
Phylogenomic Reconstruction and Metabolic Potential of the Genus Aminobacter

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Genome Sequencing
2.2. Dataset Collection
2.3. Phylogenetic Analysis of the 16S rRNA Gene
2.4. Whole Genome-Based Phylogeny
2.5. Digital DNA–DNA Hybridization (dDDH) and Average Nucleotide Identity (ANI)
2.6. Detection and Structural Analysis of Nodulation and Nitrogen-Fixation Genes
2.7. Detection and Structural Analysis of Methylotrophy Genes
2.8. Detection and Structural Analysis of Genes Implicated in Xenobiotic Degradation
3. Results
3.1. Aminobacter Anthyllidis LMG 26462T Genome Sequence
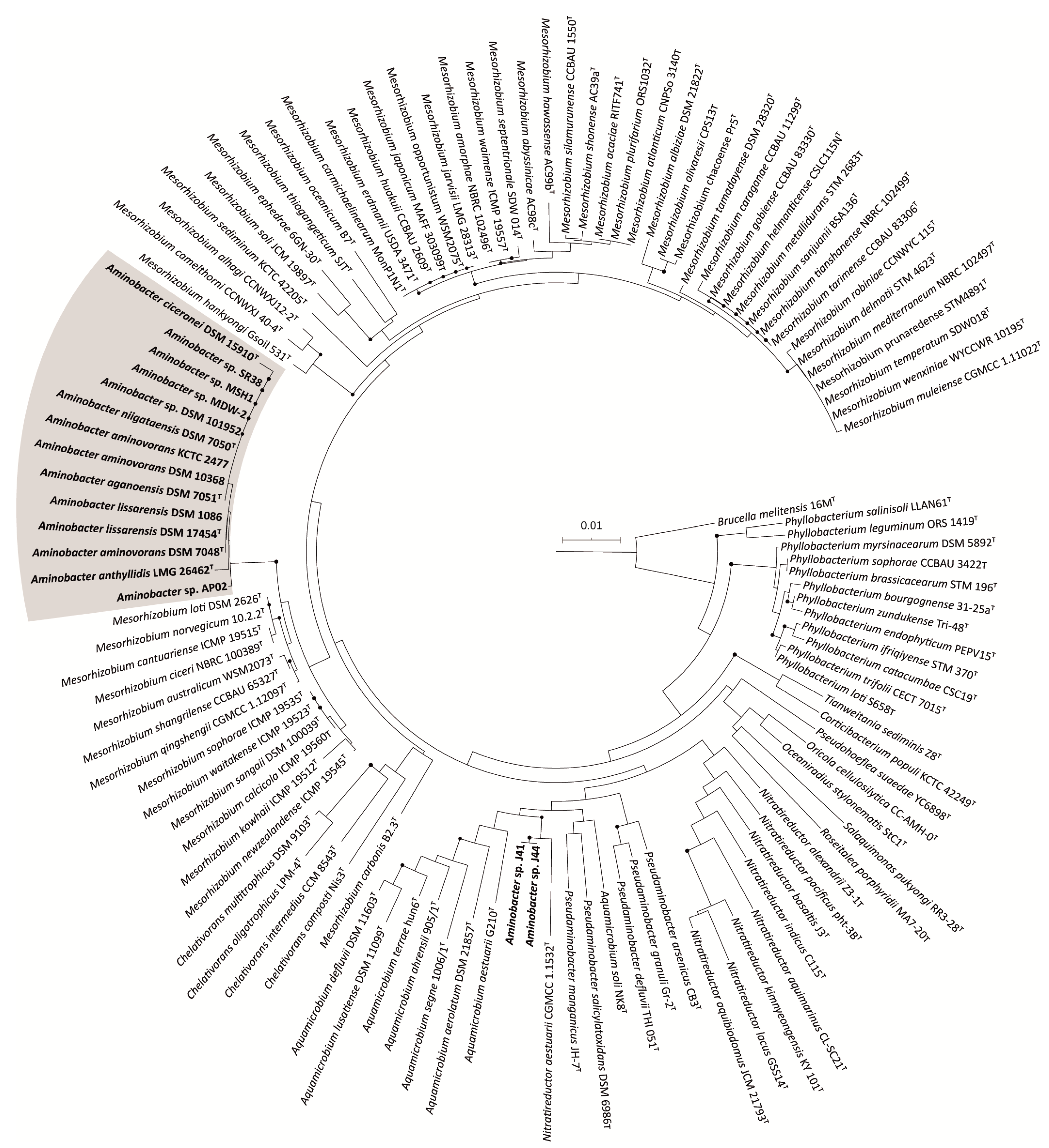
3.2. 16S rRNA Gene-Based Phylogeny of Phyllobacteriaceae
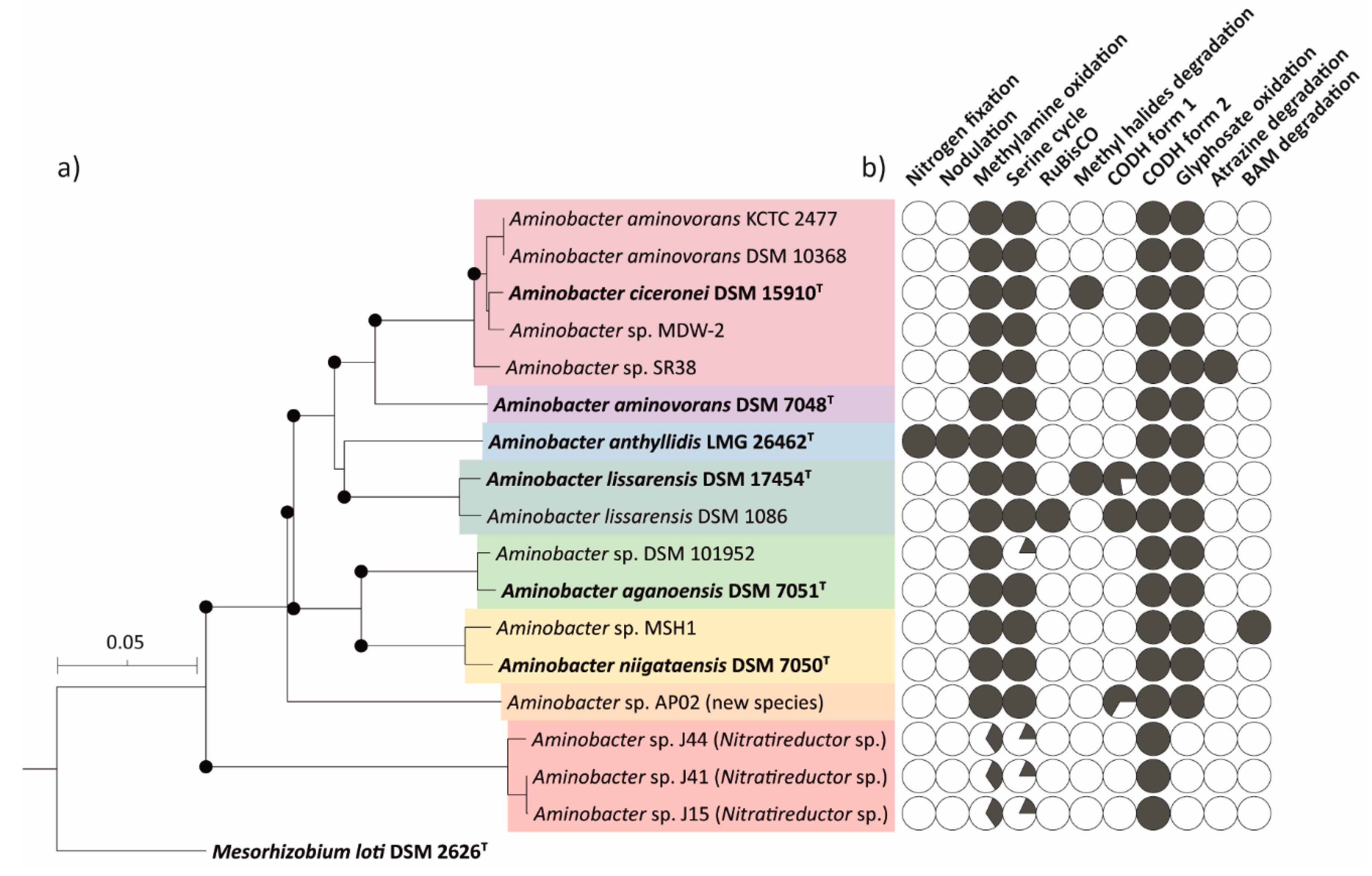
3.3. Whole Genome Phylogeny of Aminobacter Strains
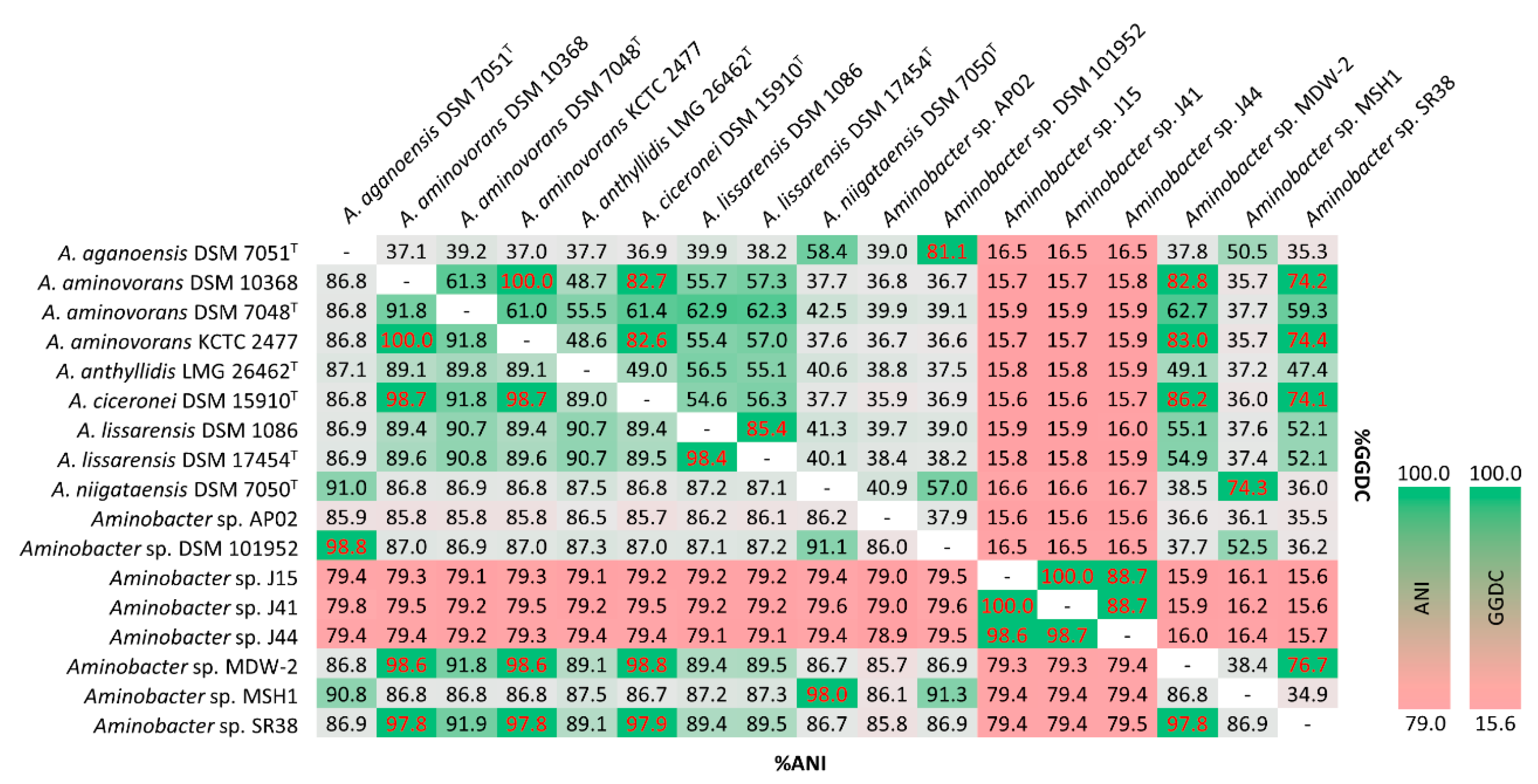
3.4. ANI and dDDH of Aminobacter Species
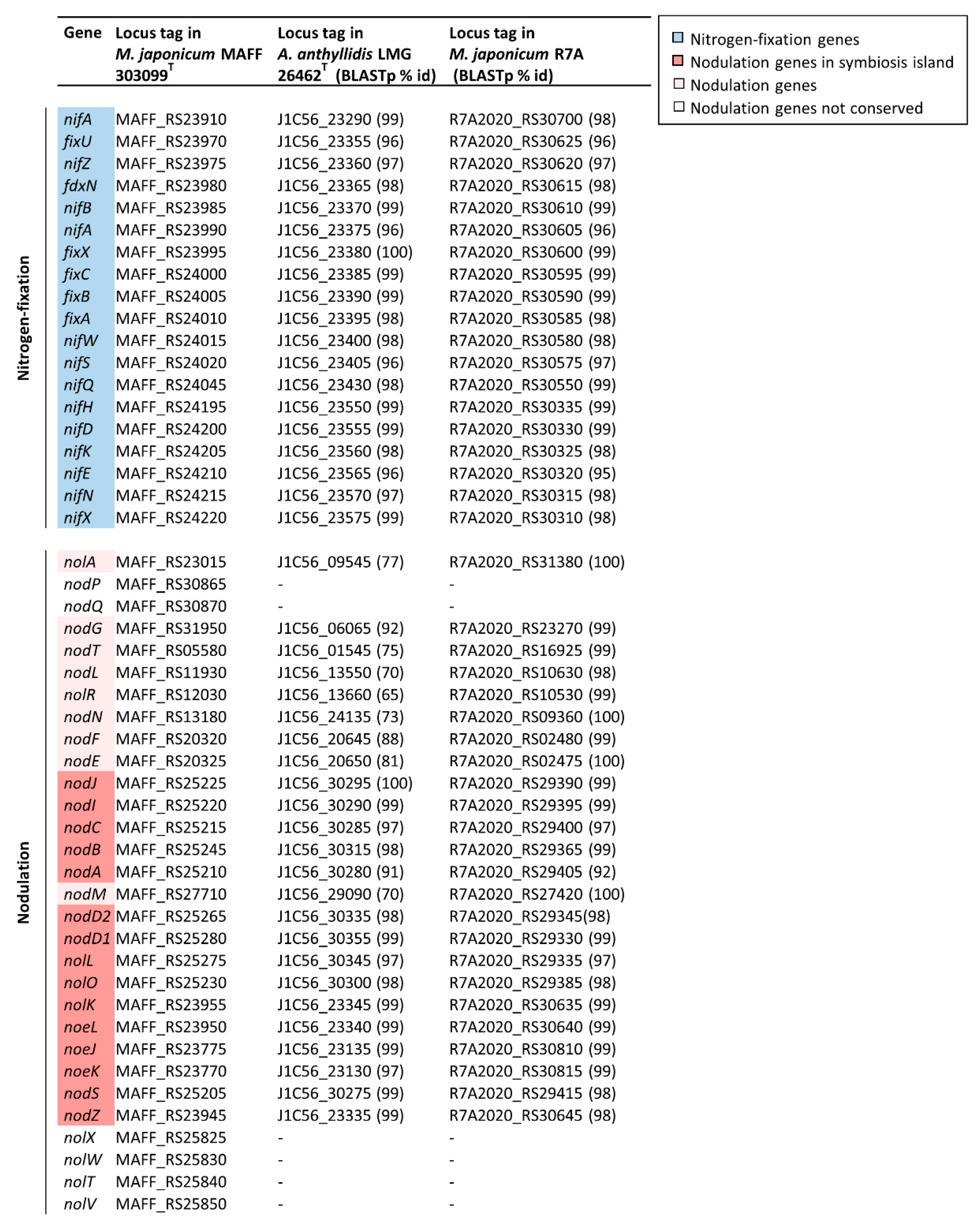
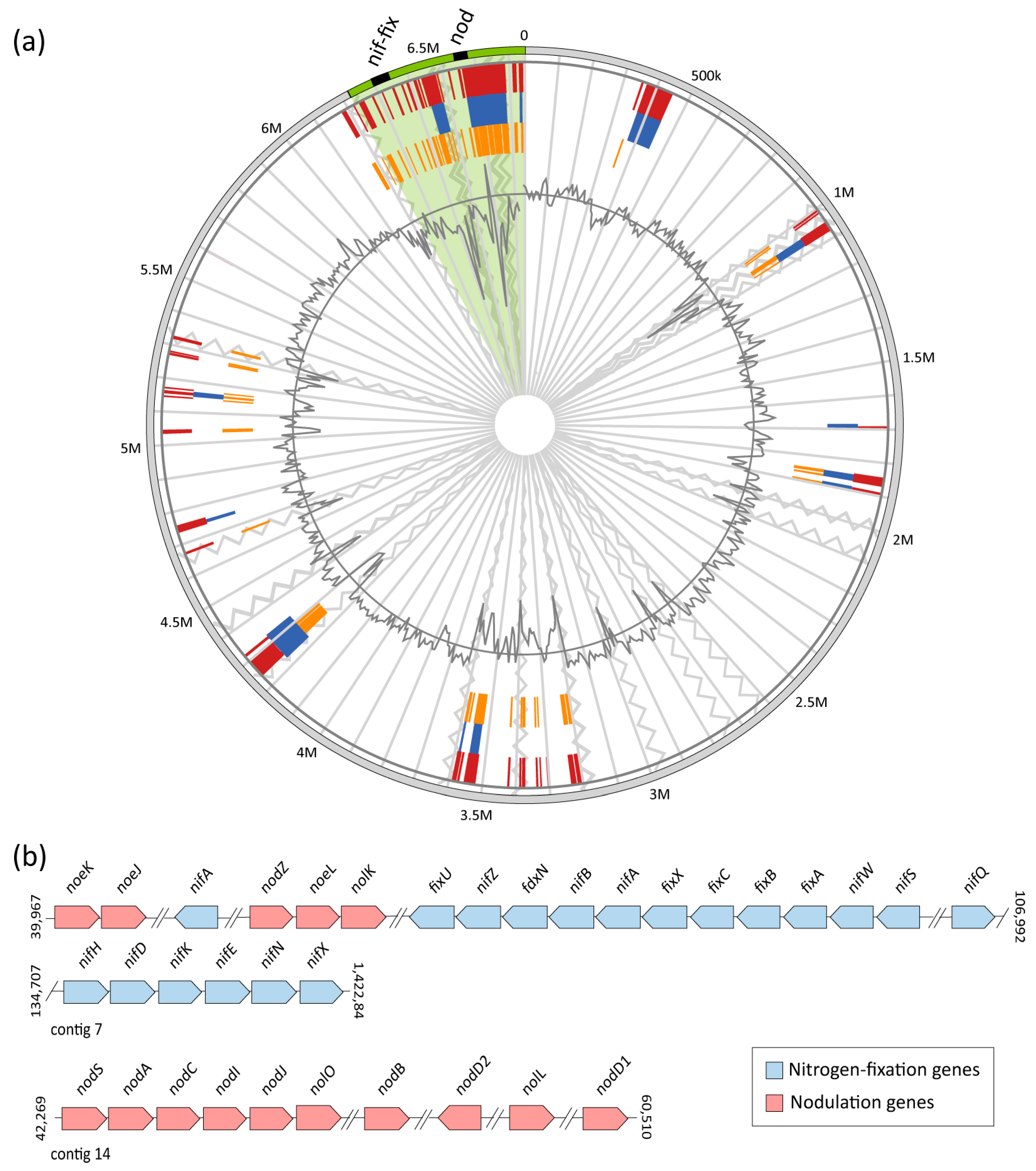
3.5. Nodulation and Nitrogen-Fixation Genes in Members of the Aminobacter Genus
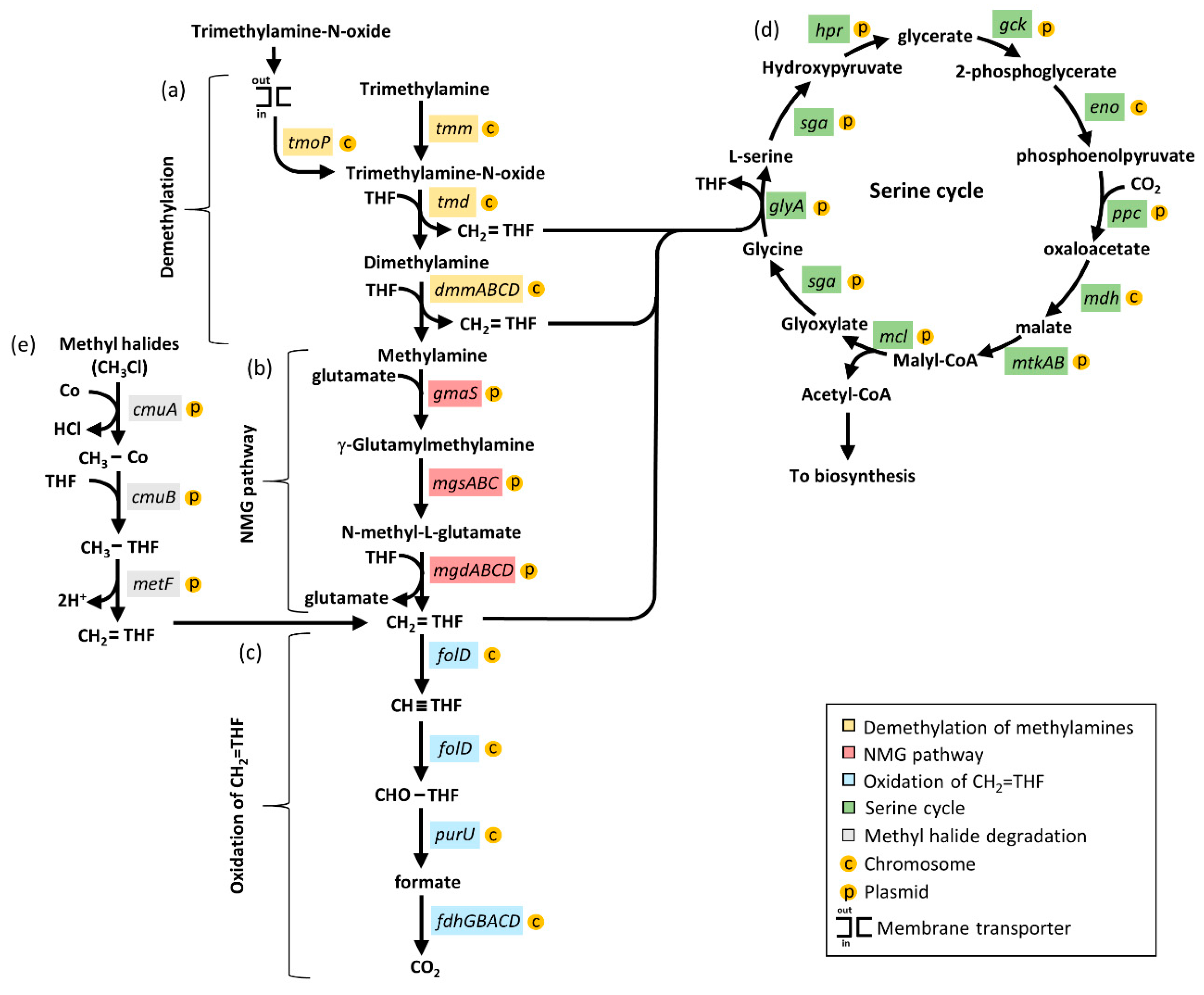
3.6. Methylotrophy Genes in Members of the Aminobacter Genus
3.7. Methyl Halide Utilisation Gene Cluster in Members of the Aminobacter Genus
3.8. Carbon Monoxide Dehydrogenase Genes in Members of the Aminobacter Genus
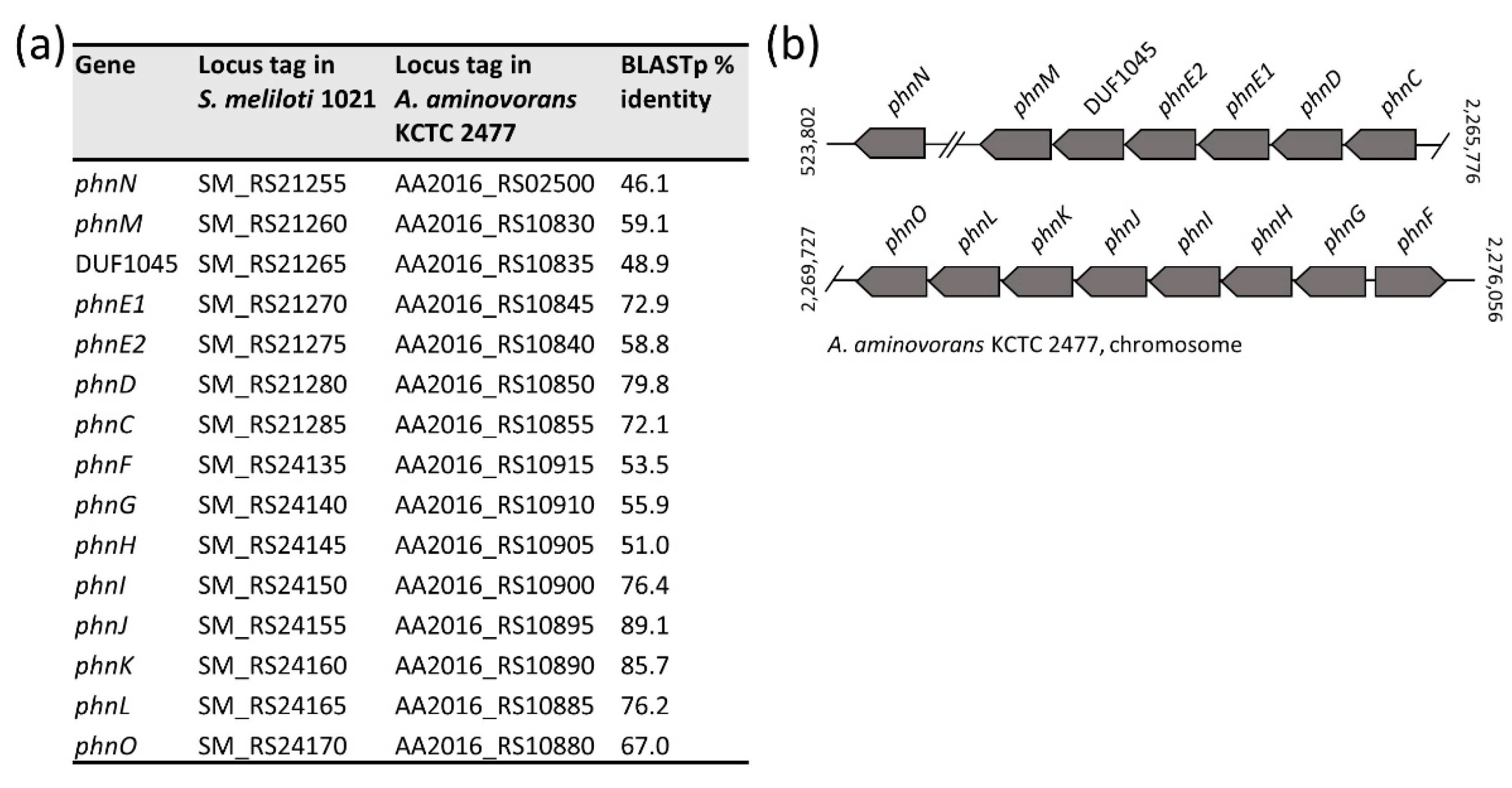
3.9. Glyphosate Oxidation Genes in Members of the Aminobacter Genus
3.10. 2,6-dichlorobenzamide and Atrazine Degradation Genes in Members of the Aminobacter Genus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Urakami, T. Aminobacter. In Bergey’s Manual of Systematics of Archaea and Bacteria, 1st ed.; Whitman, W.B., Rainey, F., Kämpfer, P., Trujillo, M., Chun, J., DeVos, P., Hedlund, B., Dedysh, S., Eds.; Wiley: Hoboken, NJ, USA, 2015; pp. 1–5. ISBN 978-1-118-96060-8. [Google Scholar]
- Urakami, T.; Araki, H.; Oyanagi, H.; Suzuki, K.-I.; Komagata, K. Transfer of Pseudomonas aminovorans (den Dooren de Jong 1926) to Aminobacter gen. nov. as Aminobacter aminovorans comb. nov. and Description of Aminobacter aganoensis sp. nov. and Aminobacter niigataensis sp. nov. Int. J. Syst. Bacteriol. 1992, 42, 84–92. [Google Scholar] [CrossRef]
- Hördt, A.; López, M.G.; Meier-Kolthoff, J.P.; Schleuning, M.; Weinhold, L.-M.; Tindall, B.J.; Gronow, S.; Kyrpides, N.; Woyke, T.; Göker, M. Analysis of 1,000+ Type-Strain Genomes Substantially Improves Taxonomic Classification of Alphaproteobacteria. Front. Microbiol. 2020, 11, 468. [Google Scholar] [CrossRef]
