
Spatial and Temporal Distribution of Bacterioplankton Molecular Ecological Networks in the Yuan River under Different Human Activity Intensity

Abstract
:1. Introduction
2. Materials and Methods
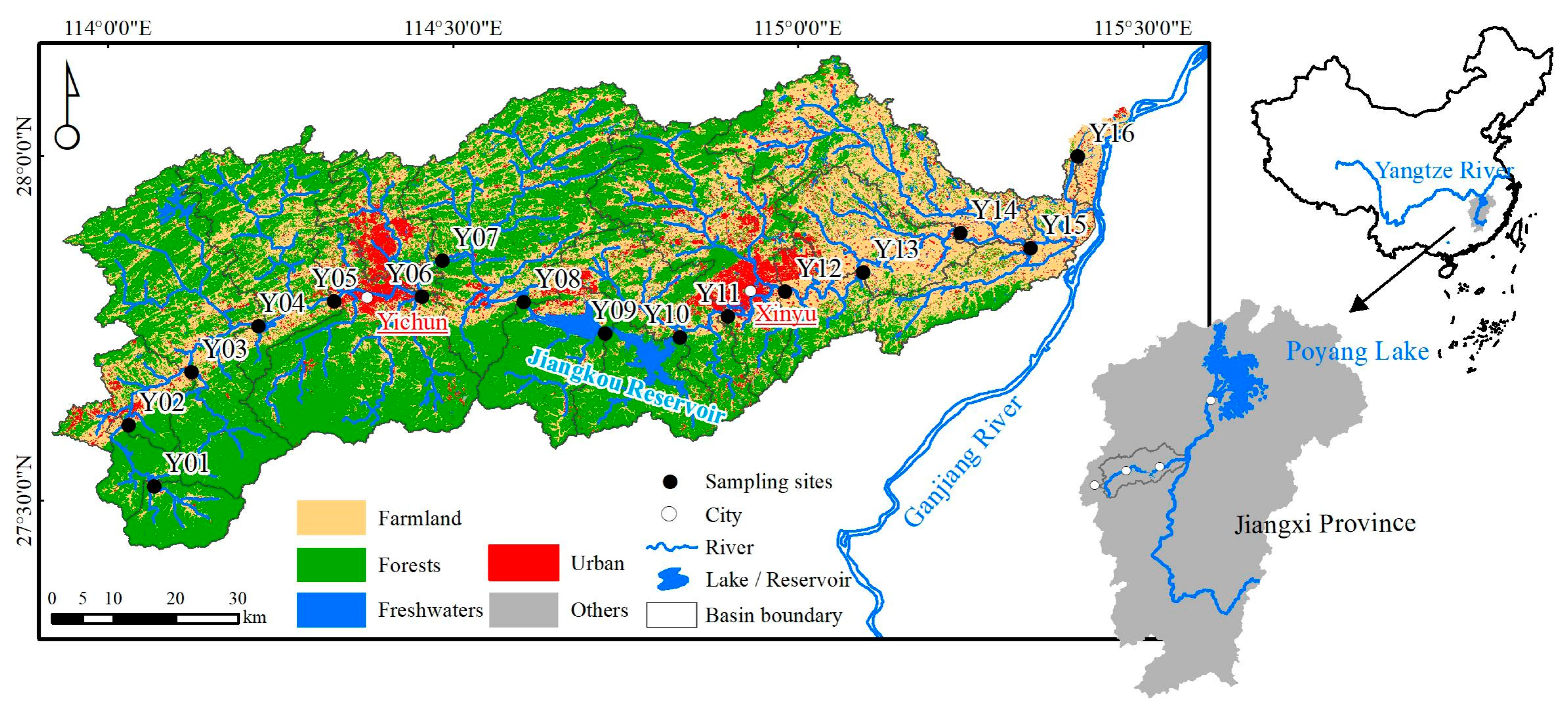
2.1. Study Area, Sampling and Physiochemical Analysis
2.2. DNA Extraction and Illumina DNA Sequencing
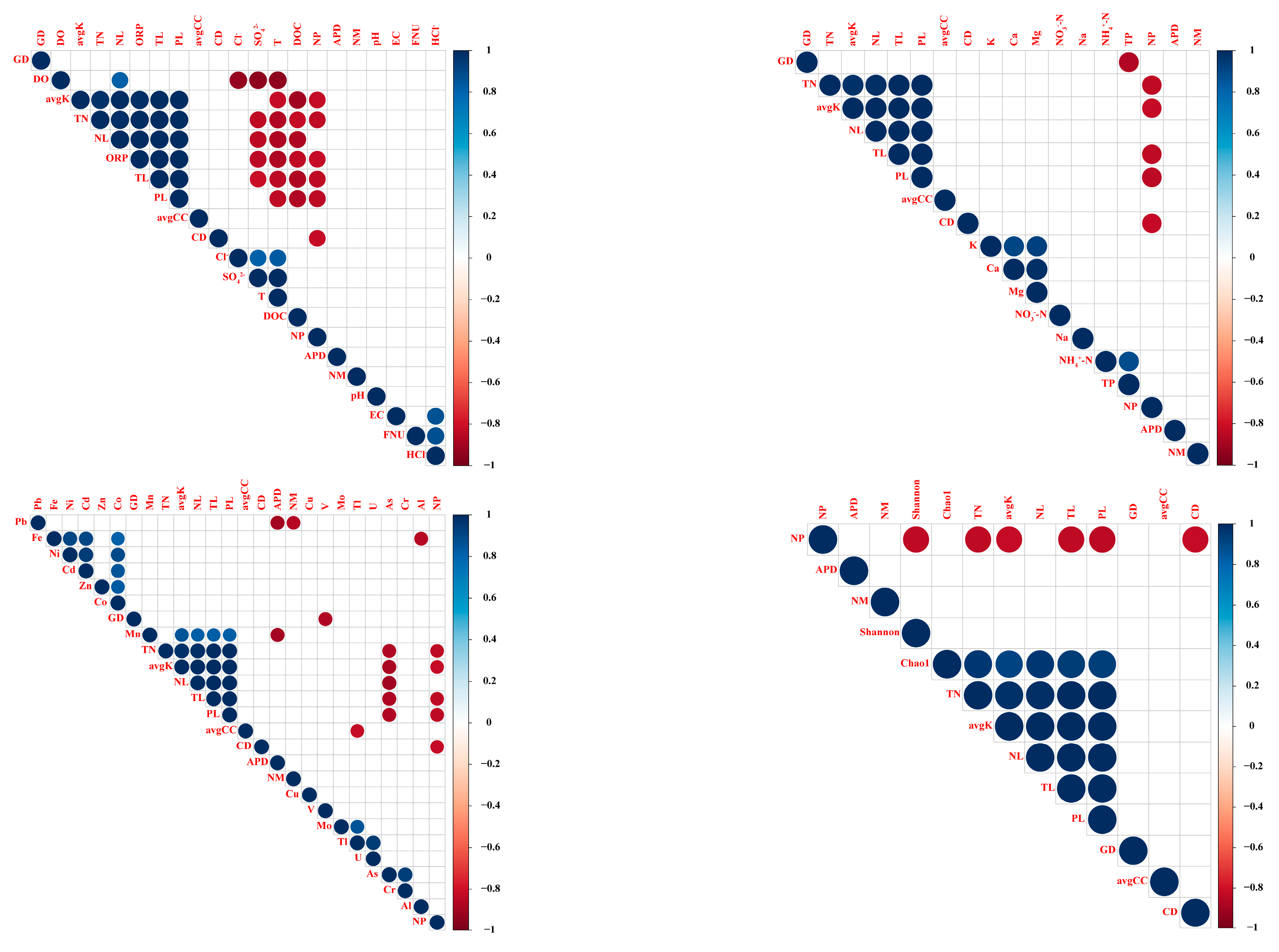
2.3. Statistical Analyses
3. Results
3.1. Quantification of the Human Activity Intensity
3.2. Taxonomic Diversity of the Bacterioplankton Community in the Yuan River
3.3. Taxonomic Composition and Functional Analysis of the Bacterioplankton Community in the Yuan River
3.4. Interactions between Bacterial Taxa in the Network
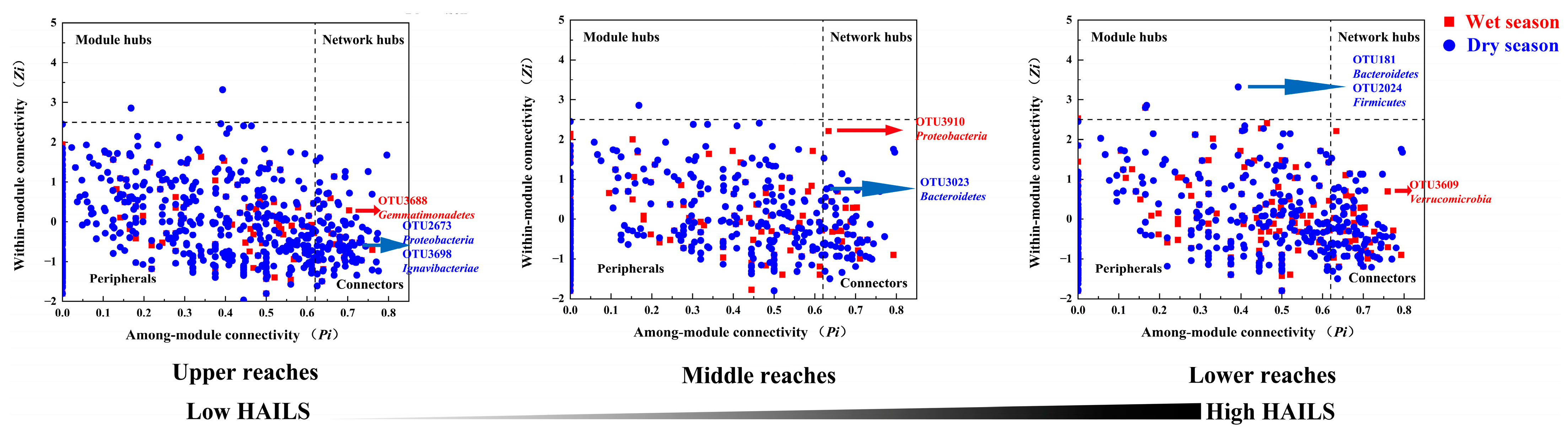
3.5. Keystone Species in Bacterial Networks
3.6. Major Modules in Bacterial Networks
4. Discussion
4.1. Human Activity Mediates the Assembly Processes of Bacterioplankton Communities via Altering Environmental Conditions
4.2. Human Activity Destabilizes Bacterioplankton Molecular Ecological Network Stability in the Yuan River
4.3. Human Activity Increase Triggers Keystone Species Change
4.4. Human Activity Reduces Ecological Niche Differentiation and These Interacts Less with Each Other in the Yuan River
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adhikari, N.P.; Liu, Y.; Liu, K.; Zhang, F.; Adhikari, S.; Chen, Y.; Liu, X. Bacterial community composition and diversity in Koshi River, the largest river of Nepal. Ecol. Indic. 2019, 104, 501–511. [Google Scholar] [CrossRef]
- Bai, Y.; Qi, W.; Liang, J.; Qu, J. Using high-throughput sequencing to assess the impacts of treated and untreated wastewater discharge on prokaryotic communities in an urban river. Appl. Microbiol. Biotechnol. 2014, 98, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fang, W.; Li, X.; Lu, W.; Li, J. Strong linkages between dissolved organic matter and the aquatic bacterial community in an urban river. Water Res. 2020, 184, 116089. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, H.; Li, X.; Su, Z.; Li, X.; Xu, M. Effects of tillage and residue incorporation on composition and abundance of microbial communities of a fluvo-aquic soil. Eur. J. Soil Biol. 2014, 65, 70–78. [Google Scholar] [CrossRef]
- Zaneveld, J.R.; McMinds, R.; Vega Thurber, R. Stress and stability: Applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2017, 2, 17121. [Google Scholar] [CrossRef]
- Yuan, M.M.; Guo, X.; Wu, L.; Zhang, Y.; Xiao, N.; Ning, D.; Shi, Z.; Zhou, X.; Wu, L.; Yang, Y.; et al. Climate warming enhances microbial network complexity and stability. Nat. Clim. Chang. 2021, 11, 343–348. [Google Scholar] [CrossRef]
