
Reversion of Gut Microbiota during the Recovery Phase in Patients with Asymptomatic or Mild COVID-19: Longitudinal Study

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
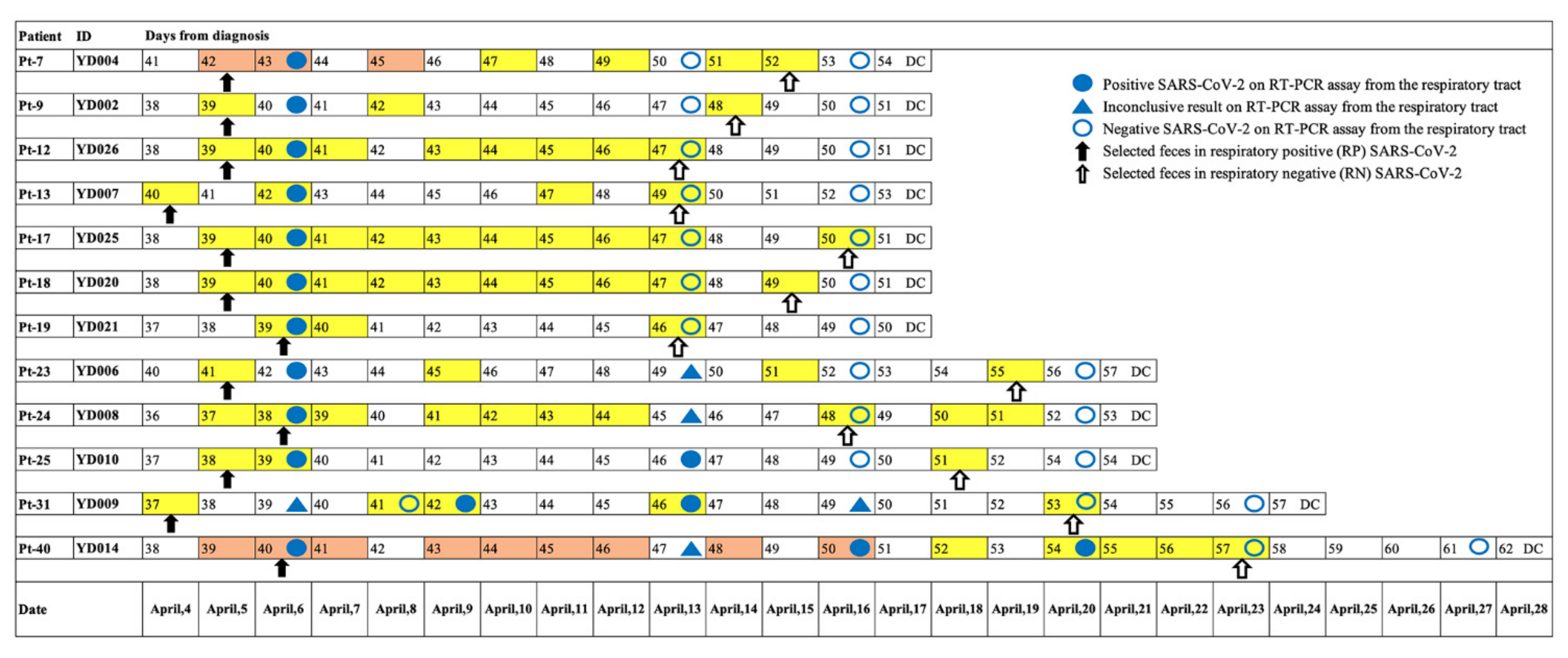
2.2. Collection of Respiratory/Fecal Samples and Clinical Data
2.3. DNA Extraction, PCR, and Sequencing of the 16S rRNA Gene
2.4. Statistical Analysis
3. Results
3.1. Clinical Characteristics of Patients with Asymptomatic or Mild COVID-19
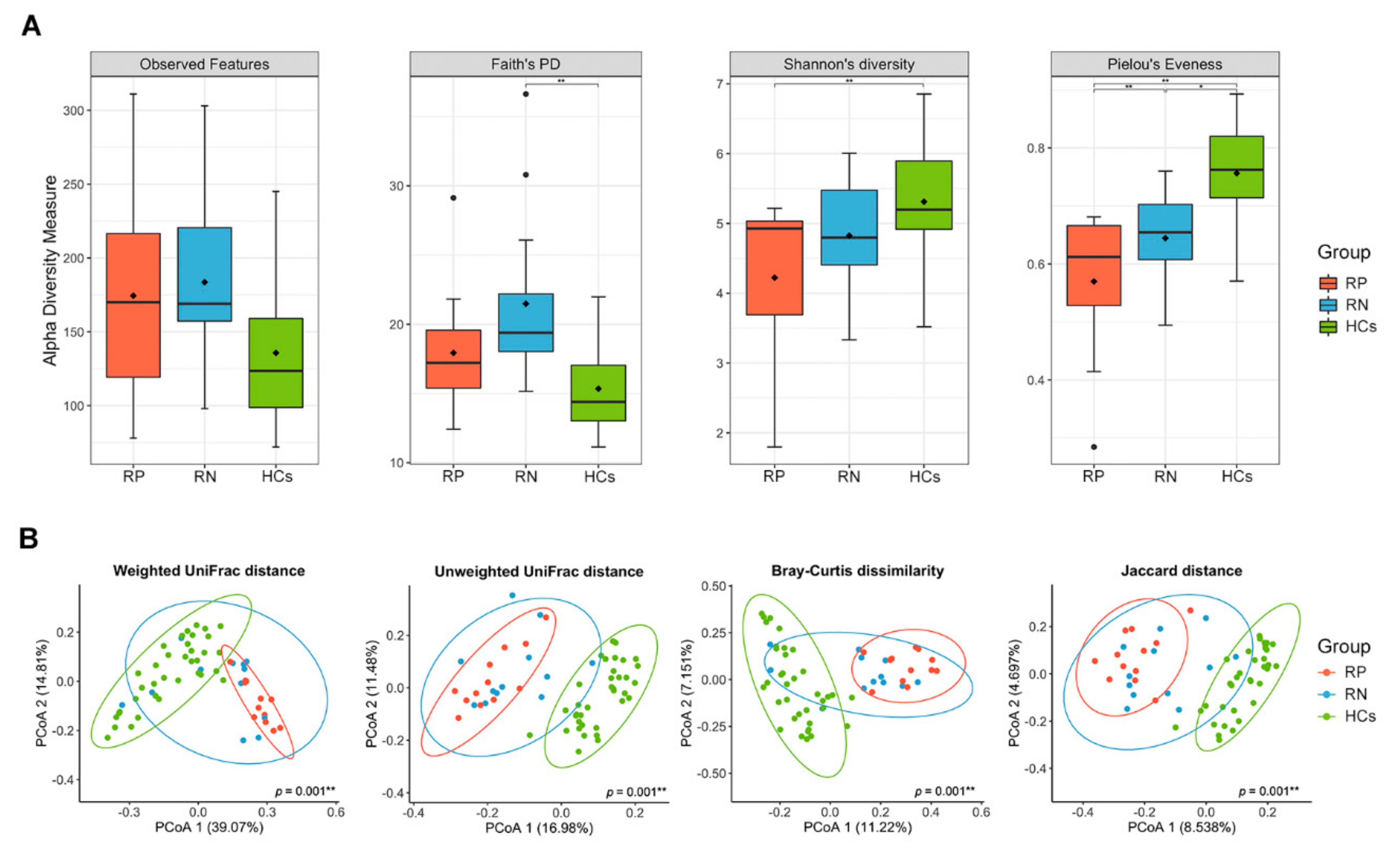
3.2. Gut Microbiota in RP SARS-CoV-2 and RN SARS-CoV-2 Significantly Differ on Measures of Alpha or Beta Diversity
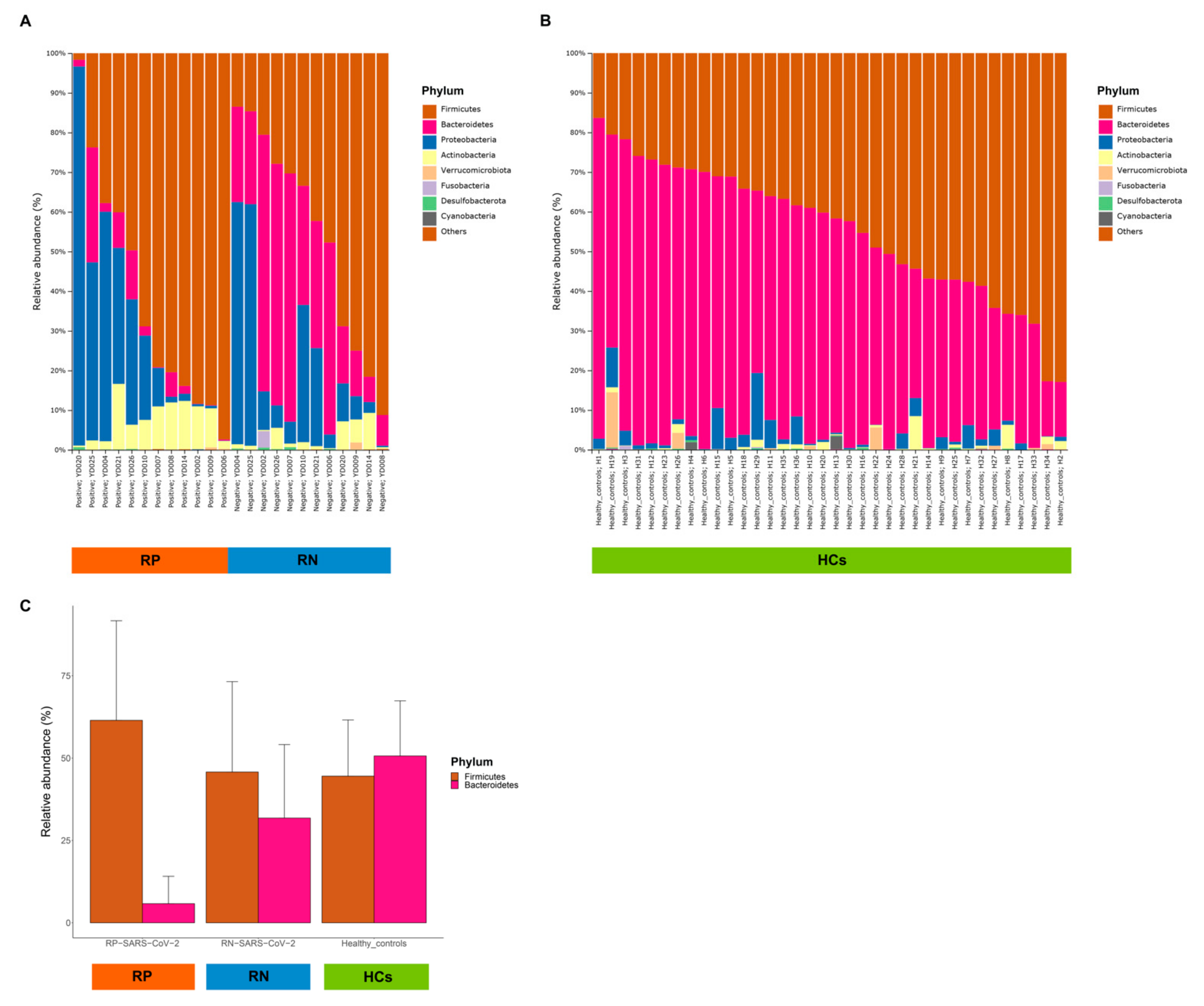
3.3. Association among Taxonomy Profiles, Infection Status of COVID-19, and Healthy Controls (HCs)
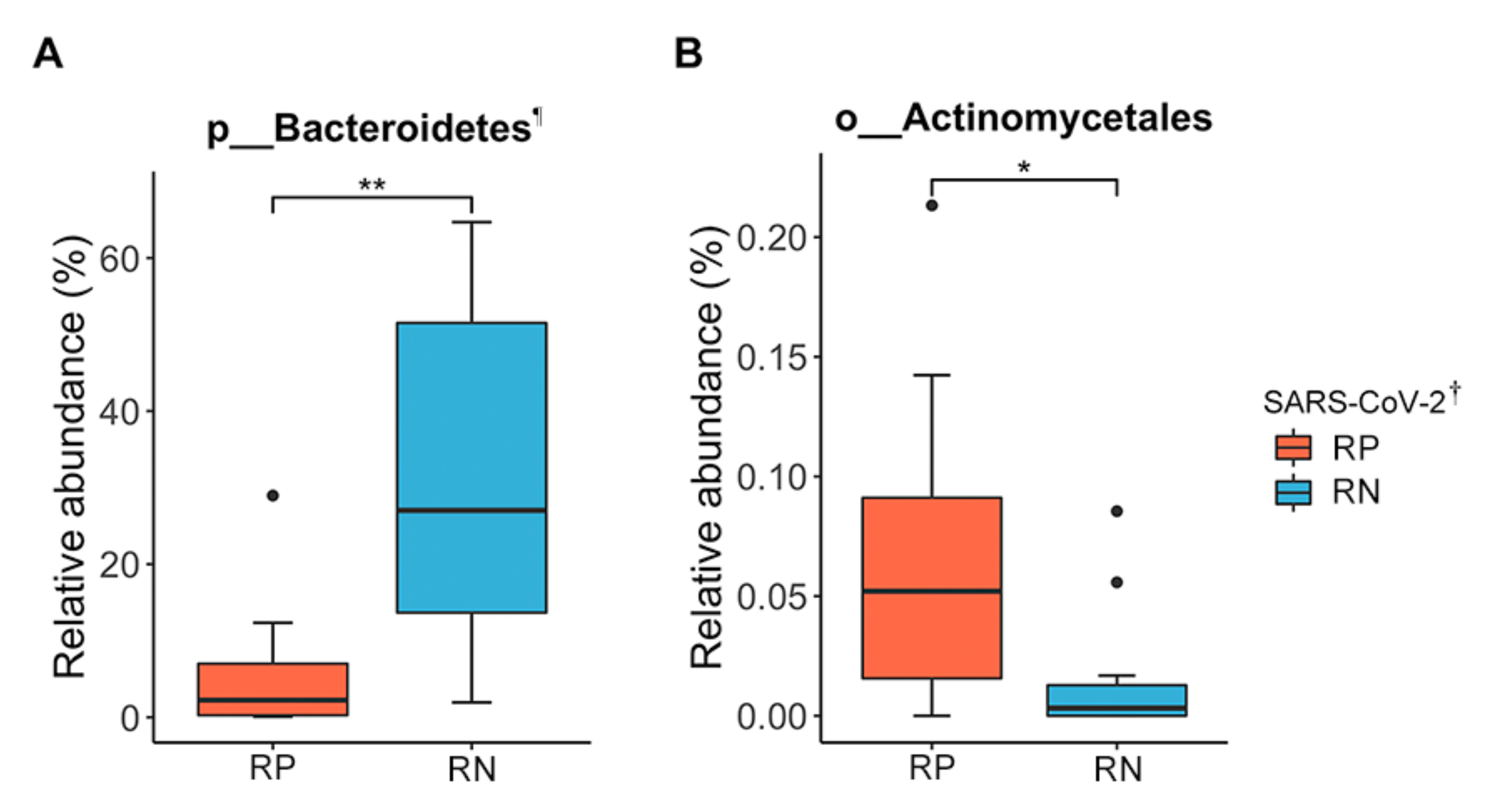
Difference in Taxonomic Composition of Gut Microbiota between RP SARS-CoV-2 and RN SARS-CoV-2 in Patients with COVID-19
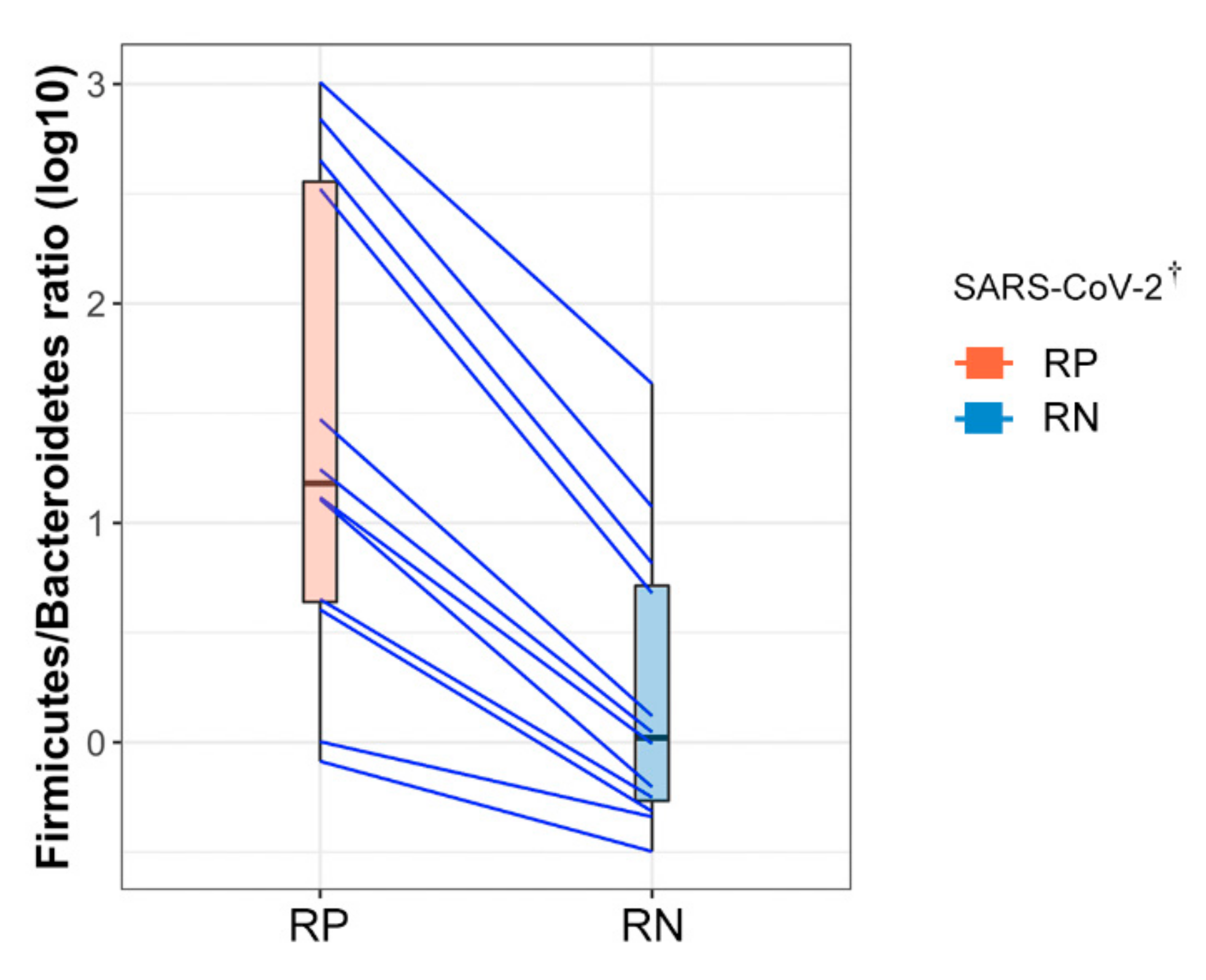
3.4. Firmicutes/Bacteroidetes (F/B) Ratio Was Markedly Increased in RP SARS-CoV-2
3.5. Comparison of Patients with COVID-19 and HCs
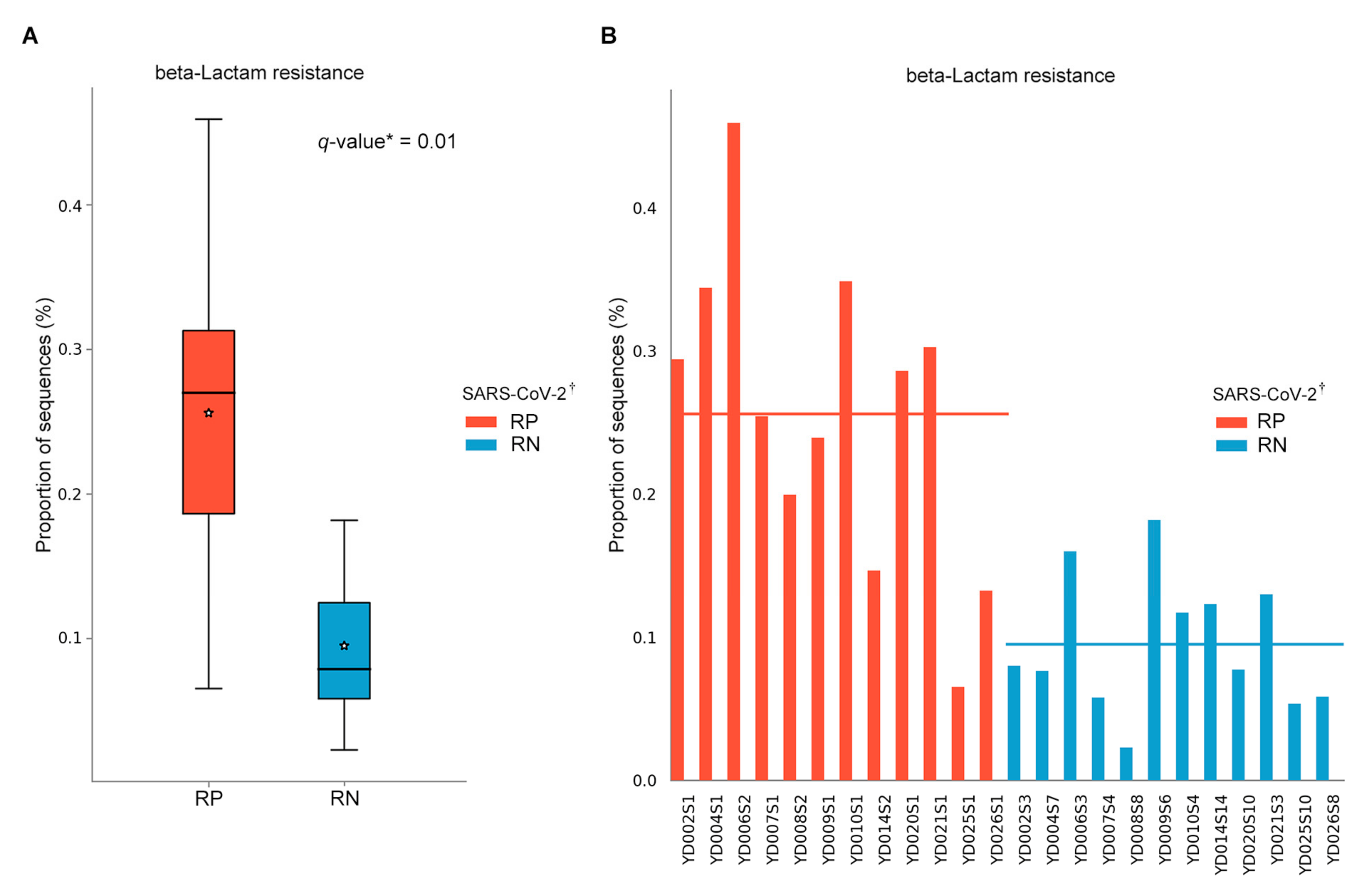
