1. Introduction
Sisal is a common name for different species and hybrid varieties in the genus
Agave (especially
Agave sisalana) cultivated worldwide for the production of hard natural fibers [
1,
2]. In Brazil, the greatest producer of sisal fibers, sisal-producing areas are concentrated in the Northeastern region of the country, especially in the state of Bahia [
3], followed by the states of Paraíba and Pernambuco [
4]. The semi-arid portion of this region, known as the
Caatinga biome, bears a high richness of highly adapted species, in all the domains of life [
5,
6]. Although the cultivated
Agave species are not native to that region, they are adapted to the specific environmental conditions of this area, including low rainfall, high temperatures, and low aboveground biomass coverage [
7]. These traits of resistance to abiotic stresses make sisal one of the few cultivation options available in the
Caatinga biome, historically neglected in infrastructure projects.
Viral infections in plants may cause damage to specific structures, such as the photosynthetic apparatus in the leaves [
8], the roots system [
9], and also in growth and development, as in early flowering, often used to accelerate yielding [
10]. In sisal, these forms of damage could harm fiber quality, which is the main commercial and valuable trait in these
Agave species. Studies in other economically important plants, such as grapevine, revealed that the viral diversity goes beyond pathogenic species, which broadens our knowledge of plant viruses [
11,
12]. Therefore, the study of neglected cultures could provide not only a better perspective of viral diversity in economically relevant individuals and related species but also a glimpse of the real circulation of viruses in the region where it is grown.
Sisal and other plants, as all known domains of life, are susceptible to viral infection. Nonetheless, the knowledge on viruses infecting non-
tequilana species in the genus
Agave is very scarce and restricted to low-throughput strategies, with only a few published articles describing infections on sisal until this date. Pinkerton and Bock (1969) [
13] indicated (but did not confirm) viruses as the causative agent of the parallel streak of sisal in Kenya; Galvez et al. (1977) [
14] described the
Necrotic streak virus in the genus
Furcraea (not to be confused with
Agave fourcroydes, one of the plant taxa in the present study), later described in-depth by Morales et al. (1992) [
15]; and Izaguirre-mayoral et al. (1995) [
16], which described the infection of
Cactus X virus on
A. sisalana. In a more recent record, Chabi-Jesus et al. (2019) [
17] described the presence of the
Citrus Chlorotic Spot virus in
Agave desmettiana individuals. Hence, the real diversity of viruses associated with sisal and its respective associated In these cases, different metagenomics strategies have been applied, including small RNA (sRNA) sequencing [
18,
19,
20,
21], DNA sequencing [
22], simultaneous extraction of microorganisms and surroundings is still unclear. As mentioned before, different works in other plant species show an abundant viral diversity, such as those observed in lilies [
20], grapevines [
23], and peaches [
24], including those of viruses infecting plant-associated microorganisms [
25,
26]. DNA and RNA followed by digestion with RNases/DNases [
27], and even amplification-based methods [
28]. Among them, the next-generation sequencing of RNA (metatranscriptomics) has been consolidated as an important unbiased strategy for virome studies [
23,
29,
30]. Indeed, this strategy allows the detection of almost all viral species, since most of the viruses are made of or produce RNA molecules during replication, which would not be detected using DNA deep sequencing.