- Artuso, I.; Turrini, P.; Pirolo, M.; Lucidi, M.; Tescari, M.; Visaggio, D.; Mansi, A.; Lugli, G.A.; Ventura, M.; Visca, P. Phylogenomic analysis and characterization of carbon monoxide utilization genes in the family Phyllobacteriaceae with reclassification of Aminobacter carboxidus (Meyer et al. 1993, Hördt et al. 2020) as Aminobacter lissarensis comb. nov. (McDonald et al. 2005). Syst. Appl. Microbiol. 2021, 44, 126199. [Google Scholar] [CrossRef] [PubMed]
- Den Dooren de Jong, L.E. Bijdrage Tot de Ken Nisvan Het Mineralisatieproces; Nijgh and van Ditmar Uitgevers-Mij: Rotterdam, The Netherlands, 1926; pp. 1–200. [Google Scholar]
- Urakami, T.; Araki, H.; Kobayashi, H. Isolation and identification of tetramethylammonium-biodegrading bacteria. J. Ferment. Bioeng. 1990, 70, 41–44. [Google Scholar] [CrossRef]
- Urakami, T.; Kobayashi, H.; Araki, H. Isolation and identification of N,N-Dimethylformamide-biodegrading bacteria. J. Ferment. Bioeng. 1990, 70, 45–47. [Google Scholar] [CrossRef]
- Hancock, T.L.C.; Costello, A.M.; Lidstrom, M.E.; Oremland, R.S. Strain IMB-1, a Novel Bacterium for the Removal of Methyl Bromide in Fumigated Agricultural Soils. Appl. Environ. Microbiol. 1998, 64, 2899–2905. [Google Scholar] [CrossRef] [Green Version]
- Coulter, C.; Hamilton, J.T.G.; McRoberts, W.C.; Kulakov, L.; Larkin, M.J.; Harper, D.B. Halomethane:Bisulfide/Halide Ion Methyltransferase, an Unusual Corrinoid Enzyme of Environmental Significance Isolated from an Aerobic Methylotroph Using Chloromethane as the Sole Carbon Source. Appl. Environ. Microbiol. 1999, 65, 4301–4312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, I.; Kampfer, P.; Topp, E.; Warner, K.L.; Cox, M.J.; Hancock, T.L.C.; Miller, L.; Larkin, M.J.; Ducrocq, V.; Coulter, C.; et al. Aminobacter ciceronei sp. nov. and Aminobacter lissarensis sp. nov., isolated from various terrestrial environments. Int. J. Syst. Evol. Microbiol. 2005, 55, 1827–1832. [Google Scholar] [CrossRef]
- Maynaud, G.; Willems, A.; Soussou, S.; Vidal, C.; Mauré, L.; Moulin, L.; Cleyet-Marel, J.-C.; Brunel, B. Molecular and phenotypic characterization of strains nodulating Anthyllis vulneraria in mine tailings, and proposal of Aminobacter anthyllidis sp. nov., the first definition of Aminobacter as legume-nodulating bacteria. Syst. Appl. Microbiol. 2012, 35, 65–72. [Google Scholar] [CrossRef]
- Meyer, O.; Schlegel, H.G. Biology of Aerobic Carbon Monoxide-Oxidizing Bacteria. Annu. Rev. Microbiol. 1983, 37, 277–310. [Google Scholar] [CrossRef]
- Anthony, C. The Biochemistry of Methylotrophs; Academic Press: London, UK; New York, NY, USA, 1982; ISBN 978-0-12-058820-6. [Google Scholar]
