
Metagenomic Insight of a Full Scale Eco-Friendly Treatment System of Textile Dye Wastewater Using Bioaugmentation of the Composite Culture CES-1

 , , ,
, , ,
Abstract
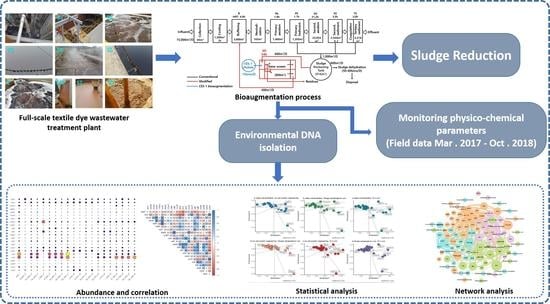
:
1. Introduction
2. Materials and Methods
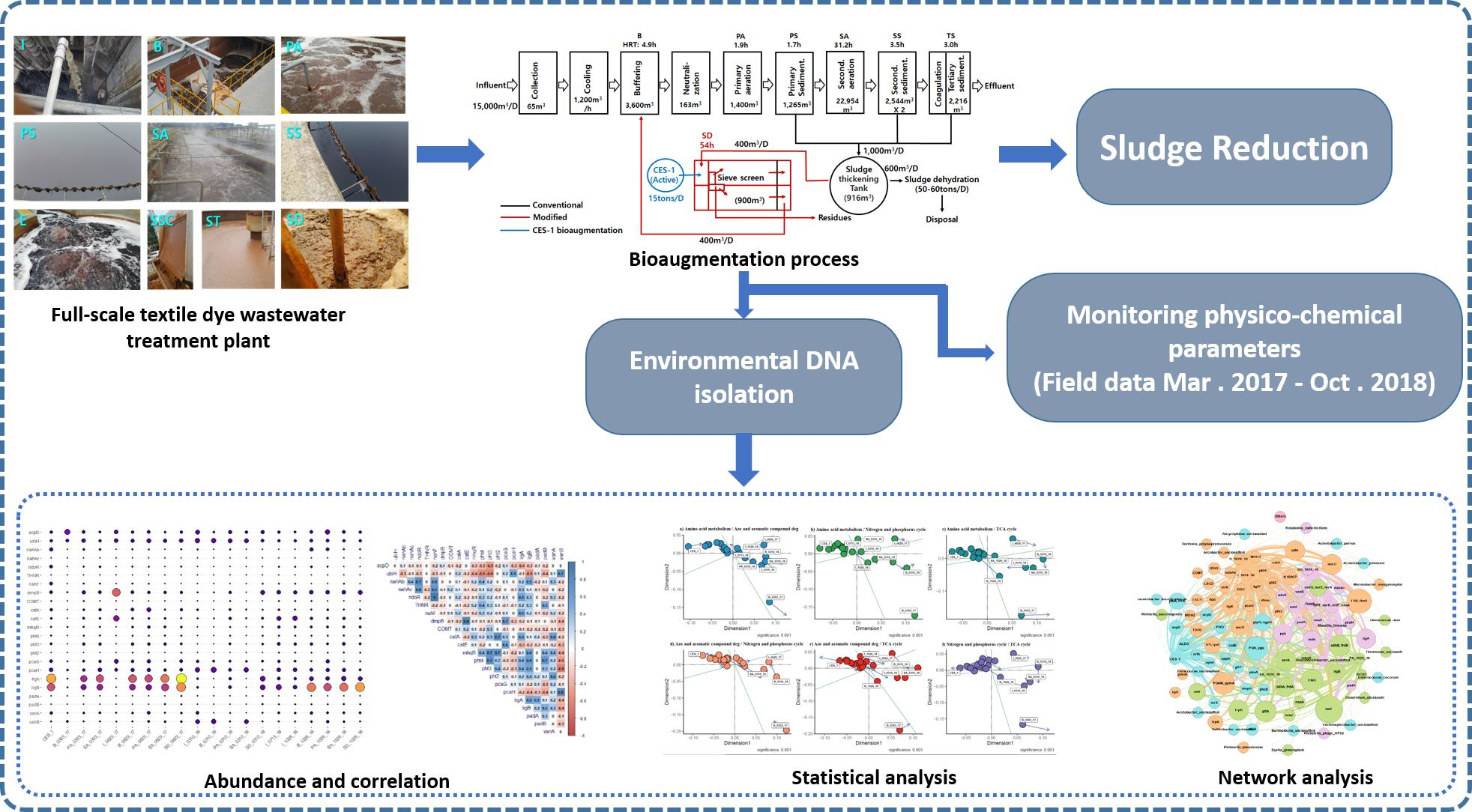
2.1. Description of Textile Dye Wastewater Treatment System and Its Operations
2.2. Monitoring the Water Quality of Dye Wastewater Treatment System
2.3. DNA Isolation from Samples and Sequencing of Metagenomes
2.4. Contig Assembly and Binning of Metagenomes
2.5. Visualization of Metabolic Pathways and Microbial Communities
3. Results and Discussion
3.1. Wastewater Treatment Monitoring during Bioaugmentation
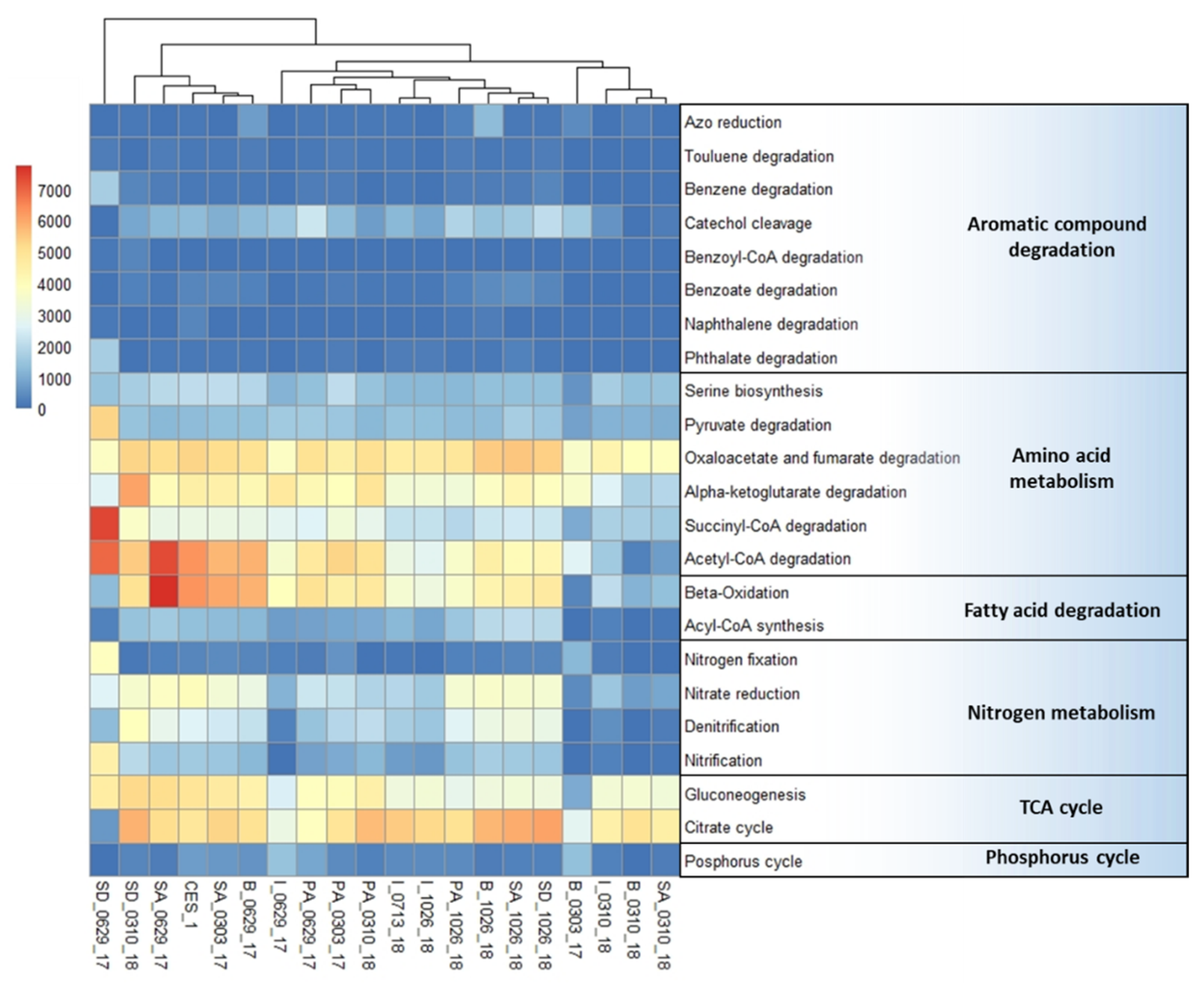
3.2. Comparative Analysis of Potential Different Metabolic Pathways Involved in Chemical Degradation Processes in the Treatment System
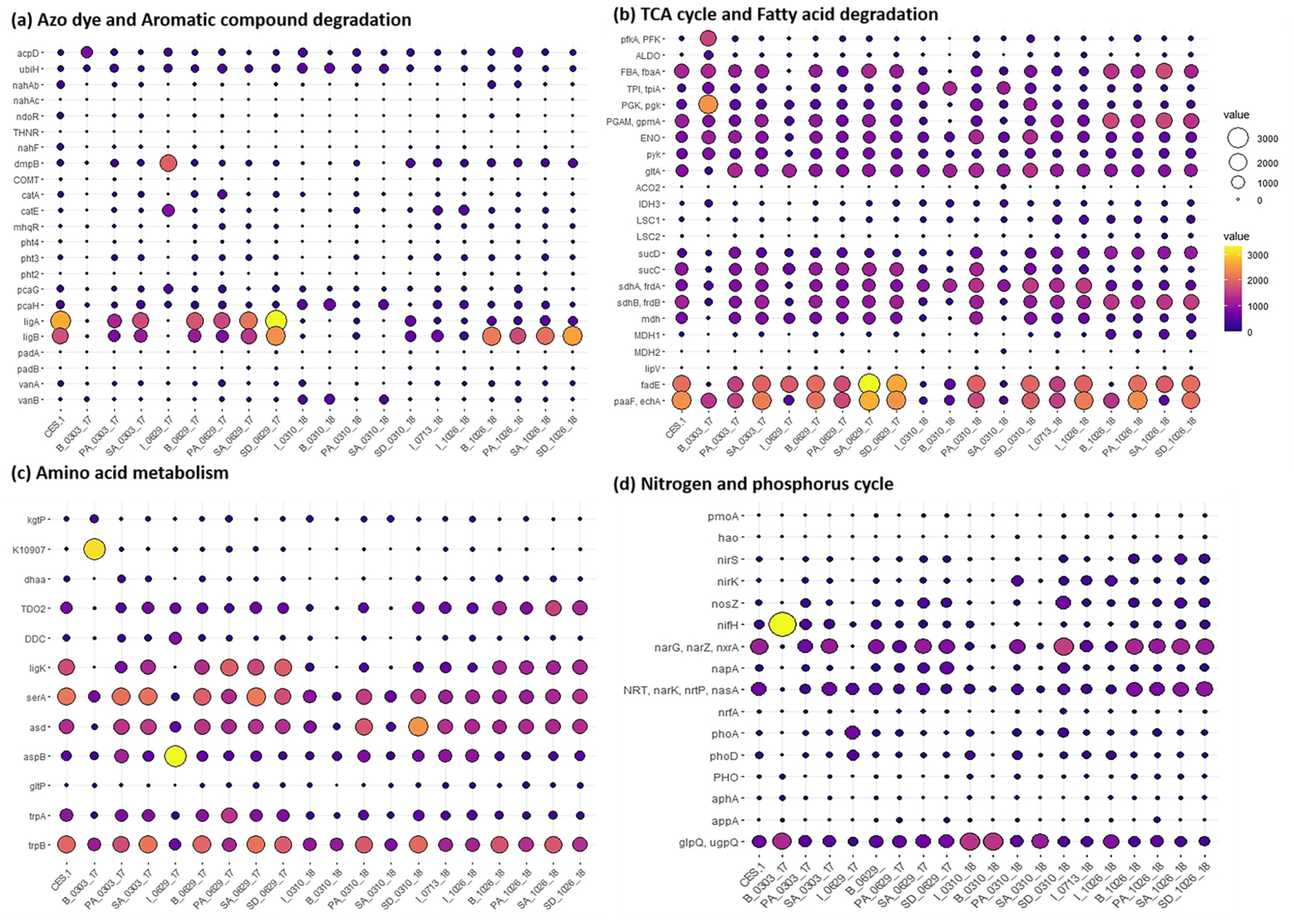
3.3. Dynamic Changes of Functional Genes for Major Metabolic Pathways in Textile Dye Wastewater Treatment Process Undergoing Bioaugmentation of CES-1 over Time
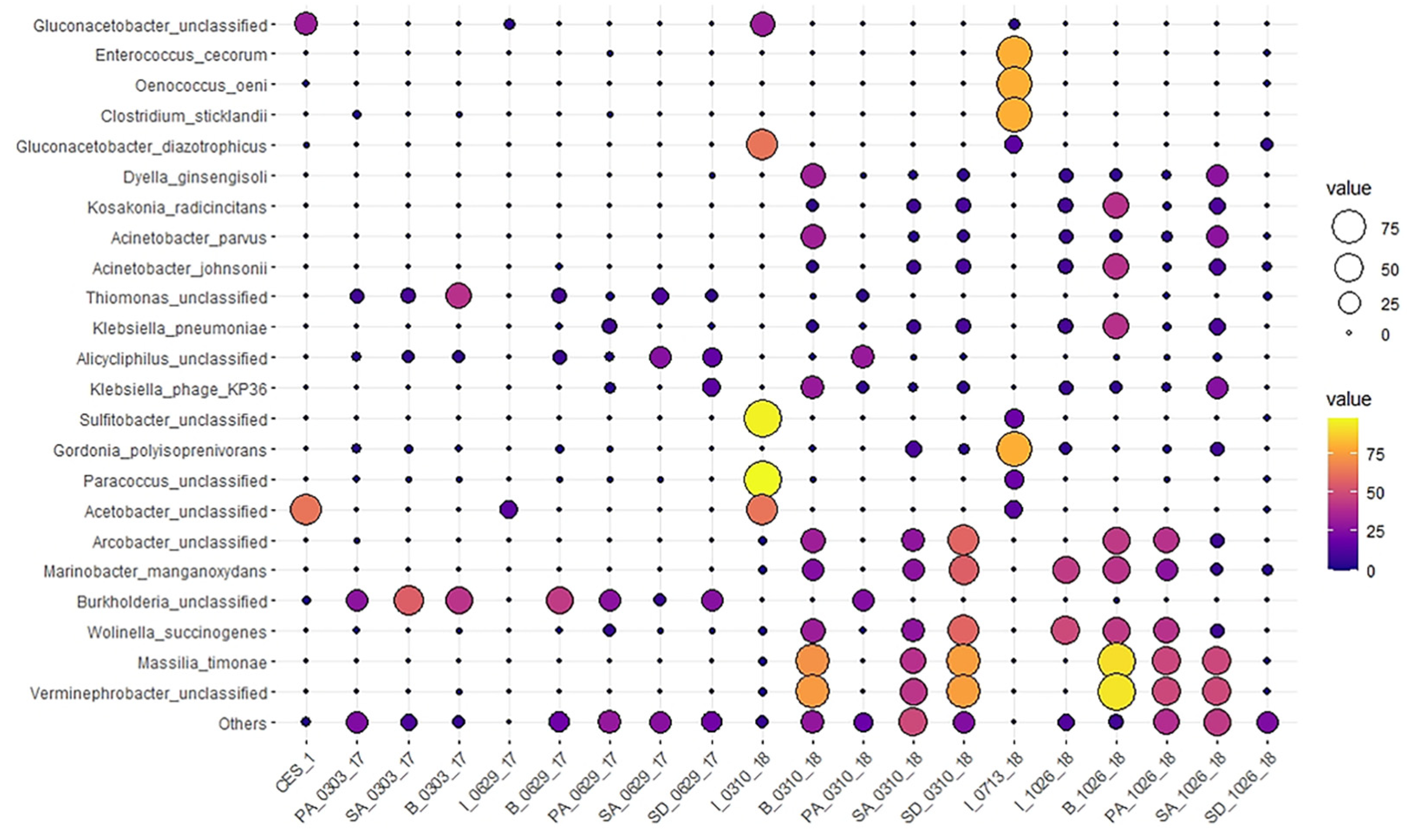