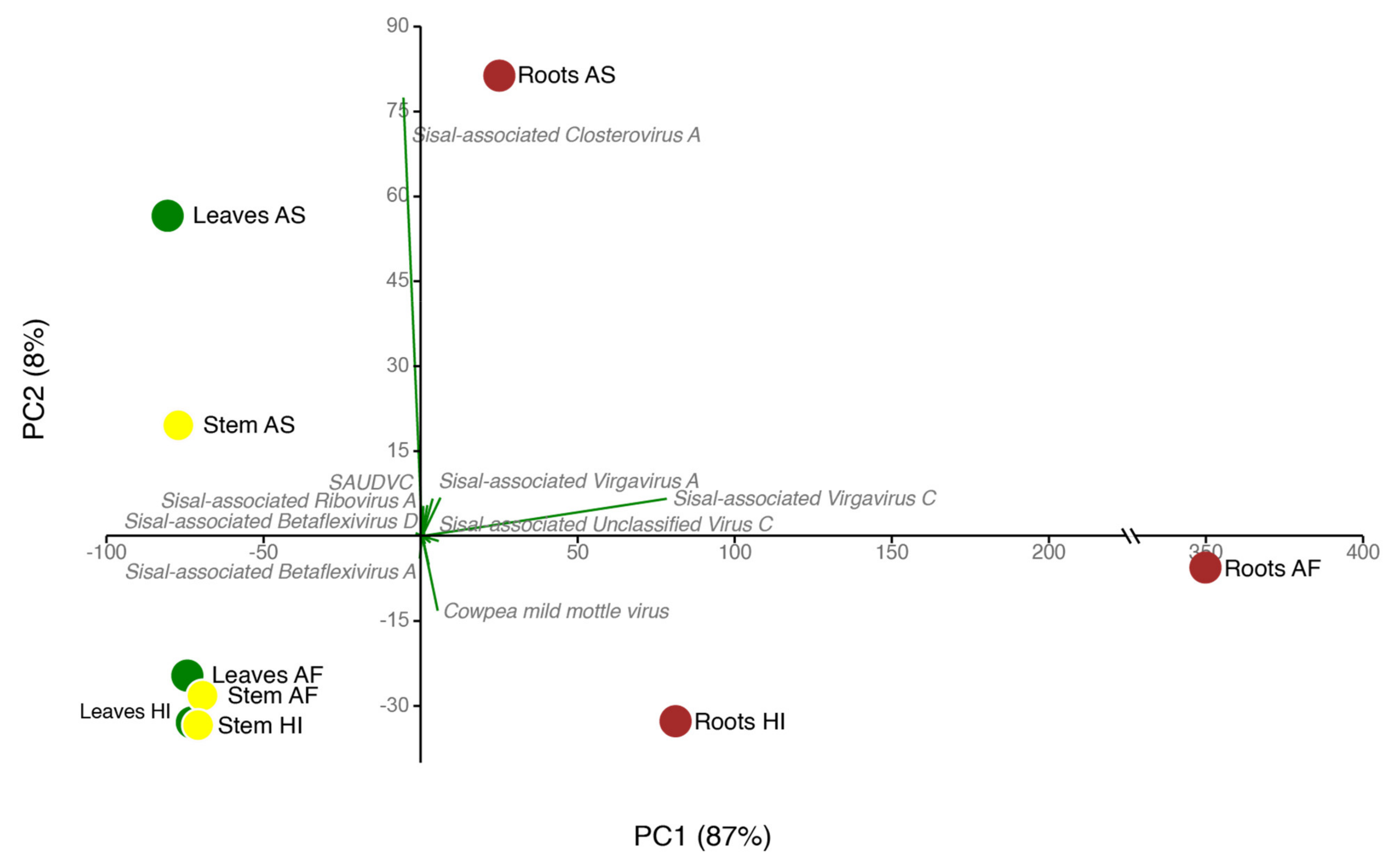
Therefore, our study focused on uncovering the viral diversity of Agave species using metatranscriptomics. We investigate three different Agave cultivars used for fiber extraction (A. fourcroydes, A. sisalana and Agave Hybrid 11648) collecting samples from three different organs (leaves, stems, and roots) in biological triplicates. Using this approach in association with DNA deep sequencing to filter out endogenous elements, we identified 25 putative viral species in asymptomatic Agave individuals, including the known species Cowpea Mild Mottle Virus (CPMMV) and 24 previously unknown viral species, belonging to at least six viral families: Alphaflexiviridae, Betaflexiviridae, Botourmiaviridae, Closteroviridae, Partitiviridae, Virgaviridae and three distinct unclassified viruses. A. fourcroydes displayed the highest diversity among the three Agave taxa while the roots were the plant organ with the highest viral diversity. We also observed discrepant abundance for the same virus among different organs, highlighting replication strategy preferences, such as observed for Sisal-associated Closterovirus A (higher in leaves and stems) and Sisal-associated Virgavirus C (higher in roots). Altogether, our results highlight both the importance of unbiased high-throughput strategies for the discovery of new viral species and also the relevance of screening asymptomatic plants for obtaining a more realistic viral diversity scenario.