3.6. Predicted Metabolic Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Tang, M.; Zheng, X.; Liu, Y.; Li, X.; Shan, H. Evidence for Gastrointestinal Infection of SARS-CoV-2. Gastroenterology 2020, 158, 1831–1833. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.S.; Hung, I.F.N.; Chan, P.P.Y.; Lung, K.C.; Tso, E.; Liu, R.; Ng, Y.Y.; Chu, M.Y.; Chung, T.W.H.; Tam, A.R.; et al. Gastrointestinal Manifestations of SARS-CoV-2 Infection and Virus Load in Fecal Samples From a Hong Kong Cohort: Systematic Review and Meta-analysis. Gastroenterology 2020, 159, 81–95. [Google Scholar] [CrossRef]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Muller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, X.; Zhu, B.; Liang, H.; Fang, C.; Gong, Y.; Guo, Q.; Sun, X.; Zhao, D.; Shen, J.; et al. Characteristics of pediatric SARS-CoV-2 infection and potential evidence for persistent fecal viral shedding. Nat. Med. 2020, 26, 502–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory Viral Infection-Induced Microbiome Alterations and Secondary Bacterial Pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildiz, S.; Mazel-Sanchez, B.; Kandasamy, M.; Manicassamy, B.; Schmolke, M. Influenza A virus infection impacts systemic microbiota dynamics and causes quantitative enteric dysbiosis. Microbiome 