3.4. Dynamic Changes of Microbial Community Structures in Textile Dye Wastewater Treatment Process under Bioaugmentation of CES-1 at Different Treatment Stages over Time
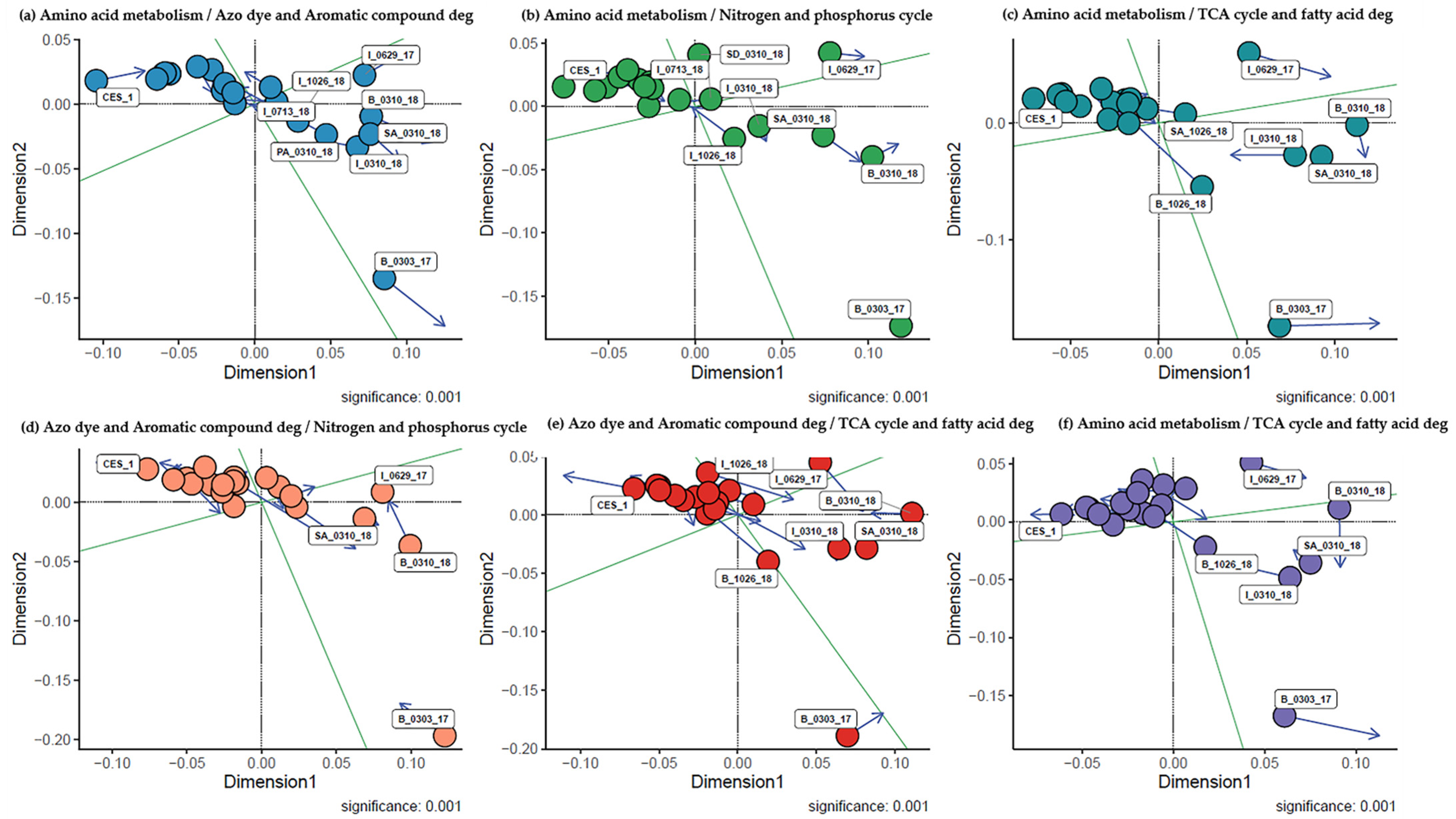
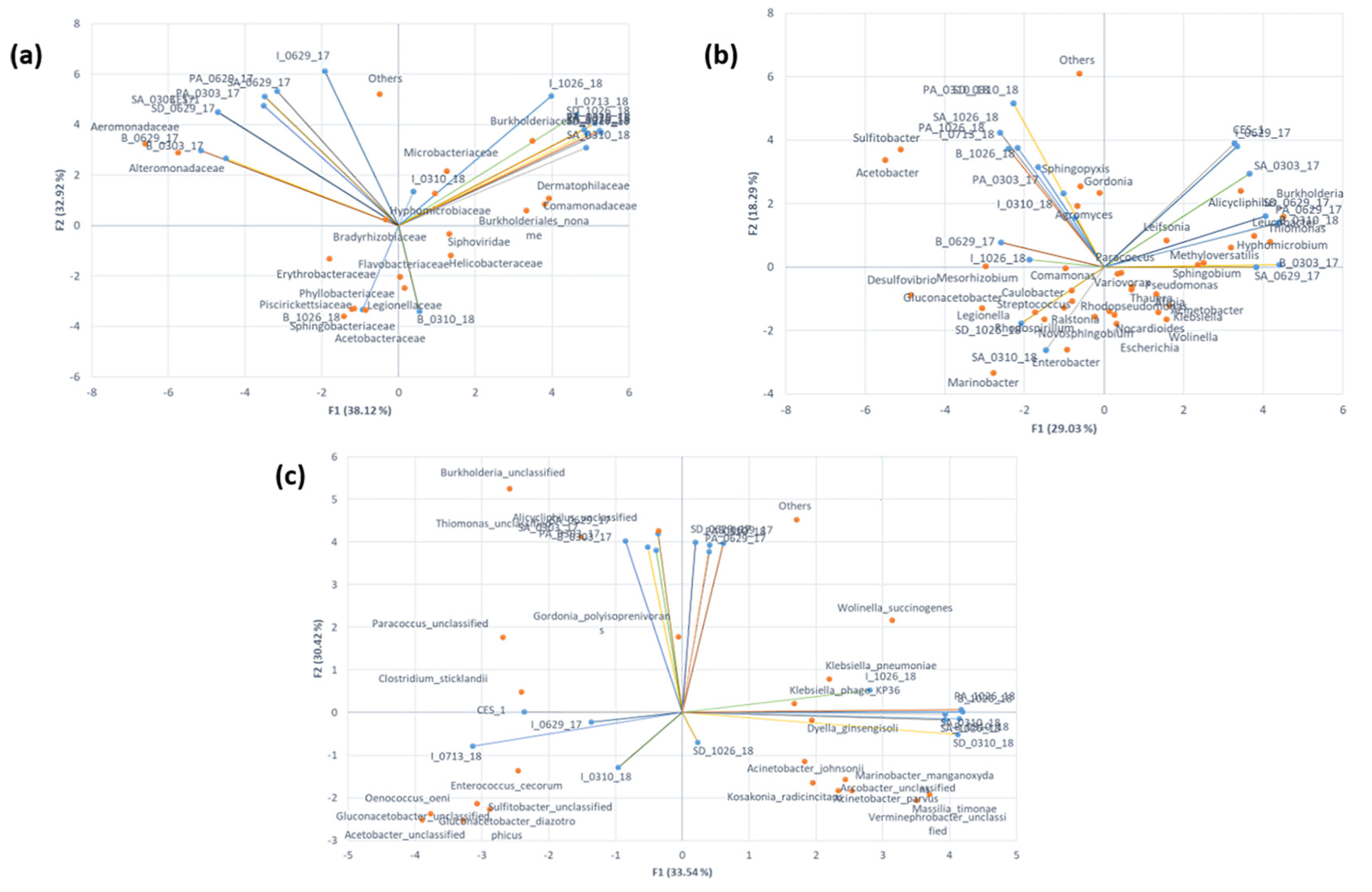
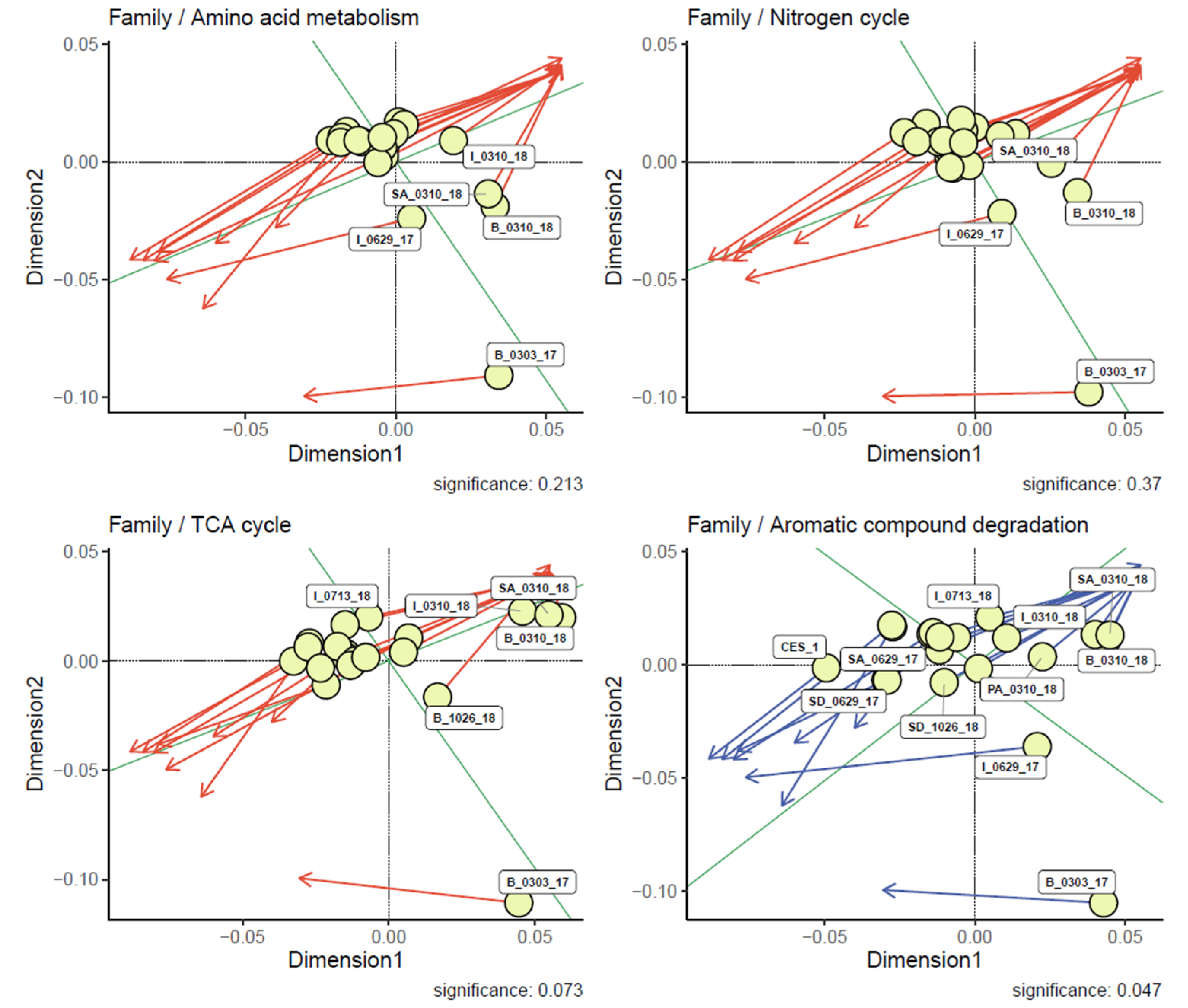
3.5. Analysis of Relationships between the Microbial Community Structures and Major Degradation Pathways Involved in the Textile Dye Treatment Process under Bioaugmentation
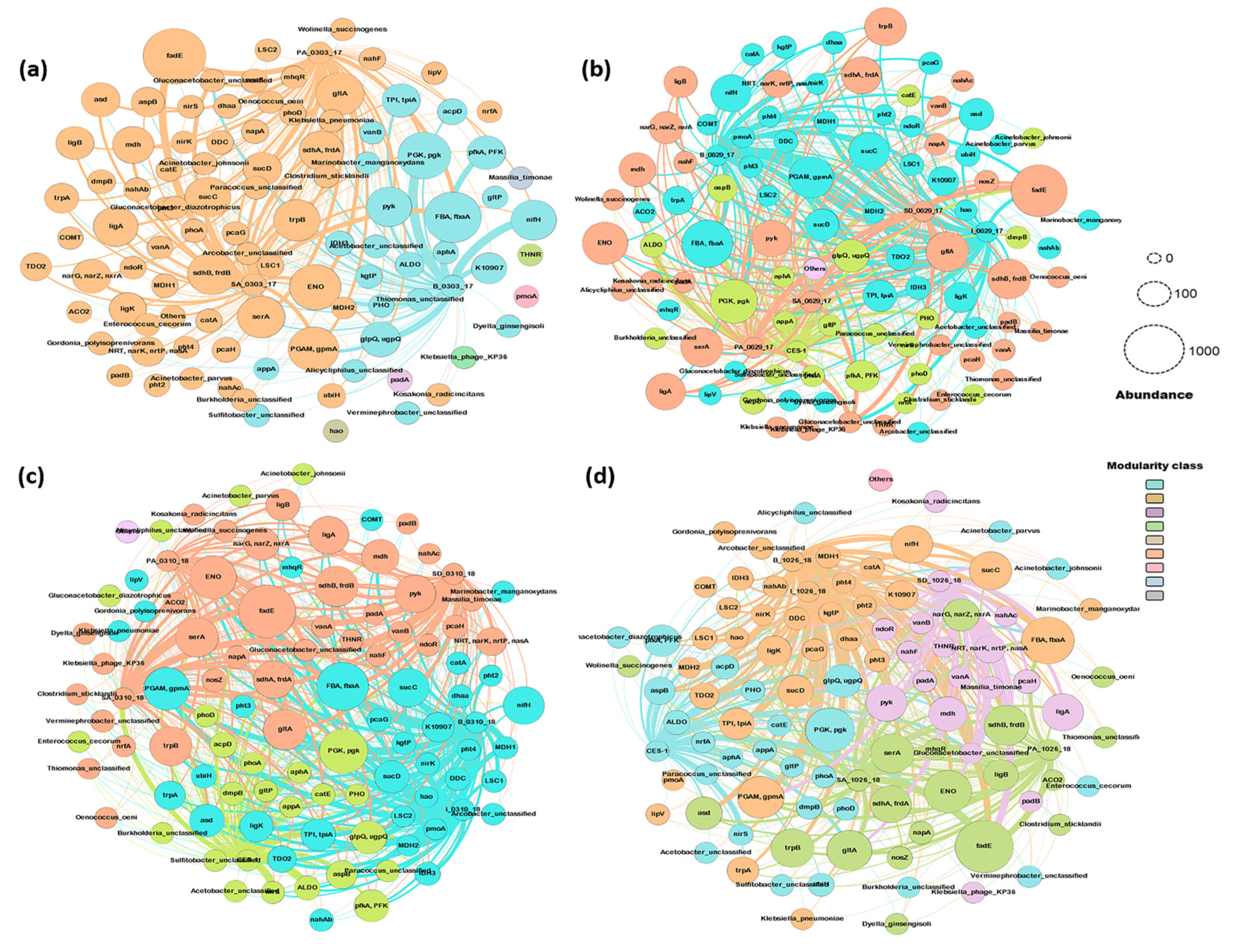
3.6. Time-Course Network Analysis among Microbial Communities, Metabolic Pathways Involved in the Degradation of Azo Dye and Other Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Šajn, N. Environmental Impact of the Textile and Clothing Industry; European Parliament: Brussels, Belgium, 2019; Volume PE 633.143. [Google Scholar]
- Sharma, S.; Bhattacharya, A. Drinking water contamination and treatment techniques. Appl. Water Sci. 2017, 7, 1043–1067. [Google Scholar] [CrossRef] [Green Version]
- Banat, I.; Nigam, P.; Singh, D.; Marchant, R. Microbial decolorization of textile-dyecontaining effluents: A review. Bioresour. Technol. 1996, 58. [Google Scholar] [CrossRef]
