
Global Genomic Analysis of SARS-CoV-2 RNA Dependent RNA Polymerase Evolution and Antiviral Drug Resistance

 ,
,  , , , , ,
, , , , ,  , , , ,
, , , ,  , , add
Show full author list
, , add
Show full author list
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Inferring Signatures of Selection in nsp12
2.2. Global Dataset, Spatiotemporal Trends and Genome Entropy
2.3. Genome Drafting Tool and Availability
2.4. Patients, Samples, and Diagnosis
2.4.1. Basel University Hospital Single Time Point Open Cohort
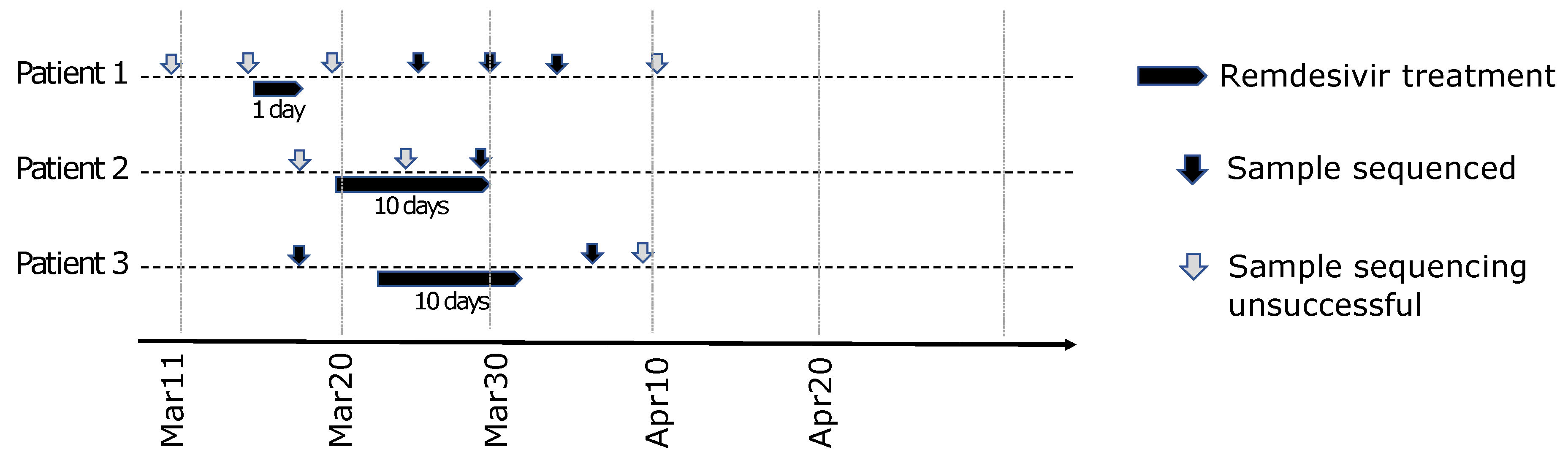
2.4.2. Longitudinal Cohort and Remdesivir Treatment
2.4.3. Diagnosis and Sequencing
2.5. Statistics and Visualization
2.5.1. Genome Diversity and Statistical Test
2.5.2. Associated Mutation Inference, Structural Fitting and Visualization
2.5.3. Stability Analysis of Mutation Combinations
3. Results
3.1. Selection Analysis of nsp12
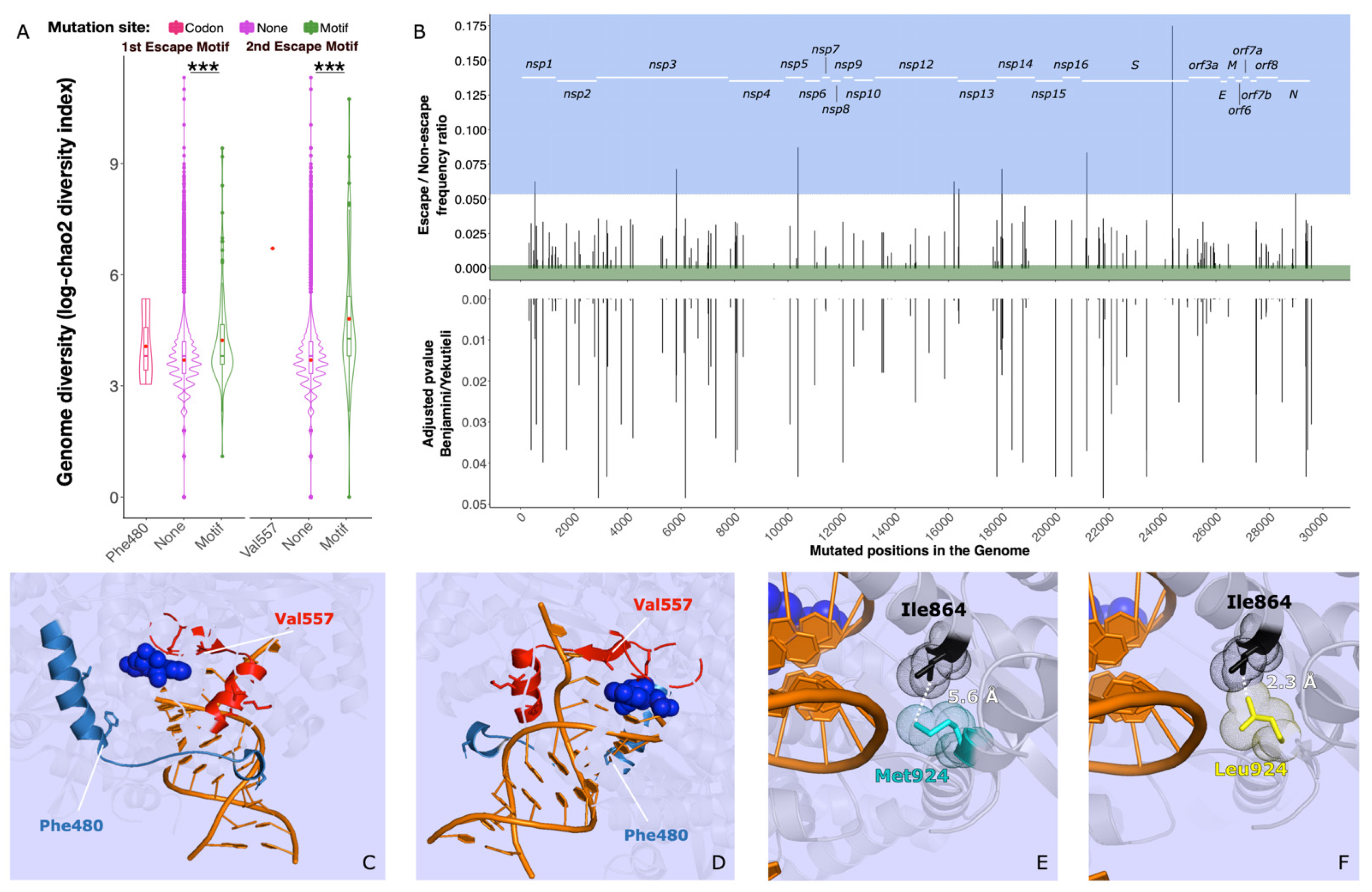
3.2. Identification of Key Residues and Motifs for Drug Binding Sites
3.3. Potential Escape Mutants in the Global Dataset
3.4. Potential Escape Mutants during Remdesivir Treatment
3.5. Associated Mutations and Stability Loss
4. Discussion
5. Conclusions
6. Ethics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, X.; Wang, X.-J. Potential Inhibitors against 2019-NCoV Coronavirus M Protease from Clinically Approved Medicines. J. Genet. Genom. 2020, 47, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.M.; Sisk, J.M.; Mingo, R.M.; Nelson, E.A.; White, J.M.; Frieman, M.B. Abelson Kinase Inhibitors Are Potent Inhibitors of Severe Acute Respiratory Syndrome Coronavirus and Middle East Respiratory Syndrome Coronavirus Fusion. J. Virol. 2016, 90, 8924–8933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stader, F.; Khoo, S.; Stoeckle, M.; Back, D.; Hirsch, H.H.; Battegay, M.; Marzolini, C. Stopping Lopinavir/Ritonavir in COVID-19 Patients: Duration of the Drug Interacting Effect. J. Antimicrob. Chemother. 2020, 75, 3084–3086. [Google Scholar] [CrossRef]
- Shannon, A.; Le, N.T.-T.; Selisko, B.; Eydoux, C.; Alvarez, K.; Guillemot, J.-C.; Decroly, E.; Peersen, O.; Ferron, F.; Canard, B. Remdesivir and SARS-CoV-2: Structural Requirements at Both Nsp12 RdRp and Nsp14 Exonuclease Active-Sites. Antiviral Res. 2020, 178, 104793. [Google Scholar] [CrossRef]
- Selisko, B.; Papageorgiou, N.; Ferron, F.; Canard, B. Structural and Functional Basis of the Fidelity of Nucleotide Selection by Flavivirus RNA-Dependent RNA Polymerases. Viruses 2018, 10, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp): A Molecular Docking Study. Life Sci. 2020, 253, 117592. [Google Scholar] [CrossRef]
- Chien, M.; Anderson, T.K.; Jockusch, S.; Tao, C.; Li, X.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide Analogues as Inhibitors of SARS-CoV-2 Polymerase, a Key Drug Target for COVID-19. J. Proteome Res. 2020, 19, 4690–4697. [Google Scholar] [CrossRef]