2018, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Chen, Y.; Wu, Z.; Chen, Y.; Gao, H.; Lv, L.; Guo, F.; Zhang, X.; Luo, R.; Huang, C.; et al. Alterations of the Gut Microbiota in Patients With Coronavirus Disease 2019 or H1N1 Influenza. Clin. Infect. Dis. 2020, 71, 2669–2678. [Google Scholar] [CrossRef]
- Ko, J.H.; Joo, E.J.; Park, S.J.; Baek, J.Y.; Kim, W.D.; Jee, J.; Kim, C.J.; Jeong, C.; Kim, Y.J.; Shon, H.J.; et al. Neutralizing Antibody Production in Asymptomatic and Mild COVID-19 Patients, in Comparison with Pneumonic COVID-19 Patients. J. Clin. Med. 2020, 9, 2668. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Lee, C.W.; Park, D.I.; Woo, H.Y.; Cheong, H.S.; Shin, H.C.; Ahn, K.; Kwon, M.J.; Joo, E.J. Detection of SARS-CoV-2 in Fecal Samples From Patients With Asymptomatic and Mild COVID-19 in Korea. Clin. Gastroenterol. Hepatol. 2020, S1542–3565, 30777-1. [Google Scholar] [CrossRef]
- Byambasuren, O.; Cardona, M.; Bell, K.; Clark, J.; McLaws, M.-L.; Glasziou, P. Estimating the extent of asymptomatic COVID-19 and its potential for community transmission: Systematic review and meta-analysis. Off. J. Assoc. Med. Microbiol. Infect. Dis. Can. 2020. [Google Scholar] [CrossRef]