- Przystaś, W.; Zabłocka-Godlewska, E.; Grabińska-Sota, E. Biological removal of azo and triphenylmethane dyes and toxicity of process by-products. Water Air Soil Pollut. 2011, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, E.J.; Wolfe, N.L. Kinetic studies of the reduction of aromatic AZO compounds in anaerobic sediment/water systems. Environ. Toxicol. Chem. 1987, 6. [Google Scholar] [CrossRef]
- Chung, K.T.; Fulk, G.E.; Egan, M. Reduction of azo dyes by intestinal anaerobes. Appl. Environ. Microbiol. 1978, 35. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Banerjee, A.; Halder, U.; Biswas, R.; Bandopadhyay, R. Degradation of synthetic azo dyes of textile industry: A sustainable approach using microbial enzymes. Water Conserv. Sci. Eng. 2017, 2, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Alabdraba, W.; Bayati, M. Biodegradation of azo dyes: A review. Int. J. Environ. Eng. Nat. Resour. 2014, 1, 179–189. [Google Scholar]
- Pandey, A.; Singh, P.; Iyengar, L. Bacterial decolorization and degradation of azo dyes. Int. Biodeterior. Biodegrad. 2007, 59, 73–84. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, W.; Liu, R.; Li, Q.; Li, B.; Wang, S.; Song, C.; Qiao, C.; Mulchandani, A. Phylogenetic diversity and metabolic potential of activated sludge microbial communities in full-scale wastewater treatment plants. Environ. Sci. Technol. 2011, 45, 7408–7415. [Google Scholar] [CrossRef]