- Jockusch, S.; Tao, C.; Li, X.; Chien, M.; Kumar, S.; Morozova, I.; Kalachikov, S.; Russo, J.J.; Ju, J. Sofosbuvir Terminated RNA Is More Resistant to SARS-CoV-2 Proofreader than RNA Terminated by Remdesivir. Sci. Rep. 2020, 10, 16577. [Google Scholar] [CrossRef]
- Bartholomeusz, A.; Tehan, B.G.; Chalmers, D.K. Comparisons of the HBV and HIV Polymerase, and Antiviral Resistance Mutations. Antivir. Ther. 2004, 9, 149–160. [Google Scholar]
- Eltahla, A.A.; Luciani, F.; White, P.A.; Lloyd, A.R.; Bull, R.A. Inhibitors of the Hepatitis C Virus Polymerase; Mode of Action and Resistance. Viruses 2015, 7, 5206–5224. [Google Scholar] [CrossRef] [Green Version]
- Bloom, J.D.; Gong, L.I.; Baltimore, D. Permissive Secondary Mutations Enable the Evolution of Influenza Oseltamivir Resistance. Science 2010, 328, 1272–1275. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Leveque, V.; Le Pogam, S.; Ma, H.; Philipp, F.; Inocencio, N.; Smith, M.; Alker, A.; Kang, H.; Najera, I.; et al. Selected Replicon Variants with Low-Level in Vitro Resistance to the Hepatitis C Virus NS5B Polymerase Inhibitor PSI-6130 Lack Cross-Resistance with R1479. Antimicrob. Agents Chemother. 2008, 52, 4356–4369. [Google Scholar] [CrossRef] [Green Version]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio 2018, 9, e00221-18. [Google Scholar] [CrossRef] [Green Version]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Spinner, C.D.; Gottlieb, R.L.; Criner, G.J.; Arribas López, J.R.; Cattelan, A.M.; Soriano Viladomiu, A.; Ogbuagu, O.; Malhotra, P.; Mullane, K.M.; Castagna, A.; et al. Effect of Remdesivir vs. Standard Care on Clinical Status at 11 Days in Patients With Moderate COVID-19: A Randomized Clinical Trial. JAMA 2020, 324, 1048–1057. [Google Scholar] [CrossRef]
- WHO Solidarity Trial Consortium; Pan, H.; Peto, R.; Henao-Restrepo, A.-M.; Preziosi, M.-P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernández García, C.; Kieny, M.-P.; et al. Repurposed Antiviral Drugs for Covid-19 - Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2020, 384, 497–511. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in Adults with Severe COVID-19: A Randomised, Double-Blind, Placebo-Controlled, Multicentre Trial. Lancet Lond. Engl. 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 Mutation Hot Spots Include a Novel RNA-Dependent-RNA Polymerase Variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Eskier, D.; Suner, A.; Karakülah, G.; Oktay, Y. Mutation Density Changes in SARS-CoV-2 Are Related to the Pandemic Stage but to a Lesser Extent in the Dominant Strain with Mutations in Spike and RdRp. PeerJ 2020, 8, e9703. [Google Scholar] [CrossRef]