- Goldford, J.E.; Lu, N.; Bajic, D.; Estrela, S.; Tikhonov, M.; Sanchez-Gorostiaga, A.; Segre, D.; Mehta, P.; Sanchez, A. Emergent simplicity in microbial community assembly. Science 2018, 361, 469–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Wang, P.; Devlin, A.T.; Xiao, S.; Ding, M. Influence of Soil and Water Conservation Measures on Soil Microbial Communities in a Citrus Orchard of Southeast China. Microorganisms 2021, 9, 319. [Google Scholar] [CrossRef]
- Fan, K.; Weisenhorn, P.; Gilbert, J.A.; Chu, H. Wheat rhizosphere harbors a less complex and more stable microbial co-occurrence pattern than bulk soil. Soil Biol. Biochem. 2018, 125, 251–260. [Google Scholar] [CrossRef]
- David, A.S.; Quintana-Ascencio, P.F.; Menges, E.S.; Thapa-Magar, K.B.; Searcy, C.A. Soil Microbiomes Underlie Population Persistence of an Endangered Plant Species. Am. Nat. 2019, 194, 488–494. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, X.; Tang, Q. Human activity intensity of land surface: Concept, methods and application in China. J. Geogr. Sci. 2015, 26, 1349–1361. [Google Scholar] [CrossRef]
- Wu, J.S.; Cao, Q.W.; Shi, S.Q.; Huang, X.L.; Lu, Z.Q. Spatio-temporal variability of habitat quality in Beijing-Tianjin-Hebei Area based on land use change. Chin. J. Appl. Ecol. 2015, 26, 3457–3466. [Google Scholar]
- Liu, T.; Zhang, A.N.; Wang, J.; Liu, S.; Jiang, X.; Dang, C.; Ma, T.; Liu, S.; Chen, Q.; Xie, S. Integrated biogeography of planktonic and sedimentary bacterial communities in the Yangtze River. Microbiome 2018, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chen, H.; Abdullah Al, M.; Ndayishimiye, J.C.; Yang, J.R.; Isabwe, A.; Luo, A.; Yang, J. Urbanization reduces resource use efficiency of phytoplankton community by altering the environment and decreasing biodiversity. J. Environ. Sci.-China 2022, 112, 140–151. [Google Scholar] [CrossRef]
- Peng, F.; Guo, Y.; Isabwe, A.; Chen, H.; Wang, Y.; Zhang, Y.; Zhu, Z.; Yang, J. Urbanization drives riverine bacterial antibiotic resistome more than taxonomic community at watershed scale. Environ. Int. 2020, 137, 105524. [Google Scholar] [CrossRef]
- Zhao, J.; Peng, W.; Ding, M.; Nie, M.; Huang, G. Effect of Water Chemistry, Land Use Patterns, and Geographic Distances on the Spatial Distribution of Bacterioplankton Communities in an Anthropogenically Disturbed Riverine Ecosystem. Front. Microbiol. 2021, 12, 1089. [Google Scholar] [CrossRef]