4. Discussion
Plants, such as all the known living organisms, are susceptible to viral infections. Some viral infections are cryptic, i.e., a given virus infects a host with no apparent symptoms [
50,
51]. In this study, we have described 25 cryptical viral species associated with three plant taxa in the genus
Agave:
Agave fourcroydes,
Agave sisalana and
Agave hybrid 11648 using RNA-sequencing and genomic DNA sequencing for curation. The use of metagenomics-based approaches for the discovery of viral species is seen in research conducted by Charon et al. [
52], and Wolf et al. [
53], and also reviewed by Greninger [
54], Maclot et al. [
55], and Shi et al. [
56]. In our methods, we also discarded false-positive results and endogenous elements (EVEs). The plants were grown in a genetic collection, located in an area with very typical climatic and soil conditions of the sisal producing areas in Brazil. To the best of our knowledge, this is the first unbiased virome study in the genus
Agave, the first virology study in
A. fourcroydes and A. hybrid 11648, as well as the first virology study in
A. sisalana since the 1995 study by Izaguirre-mayoral et al. [
16], with another two earlier studies on the viral streak of sisal [
13,
14]. Among the 25 total species described in our study, only one is a known species,
Cowpea mild mottle virus (CPMMV), and all the other 24 are new viral species. Our approach was based on the similarity of transcripts detected in leaf, stem, and root samples from these three plant taxa to viral nucleotide sequences and protein domains in public databases, followed by the verification of the origin of those sequences via genomic DNA sequencing, removal of false positives, and identity confirmation via phylogeny, when possible.
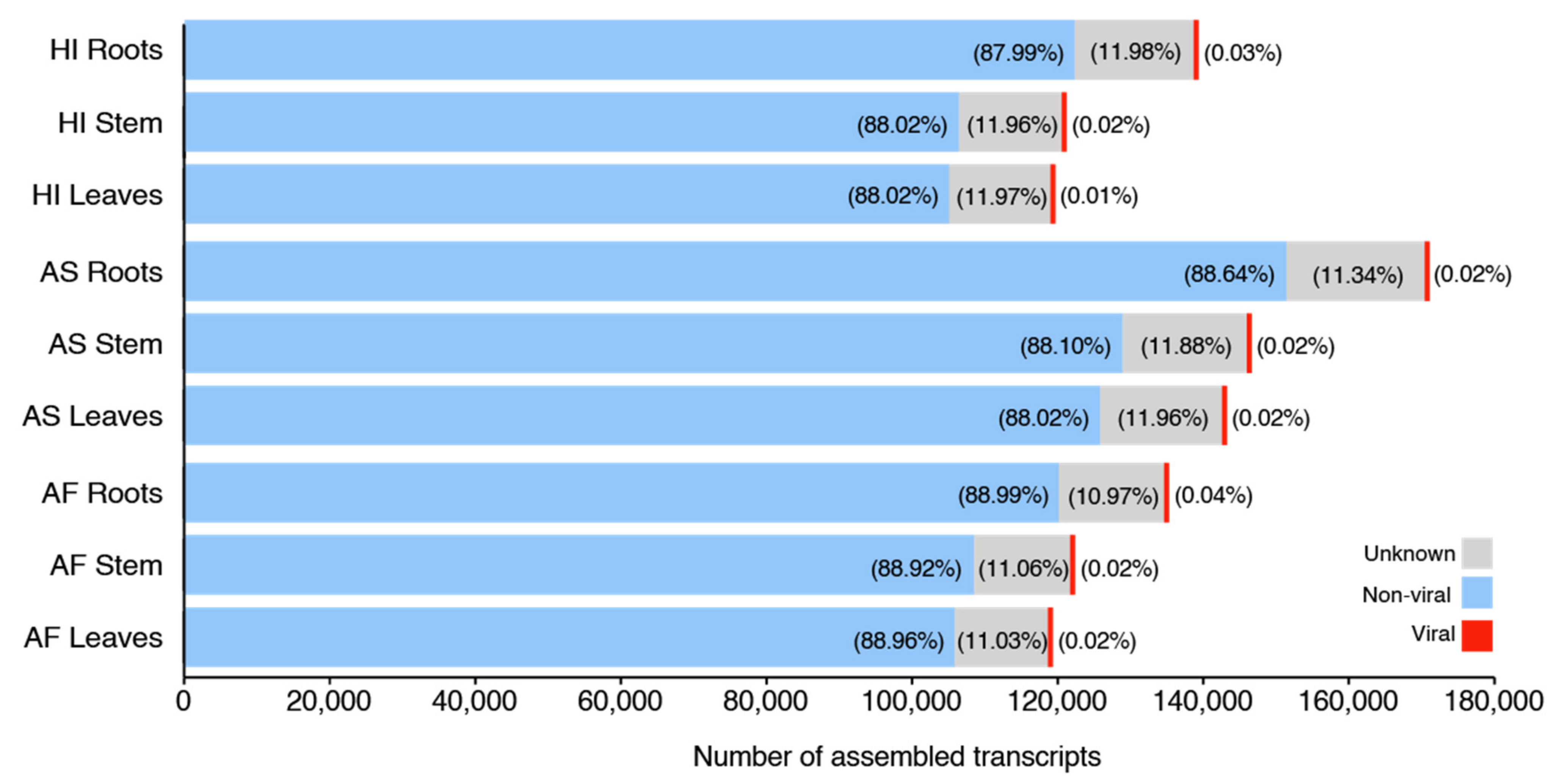
CPMMV was the only viral species detected in our samples through sequence similarity search at the nucleotide level (