- Chistoserdova, L. Modularity of methylotrophy, revisited. Environ. Microbiol. 2011, 13, 2603–2622. [Google Scholar] [CrossRef]
- Neff, J.; Holland, E.A.; Dentener, F.J.; McDowell, W.; Russell, K.M. The origin, composition and rates of organic nitrogen deposition: A missing piece of the nitrogen cycle? Biogeochemistry 2002, 57, 99–136. [Google Scholar] [CrossRef]
- Chistoserdova, L.; Kalyuzhnaya, M.G.; Lidstrom, M.E. The Expanding World of Methylotrophic Metabolism. Annu. Rev. Microbiol. 2009, 63, 477–499. [Google Scholar] [CrossRef] [Green Version]
- Latypova, E.; Yang, S.; Wang, Y.-S.; Wang, T.; Chavkin, T.A.; Hackett, M.; Schäfer, H.; Kalyuzhnaya, M.G. Genetics of the glutamate-mediated methylamine utilization pathway in the facultative methylotrophic beta-proteobacteriumMethyloversatilis universalisFAM5. Mol. Microbiol. 2010, 75, 426–439. [Google Scholar] [CrossRef]
- Butler, J.H. Better budgets for methyl halides? Nat. Cell Biol. 2000, 403, 260–261. [Google Scholar] [CrossRef] [PubMed]
- McDonald, I.R.; Warner, K.L.; McAnulla, C.; Woodall, C.; Oremland, R.S.; Murrell, J.C. A review of bacterial methyl halide degradation: Biochemistry, genetics and molecular ecology. Environ. Microbiol. 2002, 4, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Woodall, C.; Warner, K.L.; Oremland, R.S.; Murrell, J.C.; McDonald, I.R. Identification of Methyl Halide-Utilizing Genes in the Methyl Bromide-Utilizing Bacterial Strain IMB-1 Suggests a High Degree of Conservation of Methyl Halide-Specific Genes in Gram-Negative Bacteria. Appl. Environ. Microbiol. 2001, 67, 1959–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, K.L.; Larkin, M.J.; Harper, D.B.; Murrell, J.C.; McDonald, I.R. Analysis of genes involved in methyl halide degradation inAminobacter lissarensisCC495. FEMS Microbiol. Lett. 2005, 251, 45–51. [Google Scholar] [CrossRef] [Green Version]
- King, G.M.; Weber, C.F. Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat. Rev. Genet. 2007, 5, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Turrini, P.; Artuso, I.; Tescari, M.; Lugli, G.A.; Frangipani, E.; Ventura, M.; Visca, P. Draft Genome Sequence of the Carboxydotrophic Alphaproteobacterium Aminobacter carboxidus Type Strain DSM 1086. Microbiol. Resour. Announc. 2020, 9, e01170-20. [Google Scholar] [CrossRef]
- Gorodylova, N.; Michel, C.; Seron, A.; Joulian, C.; Delorme, F.; Bresch, S.; Garreau, C.; Giovannelli, F.; Michel, K. Modified zeolite-supported biofilm in service of pesticide biodegradation. Environ. Sci. Pollut. Res. 2021, 1–21. [Google Scholar] [CrossRef]