- Mishra, A.; Alnahit, A.; Campbell, B. Impact of land uses, drought, flood, wildfire, and cascading events on water quality and microbial communities: A review and analysis. J. Hydrol. 2021, 596, 125707. [Google Scholar] [CrossRef]
- Savio, D.; Sinclair, L.; Ijaz, U.Z.; Parajka, J.; Reischer, G.H.; Stadler, P.; Blaschke, A.P.; Bloschl, G.; Mach, R.L.; Kirschner, A.K.; et al. Bacterial diversity along a 2600 km river continuum. Environ. Microbiol. 2015, 17, 4994–5007. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Yu, K.; Liao, Z.; Yu, X.; Qin, Z.; Liang, J.; Wang, G.; Wu, Q.; Jiang, L. Microbiome community and complexity indicate environmental gradient acclimatisation and potential microbial interaction of endemic coral holobionts in the South China Sea. Sci. Total Environ. 2021, 765, 142690. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Li, Y.; Li, Y.; Hu, Q.; Wang, C.; Hu, J.; Zhang, W.; Wang, L.; Zhang, C.; Zhang, H. New insights into the vertical distribution and microbial degradation of microplastics in urban river sediments. Water Res. 2021, 188, 116449. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Yan, D.; Sun, R.; Song, X.; Lin, T.; Yi, Y. Community composition and correlations between bacteria and algae within epiphytic biofilms on submerged macrophytes in a plateau lake, southwest China. Sci. Total Environ. 2020, 727, 138398. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Lu, Y. Abundant fungi adapt to broader environmental gradients than rare fungi in agricultural fields. Glob. Chang. Biol. 2020, 26, 4506–4520. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Yang, T.; Bao, Y.; He, P.; Yang, K.; Mei, X.; Wei, Z.; Xu, Y.; Shen, Q.; Banerjee, S. Network analysis and subsequent culturing reveal keystone taxa involved in microbial litter decomposition dynamics. Soil Biol. Biochem. 2021, 157, 108230. [Google Scholar] [CrossRef]
- Barberan, A.; Bates, S.; Casamayor, E.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Kirkby, C.A.; Schmutter, D.; Bissett, A.; Kirkegaard, J.A.; Richardson, A.E. Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biol. Biochem. 2016, 97, 188–198. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.; Wei, G. Biogeography and ecological diversity patterns of rare and abundant bacteria in oil-contaminated soils. Mol. Ecol. 2017, 26, 5305–5317. [Google Scholar] [CrossRef]
- Wan, X.; Gao, Q.; Zhao, J.; Feng, J.; van Nostrand, J.D.; Yang, Y.; Zhou, J. Biogeographic patterns of microbial association networks in paddy soil within Eastern China. Soil Biol. Biochem. 2020, 142, 107696. [Google Scholar] [CrossRef]