- Kim, H.N.; Yun, Y.; Ryu, S.; Chang, Y.; Kwon, M.J.; Cho, J.; Shin, H.; Kim, H.L. Correlation between gut microbiota and personality in adults: A cross-sectional study. Brain Behav. Immun. 2018, 69, 374–385. [Google Scholar] [CrossRef]
- Yun, Y.; Kim, H.N.; Kim, S.E.; Heo, S.G.; Chang, Y.; Ryu, S.; Shin, H.; Kim, H.L. Comparative analysis of gut microbiota associated with body mass index in a large Korean cohort. BMC Microbiol. 2017, 17, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.N.; Joo, E.J.; Cheong, H.S.; Kim, Y.; Kim, H.L.; Shin, H.; Chang, Y.; Ryu, S. Gut Microbiota and Risk of Persistent Nonalcoholic Fatty Liver Diseases. J. Clin. Med. 2019, 8, 1089. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.; Dillon, M.; Bokulich, N.; Abnet, C.; Al-Ghalith, G.; Alexander, H.; Alm, E.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable, and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Jaccard, P. The Distribution of the Flora in the Alpine Zone.1. N. Phytol. 1912, 11, 37–50. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2: An improved and extensible approach for metagenome inference. BioRxiv 2019, 10, 672295. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candela, M.; Biagi, E.; Maccaferri, S.; Turroni, S.; Brigidi, P. Intestinal microbiota is a plastic factor responding to environmental changes. Trends Microbiol. 