- Manav, M.C.; Sofos, N.; Hove-Jensen, B.; Brodersen, D.E. The Abc of Phosphonate Breakdown: A Mechanism for Bacterial Survival. BioEssays 2018, 40, e1800091. [Google Scholar] [CrossRef]
- Hove-Jensen, B.; Zechel, D.L.; Jochimsen, B.; Genois, M.-M.; Paquet, E.R.; Laffitte, M.-C.N.; Maity, R.; Rodrigue, A.; Ouellette, M.; Masson, J.-Y. Utilization of Glyphosate as Phosphate Source: Biochemistry and Genetics of Bacterial Carbon-Phosphorus Lyase. Microbiol. Mol. Biol. Rev. 2014, 78, 176–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz-Jensen, N.; Aamand, J.; Sørensen, S.R. Bioaugmentation potential of free and formulated 2,6-dichlorobenzamide (BAM) degrading Aminobacter sp. MSH1 in soil, sand and water. AMB Express 2016, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Mudhoo, A.; Garg, V. Sorption, Transport and Transformation of Atrazine in Soils, Minerals and Composts: A Review. Pedosphere 2011, 21, 11–25. [Google Scholar] [CrossRef]
- Billet, L.; Devers, M.; Rouard, N.; Martin-Laurent, F.; Spor, A. Labour sharing promotes coexistence in atrazine degrading bacterial communities. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- T’Syen, J.; Raes, B.; Horemans, B.; Tassoni, R.; Leroy, B.; Lood, C.; Van Noort, V.; Lavigne, R.; Wattiez, R.; Kohler, H.-P.E.; et al. Catabolism of the groundwater micropollutant 2,6-dichlorobenzamide beyond 2,6-dichlorobenzoate is plasmid encoded in Aminobacter sp. MSH1. Appl. Microbiol. Biotechnol. 2018, 102, 7963–7979. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Mancabelli, L.; Van Sinderen, D.; Ventura, M. MEGAnnotator: A user-friendly pipeline for microbial genomes assembly and annotation. FEMS Microbiol. Lett. 2016, 363, fnw049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.; Lesin, V.M.; Nikolenko, S.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. Simon Fraser University Research Computing Group IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Antipov, D.; Hartwick, N.; Shen, M.; Raiko, M.; Lapidus, A.; Pevzner, P.A. plasmidSPAdes: Assembling plasmids from whole genome sequencing data. Bioinformatics 2016, 32, btw493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampfer, P.; Neef, A.; Salkinoja-Salonen, M.S.; Buss, H.-J. Chelatobacter heintzii (Auling et al. 1993) is a later subjective synonym of Aminobacter aminovorans (Urakami et al. 1992). Int. J. Syst. Evol. Microbiol. 2002, 52, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Choe, H.; Nasir, A.; Park, D.-S.; Kim, K.M. Complete Genome Sequence of Nitrilotriacetate-Degrading Aminobacter aminovorans KCTC 2477 T. Genome Announc. 2016, 4, e01363-16. [Google Scholar] [CrossRef] [Green Version]