Figures S1–S3,
Tables S3 and S4). On the other hand, searches at the protein level were able to find sequences (our remaining 24 viral species) sharing lower similarity with known references, thus being considered a new species. CPMMV was first identified by Brunt and Kenten in 1973 [
57], infecting the Cowpea (
Vigna unguiculata, hence the name) in Ghana, and after that in a broad range of other species (
Phaseolus vulgaris, Glycine max, Nicotiana clevelandii, Theobroma cacao, among others) in vitro. Symptoms included mild to severe mottle chlorosis followed by leaf necrosis, however, visually symptomless individuals, as in our case with the
Agave species, have been described in this same study. Brunt and Kenten also described how this virus was spread by sap-feeding aphids, however, their results indicate that spreading was dependent on other viruses, such as the
Potato Y virus or
Pepper Veinal Mottle virus. Following their discovery, the whitefly (
Bemisia tabacci) is now widely accepted as the sole vector of CPMMV [
58], and the occurrence of the whitefly in the state of Paraíba is also described in the literature [
59,
60,
61], which is suggestive that this insect might also act as the vector for CPMMV in this environment. Nonetheless, CPMMV is more expressed in the stems and roots in all three
Agave species, not being detected at any levels in the leaves of
A. sisalana or A. hybrid 11648, and very low levels in the leaves of
A. fourcroydes, which is in opposition to earlier findings for this species, considering the interaction of its vector (the whitefly) with plant leaves. The isolate PB:AF is also phylogenetically closer to other Brazilian isolates (highlighted in blue), considering all genomes available for this species so far (
Figure S4). Of note, CPMMV seems to be a broad range of hosts, since isolates have been identified in many hosts, such as soybean, the common bean, and papaya.