- Coyte, K.Z.; Schluter, J.; Foster, K.R. The ecology of the microbiome: Networks, competition, and stability. Science 2015, 350, 663–666. [Google Scholar] [CrossRef]
- De Vries, F.T.; Griffiths, R.I.; Bailey, M.; Craig, H.; Girlanda, M.; Gweon, H.S.; Hallin, S.; Kaisermann, A.; Keith, A.M.; Kretzschmar, M.; et al. Soil bacterial networks are less stable under drought than fungal networks. Nat. Commun. 2018, 9, 3033. [Google Scholar] [CrossRef] [Green Version]
- Zaura, E.; Keijser, B.J.; Crielaard, H.W. Defining the healthy ”core microbiome” of oral microbial communities. BMC Microbiol. 2009, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ze, X.; Le, M.F.; Duncan, S.H.; Louis, P.; Flint, H.J. Some are more equal than others: The role of "keystone" species in the degradation of recalcitrant substrates. Gut Microbes 2013, 4, 236. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zheng, X.; Zhang, H.; Xiao, F.; He, Z.; Yan, Q. Keystone taxa of water microbiome respond to environmental quality and predict water contamination. Environ. Res. 2020, 187, 109666. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Reith, F.; Dennis, P.G.; Hamonts, K.; Powell, J.R.; Young, A.; Singh, B.K.; Bissett, A. Ecological drivers of soil microbial diversity and soil biological networks in the Southern Hemisphere. Ecology 2018, 99, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Sung, J.; Kim, S.; Cabatbat, J.; Jang, S.; Jin, Y.S.; Jung, G.Y.; Chia, N.; Kim, P.J. Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis. Nat. Commun. 2017, 8, 15393. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.P.; Wang, P.; Xu, Q.Y.; Shu, W.; Ding, M.J.; Zhang, H.; Zeng, T. Influence of Land Use on Carbon, Nitrogen and Phosphorus in the Water of the Yuan River Basin. Res. Environ. Sci. 2021, 34, 1–13. [Google Scholar]
- Zeng, T.; Wang, P.; Shu, W.; Zhang, H.; Ding, M.J.; Huang, G.X. Distribution characteristics and source analysis of dissolved trace metal elements in the Yuan River. Environ. Chem. 2021, 40, 583–591. [Google Scholar]
- Xu, Q.; Wang, P.; Shu, W.; Ding, M.; Zhang, H. Influence of landscape structures on river water quality at multiple spatial scales: A case study of the Yuan river watershed, China. Ecol. Indic. 2021, 121, 107226. [Google Scholar] [CrossRef]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Peng, F.; Gao, X.; Xiao, P.; Logares, R.; Jeppesen, E.; Ren, K.; Xue, Y.; Yang, J. Low shifts in salinity determined assembly processes and network stability of microeukaryotic plankton communities in a subtropical urban reservoir. Microbiome 2021, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- Guimerà, R.; Nunes Amaral, L.A. Functional cartography of complex metabolic networks. Nature 2005, 433, 895–900. [Google Scholar] [CrossRef] [Green Version]