- Fernandez, R.P.; Carmona-Balea, A.; Cuevas, C.A.; Barrera, J.A.; Kinnison, D.E.; Lamarque, J.; Blaszczak-Boxe, C.; Kim, K.; Choi, W.; Hay, T.; et al. Modeling the sources and chemistry of polar tropospheric halogens (Cl, Br, and I) using the CAM-chem global chemistry-climate model. J. Adv. Model. Earth Syst. 2019, 11, 2259–2289. [Google Scholar] [CrossRef]
- Patil, V.P.; Sandt, J.D.; Kolle, M.; Dunkel, J. Topological mechanics of knots and tangles. Science 2020, 367, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zhao, H.; Peter, K.T.; Gonzalez, M.; Wetzel, J.; Wu, C.; Hu, X.; Prat, J.; Mudrock, E.; Hettinger, R.; et al. A ubiquitous tire rubber–derived chemical induces acute mortality in coho salmon. Science 2021, 371, 185–189. [Google Scholar] [CrossRef]
- Raper, E.; Stephenson, T.; Anderson, D.; Fisher, R.; Soares, A. Industrial wastewater treatment through bioaugmentation. Process. Saf. Environ. Prot. 2018, 118. [Google Scholar] [CrossRef] [Green Version]
- Punzi, M.; Anbalagan, A.; Börner, R.A.; Svensson, B.-M.; Jonstrup, M.; Mattiasson, B. Degradation of a textile azo dye using biological treatment followed by photo-Fenton oxidation: Evaluation of toxicity and microbial community structure. Chem. Eng. J. 2015, 270, 290–299. [Google Scholar] [CrossRef]
- Cui, D.; Li, G.; Zhao, D.; Gu, X.; Wang, C.; Zhao, M. Microbial community structures in mixed bacterial consortia for azo dye treatment under aerobic and anaerobic conditions. J. Hazard. Mater. 2012, 221–222, 185–192. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, X.; Li, Y.; Li, K.; Wang, C.; Liu, J.; Yan, D.; Liu, Y.; Yang, D.; Xing, J. Heterotrophic nitrification and aerobic denitrification by a novel Halomonas campisalis. Biotechnol. Lett. 2013, 35. [Google Scholar] [CrossRef]
- Krishnamoorthy, R.; Jose, P.A.; Ranjith, M.; Anandham, R.; Suganya, K.; Prabhakaran, J.; Thiyageshwari, S.; Johnson, J.; Gopal, N.; Kumutha, K. Decolourisation and degradation of azo dyes by mixed fungal culture consisted of Dichotomomyces cejpii MRCH 1-2 and Phoma tropica MRCH 1-3. J. Environ. Chem. Eng. 2018, 6, 588–595. [Google Scholar] [CrossRef]