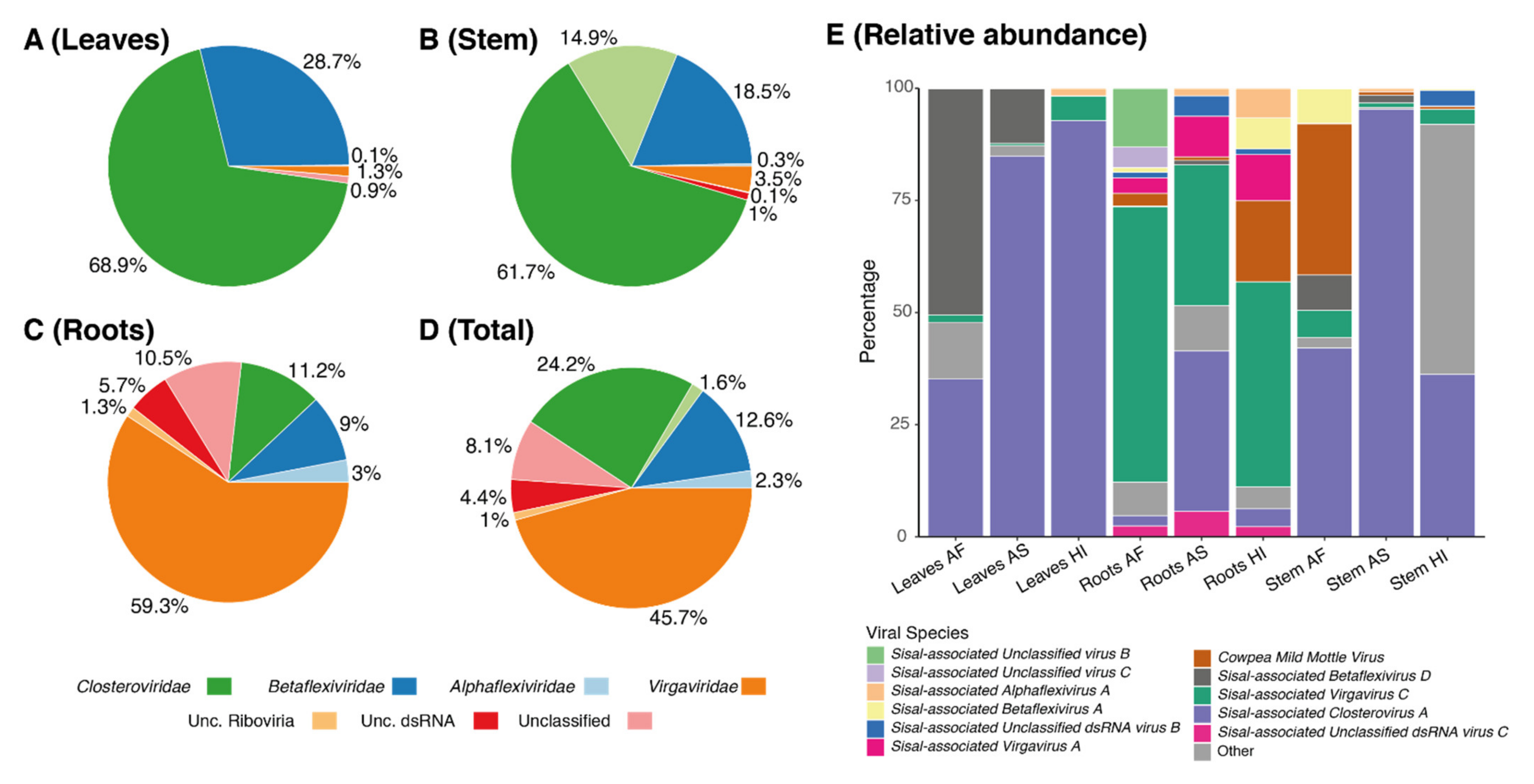
On average, each of our samples contains around 11 species unevenly distributed (
Table S9), indicating the presence of dominating species, which we describe in detail in the next paragraphs. In all the three sisal taxa, the roots were the organ with the highest number of species (richness), ranging from 12 species in the root of
A. Hybrid 11648 and 13 in the roots of
A. sisalana up to 18 species in the roots of
A. fourcroydes, the richest sample in our analysis (
Table S9). We believe this pattern is due to the diversity of associated microorganisms in the root system [
62,
63], which are, as the host plant, susceptible to viral infections. Our species have shown similarity with nine mycoviral species at the amino acid level with BLASTx. Indications that these species are also mycoviruses include not only their similarity with known species and phylogeny but also their distribution in plant organs, which is especially higher in roots for such species, making the roots significantly distinct from stem and leaf samples (