- Poudel, R.; Jumpponen, A.; Schlatter, D.; Paulitz, T.C.; Garrett, K.A. Microbiome Networks: A Systems Framework for Identifying Candidate Microbial Assemblages for Disease Management. Phytopathology 2016, 106, 1083–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tylianakis, J.M.; Morris, R.J. Ecological Networks across Environmental Gradients. Annu. Rev. Ecol. Evol. Syst. 2017, 48, 25–48. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, J.; Li, Z.; Xin, X.; Guo, Z.; Wang, D.; Li, D.; Zhao, B. Long-term phosphorus deficiency decreased bacterial-fungal network complexity and efficiency across three soil types in China as revealed by network analysis. Appl. Soil Ecol. 2020, 148, 103506. [Google Scholar] [CrossRef]
- Chen, W.; Ren, K.; Isabwe, A.; Chen, H.; Liu, M.; Yang, J. Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome 2019, 7, 138. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Pan, B.; He, H.; Zhao, G.; Hou, Y.; Zhu, P. Assembly processes and co-occurrence relationships in the bacterioplankton communities of a large river system. Ecol. Indic. 2021, 126, 107643. [Google Scholar] [CrossRef]
- Read, D.S.; Gweon, H.S.; Bowes, M.J.; Newbold, L.K.; Field, D.; Bailey, M.J.; Griffiths, R.I. Catchment-scale biogeography of riverine bacterioplankton. ISME J. 2015, 9, 516–526. [Google Scholar]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Disentangling Interactions in the Microbiome: A Network Perspective. Trends Microbiol. 2017, 25, 217–228. [Google Scholar] [CrossRef]
- Zhang, L.; Zhong, M.; Li, X.; Lu, W.; Li, J. River bacterial community structure and co-occurrence patterns under the influence of different domestic sewage types. J. Environ. Manag. 2020, 266, 110590. [Google Scholar] [CrossRef]
- Williams, R.J.; Howe, A.; Hofmockel, K.S. Demonstrating microbial co-occurrence pattern analyses within and between ecosystems. Front. Microbiol. 2014, 5, 358. [Google Scholar] [CrossRef]
- Banerjee, S.; Baah-Acheamfour, M.; Carlyle, C.N.; Bissett, A.; Richardson, A.E.; Siddique, T.; Bork, E.W.; Chang, S.X. Determinants of bacterial communities in Canadian agroforestry systems. Environ. Microbiol. 2016, 18, 1805–1816. [Google Scholar] [CrossRef]