- Meyer, O.; Stackebrandt, E.; Auling, G. Reclassification of Ubiquinone Q-10 Containing Carboxidotrophic Bacteria: Transfer of “[Pseudomonas] carboxydovorans” OM5T to Oligotropha, gen. nov., as Oligotropha carboxidovorans, comb. nov., Transfer of “[Alcaligenes] carboxydus” DSM 1086T to Carbophilus, gen. nov., as Carbophilus carboxidus, comb. nov., Transfer of “[Pseudomonas] compransoris” DSM 1231T to Zavarzinia, gen. nov., as Zavarzinia compransoris, comb. nov., and Amended Descriptions of the New Genera. Syst. Appl. Microbiol. 1993, 16, 390–395. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Z.; Jin, W.; Wang, X.; Zhang, Y.; Zhu, S.; Yu, X.; Hu, G.; Hong, Q. Degradation of methomyl by the combination of Aminobacter sp. MDW-2 and Afipia sp. MDW-3. Lett. Appl. Microbiol. 2017, 64, 289–296. [Google Scholar] [CrossRef]
- Sørensen, S.R.; Holtze, M.S.; Simonsen, A.; Aamand, J. Degradation and Mineralization of Nanomolar Concentrations of the Herbicide Dichlobenil and Its Persistent Metabolite 2,6-Dichlorobenzamide by Aminobacter spp. Isolated from Dichlobenil-Treated Soils. Appl. Environ. Microbiol. 2007, 73, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Billet, L.; Devers-Lamrani, M.; Serre, R.-F.; Julia, E.; Vandecasteele, C.; Rouard, N.; Martin-Laurent, F.; Spor, A. Complete Genome Sequences of Four Atrazine-Degrading Bacterial Strains, Pseudomonas sp. Strain ADPe, Arthrobacter sp. Strain TES, Variovorax sp. Strain 38R, and Chelatobacter sp. Strain SR38. Microbiol. Resour. Announc. 2021, 10, e01080-20. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.I.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic. Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program: Table 1. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [Green Version]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Oh, H.-S.; Park, S.-C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Hidalgo, P.; Ramírez-Bahena, M.H.; Felix, J.D.F.; Igual, J.M.; Sanjuán, J.; León-Barrios, M.; Peix, A.; Velázquez, E. Reclassification of strains MAFF 303099T and R7A into Mesorhizobium japonicum sp. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 4936–4941. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hao, B.; Li, J.; Gu, H.; Peng, J.; Xie, F.; Zhao, X.; Frech, C.; Chen, N.; Ma, B.; et al. Whole-genome sequencing of Mesorhizobium huakuii 7653R provides molecular insights into host specificity and symbiosis island dynamics. BMC Genom. 