2012, 20, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Ma, L.; Ma, Y.; Zhang, F.; Zhao, C.; Nie, Y. Insights into the role of gut microbiota in obesity: Pathogenesis, mechanisms, and therapeutic perspectives. Protein Cell 2018, 9, 397–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, A.; Cole, J.; Brenner, L.N.; Meigs, J.B.; Florez, J.C.; Mercader, J.M. Cardiometabolic Risk Factors for COVID-19 Susceptibility and Severity: A Mendelian Randomization Analysis. PLoS Med. 2021, 18, e1003553. [Google Scholar] [CrossRef]
- Freuer, D.; Linseisen, J.; Meisinger, C. Impact of body composition on COVID-19 susceptibility and severity: A two-sample multivariable Mendelian randomization study. Metabolism 2021, 118, 154732. [Google Scholar] [CrossRef]
- Castaner, O.; Goday, A.; Park, Y.M.; Lee, S.H.; Magkos, F.; Shiow, S.T.E.; Schroder, H. The Gut Microbiome Profile in Obesity: A Systematic Review. Int. J. Endocrinol. 2018, 2018, 4095789. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Deng, H.; Ren, Z.; Zhao, Y.; Yu, S.; Guo, Y.; Dai, J.; Chen, X.; Li, K.; Li, R.; et al. Dynamic Changes in the Microbiome and Mucosal Immune Microenvironment of the Lower Respiratory Tract by Influenza Virus Infection. Front. Microbiol. 2019, 10, 2491. [Google Scholar] [CrossRef] [Green Version]