- Martinot, M.; Jary, A.; Fafi-Kremer, S.; Leducq, V.; Delagreverie, H.; Garnier, M.; Pacanowski, J.; Mékinian, A.; Pirenne, F.; Tiberghien, P.; et al. Remdesivir Failure with SARS-CoV-2 RNA-Dependent RNA-Polymerase Mutation in a B-Cell Immunodeficient Patient with Protracted Covid-19. Clin. Infect. Dis. 2020, ciaa1474. [Google Scholar] [CrossRef]
- Ibrahim, E.S.; Kashef, M.T.; Essam, T.M.; Ramadan, M.A. A Degradome-Based Polymerase Chain Reaction to Resolve the Potential of Environmental Samples for 2,4-Dichlorophenol Biodegradation. Curr. Microbiol. 2017, 74, 1365–1372. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000; ISBN 978-0-19-535051-7. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watterson, G.A. On the Number of Segregating Sites in Genetical Models without Recombination. Theor. Popul. Biol. 1975, 7, 256–276. [Google Scholar] [CrossRef]
- Wilson, D.J. CRyPTIC Consortium GenomegaMap: Within-Species Genome-Wide DN/DS Estimation from over 10,000 Genomes. Mol. Biol. Evol. 2020, 37, 2450–2460. [Google Scholar] [CrossRef] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Stange, M.; Mari, A.; Roloff, T.; Seth-Smith, H.M.; Schweitzer, M.; Brunner, M.; Leuzinger, K.; Søgaard, K.K.; Gensch, A.; Tschudin-Sutter, S.; et al. SARS-CoV-2 Outbreak in a Tri-National Urban Area Is Dominated by a B.1 Lineage Variant Linked to a Mass Gathering Event. PLoS Pathog. 2021, 17, e1009374. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-Sites: Rapid Efficient Extraction of SNPs from Multi-FASTA Alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Wickham, H. Reshaping Data with the Reshape Package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating High-Throughput Genomic Analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Grolemund, G.; Wickham, H. Dates and Times Made Easy with Lubridate. J. Stat. Softw. 2011, 40, 1–25. [Google Scholar] [CrossRef]
- Wickham, H.; Chang, W. RStudio Ggplot2: Create Elegant Data Visualisations Using the Grammar of Graphics; 2016. Available online: https://cran.r-project.org/web/packages/ggplot2/index.html (accessed on 17 May 2021).
- Chao, A.; Colwell, R.K.; Lin, C.-W.; Gotelli, N.J. Sufficient Sampling for Asymptotic Minimum Species Richness Estimators. Ecology 2009, 90, 1125–1133. [Google Scholar] [CrossRef]
- Chao, A. Estimating the Population Size for Capture-Recapture Data with Unequal Catchability. Biometrics 1987, 43, 783–791. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The Control of the False Discovery Rate in Multiple Testing under Dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Fisher, R.A. Statistical Methods for Research Workers. In Breakthroughs in Statistics: Methodology and Distribution; Kotz, S., Johnson, N.L., Eds.; Springer Series in Statistics; Springer: New York, NY, USA, 1992; pp. 66–70. ISBN 978-1-4612-4380-9. [Google Scholar]