- Ye, D.; Jiang, Y.H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, A.; Ju, F.; Hou, L.; Li, J.; Yang, X.; Wang, H.; Mulla, S.I.; Sun, Q.; Burgmann, H.; Yu, C.P. Strong impact of anthropogenic contamination on the co-occurrence patterns of a riverine microbial community. Environ. Microbiol. 2017, 19, 4993–5009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lu, X.; Li, Z.; Cheng, Q.; Zhou, Y.; Lei, M. Liming alters microbial community composition and its co-occurrence patterns in Cd- and Pb-contaminated agricultural soil. Appl. Soil Ecol. 2021, 166, 104064. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Shi, S.; Nuccio, E.E.; Shi, Z.J.; He, Z.; Zhou, J.; Firestone, M.K. The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Lupatini, M.; Suleiman, A.K.A.; Jacques, R.J.S.; Antoniolli, Z.I.; de Siqueira Ferreira, A.; Kuramae, E.E.; Roesch, L.F.W. Network topology reveals high connectance levels and few key microbial genera within soils. Front. Environ. Sci. 2014, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Comte, J.M.; Del Giorgio, P.A. Linking the patterns of change in composition and function in bacterioplankton successions along environmental gradients. Ecology 2010, 91, 1466–1476. [Google Scholar] [CrossRef]
- Dopheide, A.; Lear, G.; He, Z.; Zhou, J.; Lewis, G.D.; Mormile, M.R. Functional Gene Composition, Diversity and Redundancy in Microbial Stream Biofilm Communities. PLoS ONE 2015, 10, e123179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Feng, F.; Medova, H.; Dean, J.; Kobli Ek, M. Functional type 2 photosynthetic reaction centers found in the rare bacterial phylum Gemmatimonadetes. Proc. Natl. Acad. Sci. USA 2014, 111, 7795–7800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Zhao, Q.; Guo, J.; Yan, N.; Chen, H.; Sheng, F.; Lin, Y.; An, D. The microbial community responsible for dechlorination and benzene ring opening during anaerobic degradation of 2,4,6-trichlorophenol. Sci. Total Environ. 2019, 651, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.; Fu, Y.; Jeewani, P.H.; Tang, C.; Pan, S.; Reid, B.J.; Gunina, A.; Li, Y.; Li, Y.; Cai, Y.; et al. Organic matter chemistry and bacterial community structure regulate decomposition processes in post-fire forest soils. Soil Biol. Biochem. 2021, 108311. [Google Scholar] [CrossRef]
- He, S.; Stevens, S.L.R.; Chan, L.; Bertilsson, S.; Glavina Del Rio, T.; Tringe, S.G.; Malmstrom, R.R.; McMahon, K.D. Ecophysiology of Freshwater Verrucomicrobia Inferred from Metagenome-Assembled Genomes. Msphere 2017, 2, e217–e277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Ma, X.; He, N.; Zhang, J.; Wu, J.; Liu, C. Shifts in microbial communities and networks are correlated with the soil ionome in a kiwifruit orchard under different fertilization regimes. Appl. Soil Ecol. 2020, 149, 103517. [Google Scholar] [CrossRef]