- Groves, H.T.; Cuthbertson, L.; James, P.; Moffatt, M.F.; Cox, M.J.; Tregoning, J.S. Respiratory Disease following Viral Lung Infection Alters the Murine Gut Microbiota. Front. Immunol. 2018, 9, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sencio, V.; Barthelemy, A.; Tavares, L.P.; Machado, M.G.; Soulard, D.; Cuinat, C.; Queiroz-Junior, C.M.; Noordine, M.L.; Salome-Desnoulez, S.; Deryuter, L.; et al. Gut Dysbiosis during Influenza Contributes to Pulmonary Pneumococcal Superinfection through Altered Short-Chain Fatty Acid Production. Cell Rep. 2020, 30, 2934–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [Green Version]
- de Vrese, M.; Winkler, P.; Rautenberg, P.; Harder, T.; Noah, C.; Laue, C.; Ott, S.; Hampe, J.; Schreiber, S.; Heller, K.; et al. Probiotic bacteria reduced duration and severity but not the incidence of common cold episodes in a double blind, randomized, controlled trial. Vaccine 2006, 24, 6670–6674. [Google Scholar] [CrossRef]
- Vouloumanou, E.K.; Makris, G.C.; Karageorgopoulos, D.E.; Falagas, M.E. Probiotics for the prevention of respiratory tract infections: A systematic review. Int. J. Antimicrob. Agents 2009, 34, 197.e1–197.e10. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Gollwitzer, E.S.; Pattaroni, C.; Lopez-Mejia, I.C.; Riva, E.; Pernot, J.; Ubags, N.; Fajas, L.; Nicod, L.P.; Marsland, B.J. Dietary Fiber Confers Protection against Flu by Shaping Ly6c(-) Patrolling Monocyte Hematopoiesis and CD8(+) T Cell Metabolism. Immunity 2018, 48, 992–1005.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.W.; Wu, X.X.; Jiang, X.G.; Xu, K.J.; Ying, L.J.; Ma, C.L.; Li, S.B.; Wang, H.Y.; Zhang, S.; Gao, H.N.; et al. Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: Retrospective case series. BMJ 2020, 368, m606. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | All Patients (n = 12) |
|---|---|
| Age, median (range) | 26 (18–47) |
| Male, n (%) | 8 (66.7%) |
| Days from diagnosis to quarantine, median (range) | 7 (5.0–18.0) |
| Days from diagnosis to fecal sample examination, median (range) | 38.0 (36.0–41.0) |
| Days from diagnosis to last follow up, median (range) | 53.0 (50.0–62.0) |
| Medical history, n (%) | |
| Digestive 1, n (%) | 4 (33.3%) |
| Others 2, n (%) | 3 (25.0%) |