2014, 15, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, J.; Trzebiatowski, J.R.; Cruickshank, R.W.; Gouzy, J.; Brown, S.D.; Elliot, R.M.; Fleetwood, D.J.; McCallum, N.G.; Rossbach, U.; Stuart, G.S.; et al. Comparative Sequence Analysis of the Symbiosis Island of Mesorhizobium loti Strain R7A. J. Bacteriol. 2002, 184, 3086–3095. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Romero, E.; Caballero-Mellado, J. RhizobiumPhylogenies and Bacterial Genetic Diversity. Crit. Rev. Plant Sci. 1996, 15, 113–140. [Google Scholar] [CrossRef]
- Pueppke, S.G. The Genetic and Biochemical Basis for Nodulation of Legumes by Rhizobia. Crit. Rev. Biotechnol. 1996, 16, 1–51. [Google Scholar] [CrossRef]
- Perret, X.; Staehelin, C.; Broughton, W.J. Molecular Basis of Symbiotic Promiscuity. Microbiol. Mol. Biol. Rev. 2000, 64, 180–201. [Google Scholar] [CrossRef] [Green Version]
- Vázquez, M.; Santana, O.; Quinto, C. The NodI and NodJ proteins from Rhizobium and Bradyrhizobium strains are similar to capsular polysaccharide secretion proteins from Gram-negative bacteria. Mol. Microbiol. 1993, 8, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Czarnecki, J.; Dziewit, L.; Puzyna, M.; Prochwicz, E.; Tudek, A.; Wibberg, D.; Schlüter, A.; Pühler, A.; Bartosik, D. Lifestyle-determining extrachromosomal replicon pAMV1 and its contribution to the carbon metabolism of the methylotrophic bacteriumParacoccus aminovoransJCM 7685. Environ. Microbiol. 2017, 19, 4536–4550. [Google Scholar] [CrossRef]
- King, G.M. Molecular andCulture-Based Analyses of Aerobic Carbon Monoxide Oxidizer Diversity. Appl. Environ. Microbiol. 2003, 69, 7257–7265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- T’Syen, J.; Tassoni, R.; Hansen, L.H.; Sorensen, S.J.; Leroy, B.; Sekhar, A.; Wattiez, R.; De Mot, R.; Springael, D. Identification of the Amidase BbdA That Initiates Biodegradation of the Groundwater Micropollutant 2,6-dichlorobenzamide (BAM) inAminobactersp. MSH1. Environ. Sci. Technol. 2015, 49, 11703–11713. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.K.; Hylling, O.; Ellegaard-Jensen, L.; Aamand, J.; Hansen, L.H. The genome of BAM-degrading Aminobacter Sp. MSH1 with several low copy plasmids. BioRxiv 2018, 307967. [Google Scholar] [CrossRef] [Green Version]
- Groisman, E.A.; Ochman, H. Pathogenicity Islands: Bacterial Evolution in Quantum Leaps. Cell 1996, 87, 791–794. [Google Scholar] [CrossRef] [Green Version]