- Hyndman, R.J.; Fan, Y. Sample Quantiles in Statistical Packages. Am. Stat. 1996, 50, 361–365. [Google Scholar] [CrossRef]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural Basis for Inhibition of the RNA-Dependent RNA Polymerase from SARS-CoV-2 by Remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A.; Danielson, M.L. Computer-Aided Drug Design Platform Using PyMOL. J. Comput. Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX Web Server: An Online Force Field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buß, O.; Rudat, J.; Ochsenreither, K. FoldX as Protein Engineering Tool: Better Than Random Based Approaches? Comput. Struct. Biotechnol. J. 2018, 16, 25–33. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, Y.; Ge, J.; Zheng, L.; Gao, Y.; Wang, T.; Jia, Z.; Wang, H.; Huang, Y.; Li, M.; et al. Architecture of a SARS-CoV-2 Mini Replication and Transcription Complex. Nat. Commun. 2020, 11, 5874. [Google Scholar] [CrossRef]
- Naydenova, K.; Muir, K.W.; Wu, L.-F.; Zhang, Z.; Coscia, F.; Peet, M.J.; Castro-Hartmann, P.; Qian, P.; Sader, K.; Dent, K.; et al. Structure of the SARS-CoV-2 RNA-Dependent RNA Polymerase in the Presence of Favipiravir-RTP. Proc. Natl. Acad. Sci. USA 2021, 118, e2021946118. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, J.; Wang, H.; Gao, Y.; Liu, Q.; Mu, A.; Ji, W.; Yan, L.; Zhu, Y.; Zhu, C.; et al. Structural Basis for RNA Replication by the SARS-CoV-2 Polymerase. Cell 2020, 182, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of Replicating SARS-CoV-2 Polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-Dependent RNA Polymerase from COVID-19 Virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor Binding and Priming of the Spike Protein of SARS-CoV-2 for Membrane Fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Lu, J.; Zhang, J.; Johnson, R.I.; McKay, L.G.A.; Storm, N.; Lavine, C.L.; Peng, H.; Cai, Y.; Rits-Volloch, S.; et al. A Trimeric Human Angiotensin-Converting Enzyme 2 as an Anti-SARS-CoV-2 Agent. Nat. Struct. Mol. Biol. 2021, 28, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct Conformational States of SARS-CoV-2 Spike Protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.-L.; Zhang, X.-L.; Zhao, X.-N.; Li, C.-B.; Lei, J.; Kou, Z.-Q.; Sun, W.-K.; Hang, Y.; Gao, F.; Ji, S.-X.; et al. Transmission Potential of Asymptomatic and Paucisymptomatic Severe Acute Respiratory Syndrome Coronavirus 2 Infections: A 3-Family Cluster Study in China. J. Infect. Dis. 2020, 221, 1948–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, B.H.; Poon, A.F.Y.; Whitlock, M.C. Compensatory Mutations Are Repeatable and Clustered within Proteins. Proc. R. Soc. B Biol. Sci. 2009, 276, 1823–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-NCoV) in Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Götte, M. Remdesivir Is a Direct-Acting Antiviral That Inhibits RNA-Dependent RNA Polymerase from Severe Acute Respiratory Syndrome Coronavirus 2 with High Potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef] [Green Version]