- Ju, F.; Guo, F.; Ye, L.; Xia, Y.; Zhang, T. Metagenomic analysis on seasonal microbial variations of activated sludge from a full-scale wastewater treatment plant over 4 years. Environ. Microbiol. Rep. 2014, 6, 80–89. [Google Scholar] [CrossRef]
- American Public Health Association. APHA Method 9221: Standard Methods for the Examination of Water and Wastewater; APHA: Washington, DC, USA, 2005; Volume 552.
- Kim, I.-S.; Ekpeghere, K.I.; Ha, S.-Y.; Kim, B.-S.; Song, B.; Kim, J.-T.; Kim, H.-G.; Koh, S.-C. Full-scale biological treatment of tannery wastewater using the novel microbial consortium BM-S-1. J. Environ. Sci. Health Part A Toxic/Hazard. Subst. Environ. Eng. 2014, 49. [Google Scholar] [CrossRef]
- Sul, W.J.; Kim, I.-S.; Ekpeghere, K.I.; Song, B.; Kim, B.-S.; Kim, H.-G.; Kim, J.-T.; Koh, S.-C. Metagenomic insight of nitrogen metabolism in a tannery wastewater treatment plant bioaugmented with the microbial consortium BM-S-1. J. Environ. Sci. Health Part A Toxic Hazard. Subst. Environ. Eng. 2016, 51, 1164–1172. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Wei, T.; Simko, V. R Package ‘Corrplot’: Visualization of a Correlation Matrix. 2017. Available online: https://github.com/taiyun/corrplot (accessed on 7 June 2021).