- Edziewit, L.; Eczarnecki, J.; Eprochwicz, E.; Ewibberg, D.; Eschlüter, A.; Epühler, A.; Ebartosik, D. Genome-guided insight into the methylotrophy of Paracoccus aminophilus JCM 7686. Front. Microbiol. 2015, 6, 852. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; McAleer, K.L.; Murrell, J.C. Monomethylamine as a Nitrogen Source for a Nonmethylotrophic Bacterium, Agrobacterium tumefaciens. Appl. Environ. Microbiol. 2010, 76, 4102–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Wexler, A.S.; Clegg, S.L. Atmospheric amines—Part I. A review. Atmos. Environ. 2011, 45, 524–546. [Google Scholar] [CrossRef]
- Schade, G.W.; Crutzen, P.J. Emission of aliphatic amines from animal husbandry and their reactions: Potential source of N2O and HCN. J. Atmos. Chem. 1995, 22, 319–346. [Google Scholar] [CrossRef]
- Lobert, J.M.; Scharffe, D.H.; Hao, W.-M.; Kuhlbusch, T.A.; Warneck, P.; Crutzen, P.J. Experimental evaluation of biomass burning emissions: Nitrogen and carbon containing compounds. In Global Biomass Burning: Atmospheric, Climatic, and Biospheric Implications; Levine, J.S., Ed.; MIT Press: Cambridge, MA, USA, 1991; ISBN 978-0-262-12159-0. [Google Scholar]
- Ternan, N.G.; Mc Grath, J.W.; Mc Mullan, G.; Quinn, J.P. Review: Organophosphonates: Occurrence, synthesis and biodegradation by microorganisms. World J. Microbiol. Biotechnol. 1998, 14, 635–647. [Google Scholar] [CrossRef]
- Ellegaard-Jensen, L.; Horemans, B.; Raes, B.; Aamand, J.; Hansen, L.H. Groundwater contamination with 2,6-dichlorobenzamide (BAM) and perspectives for its microbial removal. Appl. Microbiol. Biotechnol. 2017, 101, 5235–5245. [Google Scholar] [CrossRef]
- Li, Q.; Li, Y.; Zhu, X.; Cai, B. Isolation and characterization of atrazine-degrading Arthrobacter sp. AD26 and use of this strain in bioremediation of contaminated soil. J. Environ. Sci. 2008, 20, 1226–1230. [Google Scholar] [CrossRef]
- Simonsen, A.; Holtze, M.S.; Sørensen, S.R.; Sørensen, S.J.; Aamand, J. Mineralisation of 2,6-dichlorobenzamide (BAM) in dichlobenil-exposed soils and isolation of a BAM-mineralising Aminobacter sp. Environ. Pollut. 2006, 144, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nat. Cell Biol. 2000, 405, 299–304. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism Name | Genome Size (bp) | Genome Coverage (n×) | No. of Contigs | N50 (bp) | G + C (%) | ORFs (no.) | tRNA Genes (no.) | rRNA Genes (no.) | Source/Country | Accession No. | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A. aganoensis DSM 7051 T | 5,765,889 | 260 | 52 | 248,423 | 63.89 | 5586 | 46 | 3 | Soil/JP | GCF_014206975.1 | [2] |
| A. aminovorans DSM 10368 | 6,811,076 | 220 | 67 | 190,669 | 63.14 | 6605 | 48 | 3 | Soil/US | GCF_014195595.1 | [37] |
| A. aminovorans DSM 7048 T | 5,848,363 | 256 | 29 | 467,483 | 63.19 | 5658 | 47 | 3 | Soil/- | GCF_004341645.1 | [2,5] |