- Pruijssers, A.J.; George, A.S.; Schäfer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon, K.H.; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir Inhibits SARS-CoV-2 in Human Lung Cells and Chimeric SARS-CoV Expressing the SARS-CoV-2 RNA Polymerase in Mice. Cell Rep. 2020, 32, 107940. [Google Scholar] [CrossRef]
- Williamson, B.N.; Feldmann, F.; Schwarz, B.; Meade-White, K.; Porter, D.P.; Schulz, J.; van Doremalen, N.; Leighton, I.; Yinda, C.K.; Pérez-Pérez, L.; et al. Clinical Benefit of Remdesivir in Rhesus Macaques Infected with SARS-CoV-2. Nature 2020, 585, 273–276. [Google Scholar] [CrossRef]
- Lamb, Y.N. Remdesivir: First Approval. Drugs 2020, 80, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Götte, M. The Distinct Contributions of Fitness and Genetic Barrier to the Development of Antiviral Drug Resistance. Curr. Opin. Virol. 2012, 2, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F. Compensatory Mutations and Epistasis for Protein Function. Curr. Opin. Struct. Biol. 2018, 50, 18–25. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mari, A.; Roloff, T.; Stange, M.; Søgaard, K.K.; Asllanaj, E.; Tauriello, G.; Alexander, L.T.; Schweitzer, M.; Leuzinger, K.; Gensch, A.; et al. Global Genomic Analysis of SARS-CoV-2 RNA Dependent RNA Polymerase Evolution and Antiviral Drug Resistance. Microorganisms 2021, 9, 1094. https://doi.org/10.3390/microorganisms9051094
Mari A, Roloff T, Stange M, Søgaard KK, Asllanaj E, Tauriello G, Alexander LT, Schweitzer M, Leuzinger K, Gensch A, et al. Global Genomic Analysis of SARS-CoV-2 RNA Dependent RNA Polymerase Evolution and Antiviral Drug Resistance. Microorganisms. 2021; 9(5):1094. https://doi.org/10.3390/microorganisms9051094
Chicago/Turabian StyleMari, Alfredo, Tim Roloff, Madlen Stange, Kirstine K. Søgaard, Erblin Asllanaj, Gerardo Tauriello, Leila Tamara Alexander, Michael Schweitzer, Karoline Leuzinger, Alexander Gensch, and et al. 2021. "Global Genomic Analysis of SARS-CoV-2 RNA Dependent RNA Polymerase Evolution and Antiviral Drug Resistance" Microorganisms 9, no. 5: 1094. https://doi.org/10.3390/microorganisms9051094
APA StyleMari, A., Roloff, T., Stange, M., Søgaard, K. K., Asllanaj, E., Tauriello, G., Alexander, L. T., Schweitzer, M., Leuzinger, K., Gensch, A., Martinez, A. E., Bielicki, J., Pargger, H., Siegemund, M., Nickel, C. H., Bingisser, R., Osthoff, M., Bassetti, S., Sendi, P., ... Egli, A. (2021). Global Genomic Analysis of SARS-CoV-2 RNA Dependent RNA Polymerase Evolution and Antiviral Drug Resistance. Microorganisms, 9(5), 1094. https://doi.org/10.3390/microorganisms9051094