- Forsberg, K.J.; Patel, S.; Gibson, M.K.; Lauber, C.L.; Knight, R.; Fierer, N.; Dantas, G. Bacterial phylogeny structures soil resistomes across habitats. Nat. Cell Biol. 2014, 509, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Feng, J.; Yin, X.; Liu, J.; Fu, W.; Berendonk, T.U.; Zhang, T.; Li, X.; Li, B. Antibiotic resistome in landfill leachate from different cities of China deciphered by metagenomic analysis. Water Res. 2018, 134, 126–139. [Google Scholar] [CrossRef]
- Lawson, C.E.; Wu, S.; Bhattacharjee, A.S.; Hamilton, J.; McMahon, K.D.; Goel, R.; Noguera, D.R. Metabolic network analysis reveals microbial community interactions in anammox granules. Nat. Commun. 2017, 8, 15416. [Google Scholar] [CrossRef] [PubMed]
- Aghapour, A.A.; Moussavi, G.; Yaghmaeian, K. Biological degradation of catechol in wastewater using the sequencing continuous-inflow reactor (SCR). J. Environ. Health Sci. Eng. 2013, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Emmanuel, S.A.; Sul, W.J.; Seong, H.J.; Rhee, C.; Ekpheghere, K.I.; Kim, I.-S.; Kim, H.-G.; Koh, S.-C. Metagenomic analysis of relationships between the denitrification process and carbon metabolism in a bioaugmented full-scale tannery wastewater treatment plant. World J. Microbiol. Biotechnol. 2019, 35. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zheng, J.; Lu, Y.; Jia, R. Degradation and detoxification of the triphenylmethane dye malachite green catalyzed by crude manganese peroxidase from Irpex lacteus F17. Environ. Sci. Pollut. Res. Int. 2016, 23, 9585–9597. [Google Scholar] [CrossRef]
- Chatha, S.A.S.; Asgher, M.; Iqbal, H.M.N. Enzyme-based solutions for textile processing and dye contaminant biodegradation—A review. Environ. Sci. Pollut. Res. Int. 2017, 24, 14005–14018. [Google Scholar] [CrossRef]
- Liu, S.; Xu, X.; Kang, Y.; Xiao, Y.; Liu, H. Degradation and detoxification of azo dyes with recombinant ligninolytic enzymes from Aspergillus sp. with secretory overexpression in Pichia pastoris: Degradation and detoxification of dye. R. Soc. Open Sci. 2020, 7. [Google Scholar] [CrossRef]
- Seo, J.-S.; Keum, Y.-S.; Li, Q.X. Bacterial degradation of aromatic compounds. Int. J. Environ. Res. Public Health 2009, 6, 278–309. [Google Scholar] [CrossRef] [PubMed]
- Baserba, M.G.; Angelidaki, I.; Karakashev, D.B. Effect of continuous oleate addition on microbial communities involved in anaerobic digestion process. Bioresour. Technol. 2012, 106, 74–81. [Google Scholar] [CrossRef]
- Gu, M.; Yin, Q.; Wu, G. Metagenomic analysis of facilitation mechanism for azo dye reactive red 2 degradation with the dosage of ferroferric oxide. J. Water Process. Eng. 2021, 41, 102010. [Google Scholar] [CrossRef]
- Albertsen, M.; Hansen, L.B.S.; Saunders, A.; Nielsen, P.H.; Nielsen, K.L. A metagenome of a full-scale microbial community carrying out enhanced biological phosphorus removal. ISME J. 2011, 6, 1094–1106. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Yang, B.; Zhou, Y.; Sun, Y.; Ding, C. Effects of azo dye on simultaneous biological removal of azo dye and nutrients in wastewater. R. Soc. Open Sci. 2018, 5, 180795. [Google Scholar] [CrossRef] [Green Version]
- Scherr, K.E.; Lundaa, T.; Klose, V.; Bochmann, G.; Loibner, A.P. Changes in bacterial communities from anaerobic digesters during petroleum hydrocarbon degradation. J. Biotechnol. 2012, 157, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Zielinska, M.; Rusanowska, P.; Jarząbek, J.; Nielsen, J.L. Community dynamics of denitrifying bacteria in full-scale wastewater treatment plants. Environ. Technol. 2016, 37, 2358–2367. [Google Scholar] [CrossRef]
- Prasad, A.S.A.; Satyanarayana, V.S.V.; Rao, K.B. Biotransformation of direct blue 1 by a moderately halophilic bacterium Marinobacter sp. strain HBRA and toxicity assessment of degraded metabolites. J. Hazard. Mater. 2013, 262, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-M.; Chen, W.-M.; Chen, B.-Y.; Chang, C.-T.; Hsueh, C.-C.; Ding, Y.; Lin, K.-L.; Xu, H. Comparative study on characteristics of azo dye decolorization by indigenous decolorizers. Bioresour. Technol. 2010, 101, 2651–2656. [Google Scholar] [CrossRef]
- Yang, X.; López-Grimau, V.; Vilaseca, M.; Crespi, M. Treatment of textile wastewater by CAS, MBR, and MBBR: A comparative study from technical, economic, and environmental perspectives. Water 2020, 12, 1306. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, W.; Ma, J.; Cai, J.; Li, S.; Zhu, X.; Yang, G.; Zhao, X. Biodegradation of azo dye disperse orange S-RL by a newly isolated strain Acinetobacter sp. SRL8. Water Environ. Res. 2015, 87, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Winsor, G.; Lam, D.K.W.; Fleming, L.; Lo, R.; Whiteside, M.D.; Yu, N.; Hancock, R.; Brinkman, F.S.L. Pseudomonas genome database: Improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 2011, 39, D596–D600. [Google Scholar] [CrossRef]
- Kudlich, M.; Bishop, P.L.; Knackmuss, H.-J.; Stolz, A. Simultaneous anaerobic and aerobic degradation of the sulfonated azo dye mordant yellow 3 by immobilized cells from a naphthalenesulfonate-degrading mixed culture. Appl. Microbiol. Biotechnol. 1996, 46, 597–603. [Google Scholar] [CrossRef]
- Krishnamoorthy, R.; Choudhury, A.R.; Jose, P.A.; Suganya, K.; Senthilkumar, M.; Prabhakaran, J.; Gopal, N.O.; Choi, J.; Kim, K.; Anandham, R.; et al. Long-term exposure to azo dyes from textile wastewater causes the abundance of Saccharibacteria population. Appl. Sci. 2021, 11, 379. [Google Scholar] [CrossRef]