| A. aminovorans KCTC 2477 | 6,890,726 | 144 | 1 + 4 a | 63.14 | 6613 | 54 | 3 | Soil/US | GCF_001605015.1 | [38] | |
| A. anthyllidis LMG 26462 T | 6,717,907 | 113 | 30 | 670,596 | 62.58 | 6486 | 51 | 3 | Root nodule/FR | GCF_018555685.1 | [11] |
| A. ciceronei DSM 15910 T | 6,774,758 | 214 | 96 | 156,335 | 63.07 | 6611 | 51 | 3 | Soil/US | GCF_014138625.1 | [10] |
| A. lissarensis DSM 1086 | 6,291,275 | 65 | 31 | 458,931 | 62.96 | 6023 | 49 | 3 | Soil/RU | GCF_014863355.1 | [3,4,39] |
| A. lissarensis DSM 17454 T | 6,541,127 | 229 | 45 | 317,634 | 62.69 | 6190 | 46 | 3 | Soil/IE | GCF_014207495.1 | [10] |
| A. niigataensis DSM 7050 T | 5,287,613 | 284 | 26 | 468,746 | 63.4 | 5083 | 48 | 3 | Soil/JP | GCF_014200015.1 | [2] |
| Aminobacter sp. AP02 | 5,603,683 | 210 | 72 | 253,161 | 62.05 | 5360 | 48 | 5 | Populus root/US | GCF_003148805.1 | - |
| Aminobacter sp. DSM 101952 | 5,233,617 | 286 | 36 | 512,353 | 63.81 | 5047 | 48 | 3 | - | GCF_014201895.1 | - |
| Aminobacter sp. J15 | 4,216,557 | 293 | 122 | 94,548 | 63.32 | 3992 | 49 | 3 | - | GCF_007829635.1 | - |
| Aminobacter sp. J41 | 4,234,633 | - | 113 | 102,055 | 63.34 | 4011 | 52 | 3 | - | GCF_000526635.1 | - |
| Aminobacter sp. J44 | 4,186,064 | 250 | 90 | 92,806 | 63.4 | 3962 | 51 | 5 | - | GCF_007829415.1 | - |
| Aminobacter sp. MDW-2 | 6,607,828 | 200 | 1 + 3 a | 63.19 | 6400 | 52 | 6 | Soil/CN | GCF_014250155.1 | [40] | |
| Aminobacter sp. MSH1 | 6,321,606 | 900 | 1 + 7 a | 62.89 | 6180 | 52 | 6 | Soil/DK | GCF_003063555.1 | [41] | |
| Aminobacter sp. SR38 | 7,367,353 | 290 | 1 + 8 a | 62.89 | 7146 | 53 | 8 | Soil/FR | GCF_014843375.1 | [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Artuso, I.; Turrini, P.; Pirolo, M.; Lugli, G.A.; Ventura, M.; Visca, P. Phylogenomic Reconstruction and Metabolic Potential of the Genus Aminobacter. Microorganisms 2021, 9, 1332. https://doi.org/10.3390/microorganisms9061332
Artuso I, Turrini P, Pirolo M, Lugli GA, Ventura M, Visca P. Phylogenomic Reconstruction and Metabolic Potential of the Genus Aminobacter. Microorganisms. 2021; 9(6):1332. https://doi.org/10.3390/microorganisms9061332
Chicago/Turabian StyleArtuso, Irene, Paolo Turrini, Mattia Pirolo, Gabriele Andrea Lugli, Marco Ventura, and Paolo Visca. 2021. "Phylogenomic Reconstruction and Metabolic Potential of the Genus Aminobacter" Microorganisms 9, no. 6: 1332. https://doi.org/10.3390/microorganisms9061332
APA StyleArtuso, I., Turrini, P., Pirolo, M., Lugli, G. A., Ventura, M., & Visca, P. (2021). Phylogenomic Reconstruction and Metabolic Potential of the Genus Aminobacter. Microorganisms, 9(6), 1332. https://doi.org/10.3390/microorganisms9061332