Figure 5E). The mycoviruses sharing similarities with our species are
Agaricus bisporus virus 5 and
Agaricus bisporus virus 6 [
64],
Alternaria alternata virus 1 [
65],
Aspergillus foetidus dsRNA mycovirus [
66],
Aspergillus heteromorphus alternavirus 1 [
67],
Podosphaera prunicola tobamo-like virus [
68],
Macrophomina phaseolina tobamo-like virus [
69],
Botryosphaeria dothidea tobamo-like virus (unpublished)
, and
Stemphylium lycopersici mycovirus (unpublished) (
Table 1). The occurrence of viral species showing similarity with
Aspergillus mycoviruses, and their pattern of expression (higher in stems and roots, but also present in leaves) leads us to hypothesize that
Aspergillus welwitschiae, a fungal species which causes the bole rot of sisal [
70,
71] is also part of the healthy microbiome of sisal, causing disease through imbalances in plant metabolism, rather than infecting vulnerable plants from spores in the environment. Such a pattern has been described in peppermint by Dakin et al. (2010) [
72]. Nonetheless, it is also possible that this species is infecting the plant host, and not some associated fungal species, following the theories that mycoviruses originated from plant viruses [
73] and that these mycoviruses can replicate in plant cells [
74]. We can also hypothesize that such an infection in
A. welwitschiae could modulate pathogenicity by stimulating it, or causing hypovirulence, as seen in Nuss (2005) [
75]. The latter could be a highly promising treatment to the bole rot of sisal if properly managed.
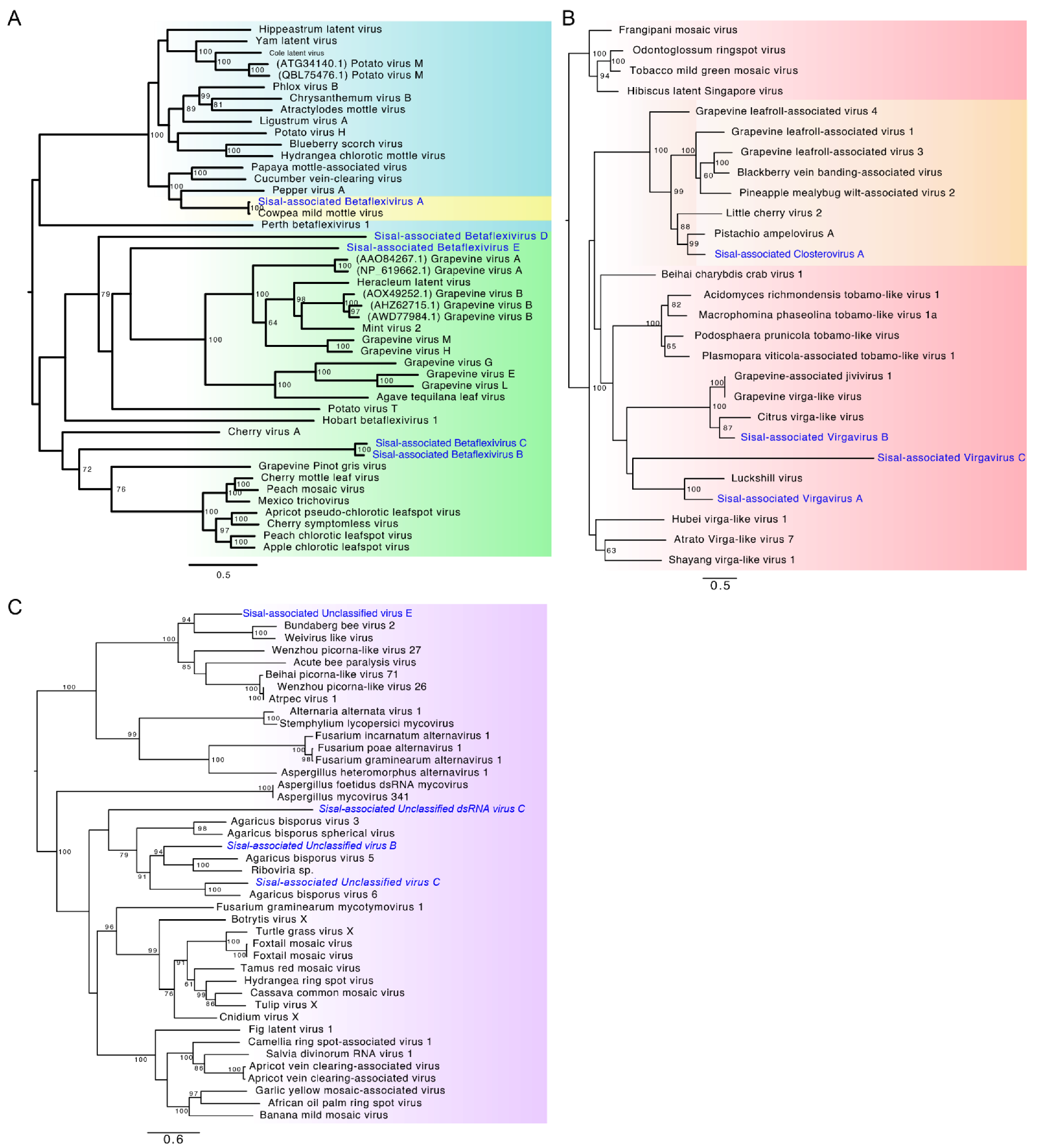
Identification of our likely mycoviral species reveals four species, out of nine, belonging to the family Virgaviridae, the family with the second-highest number of represented contigs (
Figure 5E). This viral family is commonly described as only infecting plants [
76]; however, the first mycovirus belonging to this family was described by [
69] and later by Pandey et al. (2018) [
68], and, thus, corroborating our findings. As also described by Pandey et al. (2018) [
68],
Podosphaera prunicola tobamo-like virus shows similarity with
Macrophomina phaseolina tobamo-like virus, such as our proposed new species,
