M.; et al. Inhibition of nuclear factor-kappa B activation decreases survival of Mycobacterium tuberculosis in human macrophages. PLoS ONE 2013, 8, e61925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.H.; Dadgostar, H.; Cheng, Q.; Shu, J.; Cheng, G. NF-κB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 9136–9141. [Google Scholar] [CrossRef] [Green Version]
- AlQasrawi, D.; Naser, S.A. Nicotine Modulates MyD88-Dependent Signaling Pathway in Macrophages during Mycobacterial Infection. Microorganisms 2020, 8, 1804. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Mishra, B.B.; Jordao, L.; Elliott, E.; Anes, E.; Griffiths, G. NF-κB activation controls phagolysosome fusion-mediated killing of mycobacteria by macrophages. J. Immunol. 2008, 181, 2651–2663. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, M.G.; Gonzalez, A.P.; Anes, E.; Griffiths, G. Role of lipids in killing mycobacteria by macrophages: Evidence for NF-κB-dependent and-independent killing induced by different lipids. Cell. Microbiol. 2009, 11, 406–420. [Google Scholar] [CrossRef]
- Rode, A.K.O.; Kongsbak, M.; Hansen, M.M.; Lopez, D.V.; Levring, T.B.; Woetmann, A.; Ødum, N.; Bonefeld, C.M.; Geisler, C. Vitamin D counteracts Mycobacterium tuberculosis-induced cathelicidin downregulation in dendritic cells and allows Th1 differentiation and IFNγ secretion. Front. Immunol. 2017, 8, 656. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer Sequence (5′→3′) | Reverse Primer Sequence (5′→3′) |
|---|---|---|
| GAPDH | 5′-CTTTTGCAGACCACAGTCCATG-3′ | 5′-TTTTCTAGACGGCAGGTCAGG-3′ |
| CHRNA7 | 5′-GCTCGTCACGTGGAGAGG-3′ | 5′-GGGAGGCAGTGGCTTTACC-3′ |
| REL-B | 5′-CGCGATCGTCCACCAGA-3′ | 5′-AGACTACTCCGGGACCGC-3′ |
| NFκB2 | 5′-GCCCACCCCCATTTAGATCTG-3′ | 5′-TGAGAGCATCTGCGAGCATAC-3′ |
| Bcl-2 | 5′-CGCGATCGTCCACCAGA-3′ | 5′-AGACTACTCCGGGACCGC-3′ |
| LC-3b | 5′-CCACAGCTAGCAGCTGAACT-3′ | 5′-GGCTGTCTGGTGATTCCTGTAAA-3′ |
| Cathelicidin | 5′-AGGGATGGGTGGATCAGGAA-3′ | 5′-CGAAGCACAGCTTCCTTGTAG-3′ |
| Treatment Group | Phosphorylated NF-κB Activity Score ± SD (%) | Fold Change in Phosphorylated NF-κB Activity |
|---|---|---|
| Control | 35.44 ± 1.07 | - |
| Nicotine (4 µg/mL) | 68.78 ± 6.1 | 2.44 |
| MAP | 44.45 ± 2.7 | 1.25 |
| MAP + Nicotine (4 µg/mL) | 65.20 ± 10.8 | 1.89 |
| MTB | 41.29 ± 4.4 | 1.16 |
| MTB + Nicotine (4 µg/mL) | 73.12 ± 23.3 | 2.06 |
| LPS | 23.70 ± 2.4 | 0.66 |
| LPS + Nicotine (4 µg/mL) | 58.41 ± 12.1 | 1.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AlQasrawi, D.; Naser, E.; Naser, S.A. Nicotine Increases Macrophage Survival through α7nAChR/NF-κB Pathway in Mycobacterium avium paratuberculosis Infection. Microorganisms 2021, 9, 1086. https://doi.org/10.3390/microorganisms9051086
AlQasrawi D, Naser E, Naser SA. Nicotine Increases Macrophage Survival through α7nAChR/NF-κB Pathway in Mycobacterium avium paratuberculosis Infection. Microorganisms. 2021; 9(5):1086. https://doi.org/10.3390/microorganisms9051086
Chicago/Turabian StyleAlQasrawi, Dania, Ebraheem Naser, and Saleh A. Naser. 2021. "Nicotine Increases Macrophage Survival through α7nAChR/NF-κB Pathway in Mycobacterium avium paratuberculosis Infection" Microorganisms 9, no. 5: 1086. https://doi.org/10.3390/microorganisms9051086
APA StyleAlQasrawi, D., Naser, E., & Naser, S. A. (2021). Nicotine Increases Macrophage Survival through α7nAChR/NF-κB Pathway in Mycobacterium avium paratuberculosis Infection. Microorganisms, 9(5), 1086. https://doi.org/10.3390/microorganisms9051086