- Chun, S.; Cui, Y.; Baek, S.H.; Ahn, C.; Oh, H. Seasonal succession of microbes in different size-fractions and their modular structures determined by both macro- and micro-environmental filtering in dynamic coastal waters. Sci. Total Environ. 2021, 784, 147046. [Google Scholar] [CrossRef]
- Sanfilippo, G.E.; Homola, J.J.; Ross, J.; Kanefsky, J.; Kimmel, J.; Marsh, T.L.; Scribner, K.T. Watershed-scale landuse is associated with temporal and spatial compositional variation in Lake Michigan tributary bacterial communities. J. Gt. Lakes Res. 2021, 47, 862–874. [Google Scholar] [CrossRef]
- Gifford, S.M.; Sharma, S.; Booth, M.; Moran, M.A. Expression patterns reveal niche diversification in a marine microbial assemblage. ISME J. 2013, 7, 281–298. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, F.H.; Meirelles, P.M.; Moreira, A.P.B.; Paranhos, R.P.; Dutilh, B.E.; Thompson, F.L.; Moustafa, A. Niche distribution and influence of environmental parameters in marine microbial communities: A systematic review. PeerJ 2015, 3, e1008. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.J.; Liu, J.J.; Wei, D.; Zhu, P.; Cui, X.A.; Zhou, B.K.; Chen, X.L.; Jin, J.; Liu, X.B.; Wang, G.H. Comparison on fungal molecular ecological networks of agricultural soils with different latitudes in the black soil region of Northeast China. Chin. J. Appl. Ecol. 2018, 29, 3802–3810. [Google Scholar]
- Pernthaler, J. Competition and niche separation of pelagic bacteria in freshwater habitats. Environ. Microbiol. 2017, 19, 2133–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.E.; Newton, R.J.; Mcmahon, K.D. Evidence for structuring of bacterial community composition by organic carbon source in temperate lakes. Environ. Microbiol. 2010, 11, 2463–2472. [Google Scholar] [CrossRef]
- Niño-García, J.P.; Ruiz-González, C.; Del Giorgio, P.A. Interactions between hydrology and water chemistry shape bacterioplankton biogeography across boreal freshwater networks. ISME J. 2016, 10, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judd, K.E.; Kling, C. Variation in dissolved organic matter controls bacterial production and community composition. Ecology 2006, 87, 2068–2079. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network Indexes | Wet Season | ||
|---|---|---|---|
| Upper Reaches | Middle Reaches | Lower Reaches | |
| Total nodes (TNs) | 260 | 257 | 317 |
| Total links (TLs) | 1389 | 896 | 1675 |
| Negative links (NLs) | 480 | 316 | 493 |