| BMI (kg/m2), mean ± SD | 23 ± 6.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-N.; Joo, E.-J.; Lee, C.-W.; Ahn, K.-S.; Kim, H.-L.; Park, D.-I.; Park, S.-K. Reversion of Gut Microbiota during the Recovery Phase in Patients with Asymptomatic or Mild COVID-19: Longitudinal Study. Microorganisms 2021, 9, 1237. https://doi.org/10.3390/microorganisms9061237
Kim H-N, Joo E-J, Lee C-W, Ahn K-S, Kim H-L, Park D-I, Park S-K. Reversion of Gut Microbiota during the Recovery Phase in Patients with Asymptomatic or Mild COVID-19: Longitudinal Study. Microorganisms. 2021; 9(6):1237. https://doi.org/10.3390/microorganisms9061237
Chicago/Turabian StyleKim, Han-Na, Eun-Jeong Joo, Chil-Woo Lee, Kwang-Sung Ahn, Hyung-Lae Kim, Dong-Il Park, and Soo-Kyung Park. 2021. "Reversion of Gut Microbiota during the Recovery Phase in Patients with Asymptomatic or Mild COVID-19: Longitudinal Study" Microorganisms 9, no. 6: 1237. https://doi.org/10.3390/microorganisms9061237
APA StyleKim, H. -N., Joo, E. -J., Lee, C. -W., Ahn, K. -S., Kim, H. -L., Park, D. -I., & Park, S. -K. (2021). Reversion of Gut Microbiota during the Recovery Phase in Patients with Asymptomatic or Mild COVID-19: Longitudinal Study. Microorganisms, 9(6), 1237. https://doi.org/10.3390/microorganisms9061237