- Praus, P. Principal component weighted index for wastewater quality monitoring. Water 2019, 11, 2376. [Google Scholar] [CrossRef] [Green Version]
- Unnikrishnan, S.; Khan, M.H.; Ramalingam, K. Dye-tolerant marine Acinetobacter baumannii-mediated biodegradation of reactive red. Water Sci. Eng. 2018, 11, 265–275. [Google Scholar] [CrossRef]
- Kim, I.-S.; Ekpeghere, K.; Ha, S.-Y.; Kim, S.-H.; Kim, B.-S.; Song, B.; Chun, J.; Chang, J.-S.; Kim, H.-G.; Koh, S.-C. An eco-friendly treatment of tannery wastewater using bioaugmentation with a novel microbial consortium. J. Environ. Sci. Health Part A Toxic Hazard. Subst. Environ. Eng. 2013, 48. [Google Scholar] [CrossRef]
- Ajibade, F.O.; Wang, H.-C.; Guadie, A.; Ajibade, T.F.; Fang, Y.-K.; Sharif, H.M.A.; Liu, W.-Z.; Wang, A.-J. Total nitrogen removal in biochar amended non-aerated vertical flow constructed wetlands for secondary wastewater effluent with low C/N ratio: Microbial community structure and dissolved organic carbon release conditions. Bioresour. Technol. 2021, 322, 124430. [Google Scholar] [CrossRef]
- Guo, J.; Li, J.; Chen, H.; Bond, P.; Yuan, Z. Metagenomic analysis reveals wastewater treatment plants as hotspots of antibiotic resistance genes and mobile genetic elements. Water Res. 2017, 123, 468–478. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 March 2017 (67 Days Before Augmentation) | 29 June 2017 (50 Days After Augmentation) | 10 March 2018 (300 Days After Augmentation) | 26 October 2018 (531 Days After Augmentation) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Influent [I] | Effluent [E] | Removal Rate (%) | Influent [I] | Buffering Tank [B] | Effluent [E] | Sludge Digestion [SD] | Removal Rate (%) | Influent [I] | Buffering Tank [B] | Effluent [E] | Sludge Digestion [SD] | Removal Rate (%) | Influent [I] | Buffering Tank [B] | Effluent [E] | Sludge Digestion [SD] | Removal Rate (%) | |
| COD (mg/L) | 584 ± 8.5 c | 20.6 ± 0.4 a | 94.9 | 674 ± 51.6 d | 466 ± 24.7 c | 14.5 ± 0.3 a | 141.5 ± 57 b | 97.8 | 472 ± 3.5 d | 440 ± 13 c | 21.1 ± 0.4 a | 2228 ± 3.5 e | 95.5 | 237 ± 79 b | 2112 ± 65 d | 7.3 ± 0.9 a | 1678 ± 76 c | 97.3 |
| T-N (mg/L) | 36.2 ± 1.3 b | 18.8 ± 0.8 a | 48 | 23 ± 0 c | 33.4 ± 0.8 d | 8.6 ± 0.4 b | 22.5 ± 0.1 c | 62.6 | 51.2 ± 0.1 c | 48.8 ± 2.7 c | 9.7 ± 1.4 a | 30.5 ± 1.3 b | 80.9 | 89.4 ± 0.74 e | 81.4 ± 0.96 d | 21.5 ± 3.1 a | 62.3 ± 0.5 c | 75.9 |
| T-P (mg/L) | 5.1 ± 0.5 b | 0.43 ± 0.01 a | 91.5 | 2.4 ± 0.1 b | 1.9 ± 0 b | 0.1 ± 0 c | 6.0 ± 1.2 a | 95.8 | 3.3 ± 0.1 c | 4.5 ± 0.4 d | 0.1 ± 0.04 a | 1.9 ± 0.09 b | 96.2 | 7 ± 0.1 d | 3.9 ± 1.2 d | 2.1 ± 0.41 bc | 2.9 ± 0.56 c | 70 |
| NH3 (mg/L) | 14.8 ± 0.5 b | 8.9 ± 0.4 a | 43.6 | 10.5 ± 0.1 c | 9.6 ± 0.1 b | 6.1 ± 0.2 a | 9.5 ± 0.1 b | 41.9 | 7.2 ± 0.9 c | 9.5 ± 0.5 c | 4.1 ± 0 a | 10.8 ± 0.5 c | 48.89 | 16 ± 0.2 e | 13.9 ± 0.1 c | 6 ± 0.7 a | 17.9 ± 0.4 f | 62.5 |
| NO2− (mg/L) | 1.7 c | 0.3 a | 82.3 | 1.43 d | 1.01 b | 1.4 c,d | 0.9 b | 2 | 1.92 c | 0.95 b | 0.17 a | 0.9 b | 91.1 | 2.5 ± 0.6 c | 1.6 ± 0.23 b | 0.4 ± 0.1 a | 2.8 ± 0.1 c | 84 |
| NO3− (mg/L) | 7.7 ± 0.2 c | 0.8 ± 0.07 a | 89.6 | 7.1 ± 0.5 d | 5.4 ± 0 c | 2.8 ± 0.4 b | 14.6 ± 0.2 e | 60.5 | 3.5 ± 0.14 b | 4.6 ± 0.6 c | 1.8 ± 0.4 a | 1.5 ± 0.03 d | 47.7 | 11.7 ± 0.4 c | 7.74 ± 0.2 b | 2.1 ± 0.2 a | 3.59 ± 0.5 a | 82 |
| PO4 3−(mg/L) | 3.4 ± 0 c | 0.2 ± 0.01 a | 94.1 | 3.54 ± 0.2 d | 4.3 ± 0.05 e | 0.16 ± 0 a | 3.41 ± 0.05 c | 95.4 | 3.9 ± 0.3 c | 0.8 ± 0.1 a | 0.2 ± 0.01 a | 4.3 ± 0.6 c | 94.3 | 5 ± 0.3 e | 1.5 ± 2.8 b | 0.9 ± 0.04 a | 0.0 ± 0.2 d | 82 |
| SS (mg/L) | - | - | - | 123 | - | 45 | - | 63.4 | 776 | - | 97 | - | 87.5 | 478 | - | - | 89 | 81.3 |
| Pt-Co(PCU) | - | - | - | 347.9 ± 0.2 | - | - | 76.7 ± 1.1 | 77.9 | 305.1 ± 1.3 | - | - | 30.4 ± 1.5 | 90 | 361.1 ± 0.4 | - | - | 21.4 ± 0 | 94 |
| Sampling Period | Influent (M3) | Influent | Effluent | Sludge | Total Influent | Sludge | Sludge Reduction *** |
|---|---|---|---|---|---|---|---|
| (ton) | Per ton of Influent | % | |||||
| COD | COD | COD | COD | ||||
| (g/m3) | (g/m3) | (ton) | |||||
| 2017. 3 * | 411,588 | 664 | 34 | 1690 | 273 | 6.18 | − |
| 2018. 10 ** | 385,158 | 560 | 33 | 1007 | 217 | 5.00 | 22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santhanarajan, A.E.; Sul, W.-J.; Yoo, K.-J.; Seong, H.-J.; Kim, H.-G.; Koh, S.-C. Metagenomic Insight of a Full Scale Eco-Friendly Treatment System of Textile Dye Wastewater Using Bioaugmentation of the Composite Culture CES-1. Microorganisms 2021, 9, 1503. https://doi.org/10.3390/microorganisms9071503
Santhanarajan AE, Sul W-J, Yoo K-J, Seong H-J, Kim H-G, Koh S-C. Metagenomic Insight of a Full Scale Eco-Friendly Treatment System of Textile Dye Wastewater Using Bioaugmentation of the Composite Culture CES-1. Microorganisms. 2021; 9(7):1503. https://doi.org/10.3390/microorganisms9071503
Chicago/Turabian StyleSanthanarajan, Aalfin Emmanuel, Woo-Jun Sul, Keun-Je Yoo, Hoon-Je Seong, Hong-Gi Kim, and Sung-Cheol Koh. 2021. "Metagenomic Insight of a Full Scale Eco-Friendly Treatment System of Textile Dye Wastewater Using Bioaugmentation of the Composite Culture CES-1" Microorganisms 9, no. 7: 1503. https://doi.org/10.3390/microorganisms9071503
APA StyleSanthanarajan, A. E., Sul, W. -J., Yoo, K. -J., Seong, H. -J., Kim, H. -G., & Koh, S. -C. (2021). Metagenomic Insight of a Full Scale Eco-Friendly Treatment System of Textile Dye Wastewater Using Bioaugmentation of the Composite Culture CES-1. Microorganisms, 9(7), 1503. https://doi.org/10.3390/microorganisms9071503