| Positive links (PL) | 909 | 580 | 1182 |
| Negative/positive (NP) | 0.528 | 0.545 | 0.417 |
| R square of power-law (R) | 0.25 | 0.639 | 0.488 |
| Average degree (avgK) | 10.685 | 6.973 | 10.568 |
| Average clustering coefficient (avgCC) | 0.342 | 0.299 | 0.264 |
| Average path distance (APD) | 3.814 | 4.509 | 3.49 |
| Centralization of degree (CD) | 0.067 | 0.071 | 0.068 |
| Graph density (GD) | 0.041 | 0.027 | 0.033 |
| Modularity (M) | 0.622 | 0.635 | 0.505 |
| Number of modules (NMs) | 12 | 31 | 10 |
| Network Indexes | Dry Season | ||
|---|---|---|---|
| Upper Reaches | Middle Reaches | Lower Reaches | |
| Total nodes (TN) | 1010 | 534 | 524 |
| Total links (TL) | 1.964 | 4638 | 4811 |
| Negative links (NLs) | 4605 | 1452 | 1560 |
| Positive links (PLs) | 1.4359 | 3186 | 3251 |
| Negative/positive (NP) | 0.321 | 0.456 | 0.480 |
| R square of power-law (R) | 0.306 | 0.431 | 0.433 |
| Average degree (avgK) | 37.552 | 17.371 | 18.363 |
| Average clustering coefficient (avgCC) | 0.426 | 0.416 | 0.221 |
| Average path distance (APD) | 3.483 | 3.857 | 3.21 |
| Centralization of degree (CD) | 0.117 | 0.075 | 0.061 |
| Graph density (GD) | 0.037 | 0.033 | 0.035 |
| Modularity (M) | 0.556 | 0.595 | 0.410 |
| Number of modules (NMs) | 26 | 29 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, B.; Wang, P.; Devlin, A.T.; Chen, L.; Xia, Y.; Zhang, H.; Nie, M.; Ding, M. Spatial and Temporal Distribution of Bacterioplankton Molecular Ecological Networks in the Yuan River under Different Human Activity Intensity. Microorganisms 2021, 9, 1532. https://doi.org/10.3390/microorganisms9071532
Wu B, Wang P, Devlin AT, Chen L, Xia Y, Zhang H, Nie M, Ding M. Spatial and Temporal Distribution of Bacterioplankton Molecular Ecological Networks in the Yuan River under Different Human Activity Intensity. Microorganisms. 2021; 9(7):1532. https://doi.org/10.3390/microorganisms9071532
Chicago/Turabian StyleWu, Bobo, Peng Wang, Adam T. Devlin, Lu Chen, Yang Xia, Hua Zhang, Minghua Nie, and Mingjun Ding. 2021. "Spatial and Temporal Distribution of Bacterioplankton Molecular Ecological Networks in the Yuan River under Different Human Activity Intensity" Microorganisms 9, no. 7: 1532. https://doi.org/10.3390/microorganisms9071532
APA StyleWu, B., Wang, P., Devlin, A. T., Chen, L., Xia, Y., Zhang, H., Nie, M., & Ding, M. (2021). Spatial and Temporal Distribution of Bacterioplankton Molecular Ecological Networks in the Yuan River under Different Human Activity Intensity. Microorganisms, 9(7), 1532. https://doi.org/10.3390/microorganisms9071532