Sisal-associated Virgavirus C, which is one of the most highly expressed and dominating viral species in our samples (
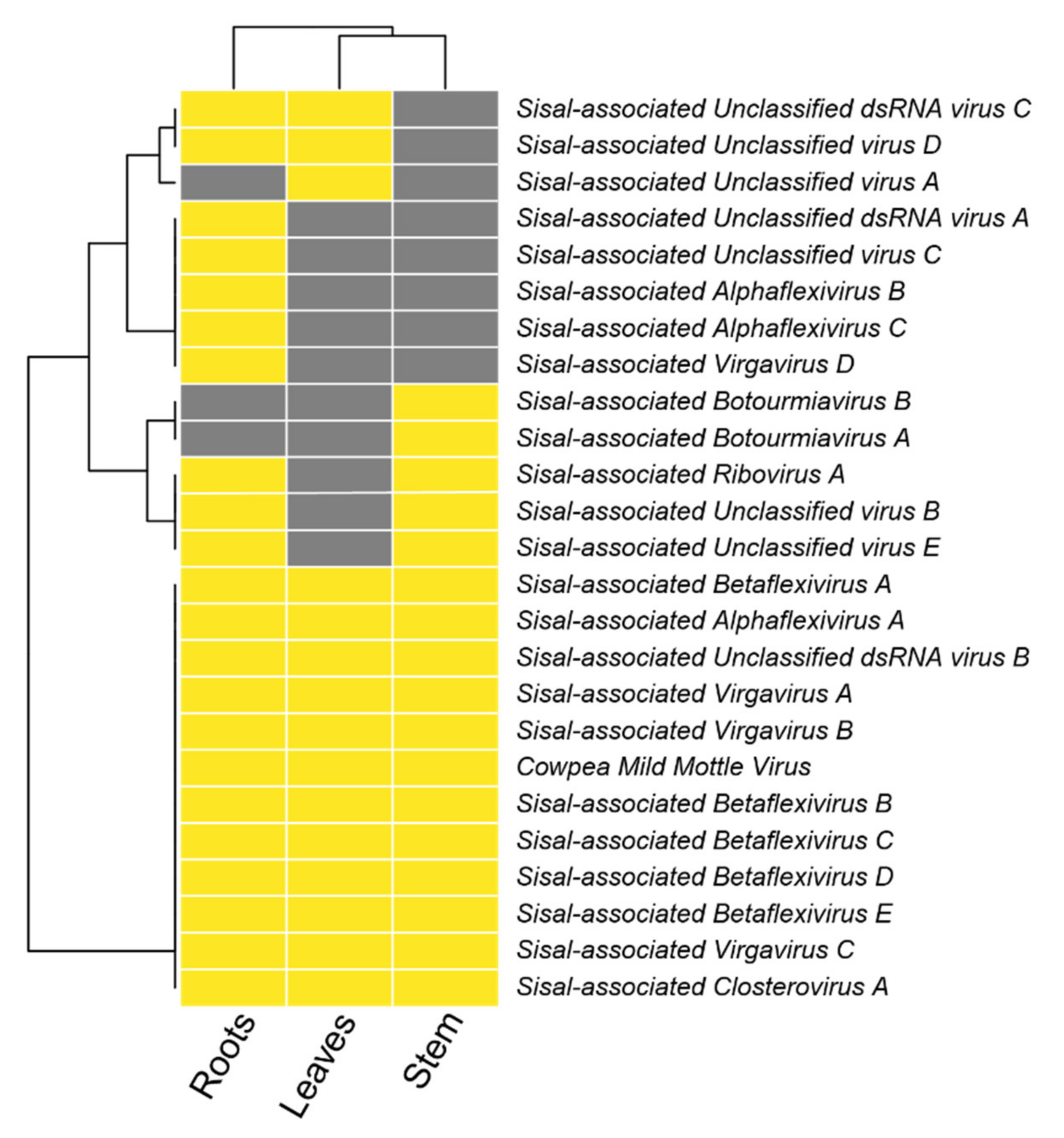
Figure 5E and
Figure 6), especially in the roots (
Figure 5E), while
Sisal-associated Virgavirus D shares similarity with
Podosphaera prunicola tobamo-like virus, unique to the roots. These results reinforce the assertion that the higher species richness in the root system is related to the microbial species associated with sisal varieties. Furthermore, in the family
Virgaviridae, Sisal-associated Virgavirus B shares similarity with “
Citrus virga-like virus” [
77], which is a plant-infecting species, and is expressed in stems and roots of
A. sisalana and
A. hybrid 11648 and leaves of
A. sisalana). This viral species was, as seen in Matsumura et al. (2017) [
77], isolated from the city of Comendador Gomes, in the state of Minas Gerais, Brazil. This location is in the
Cerrado biome, another endangered Brazilian biome that shares similarities with the
Caatinga biome where our samples came from, including moderately low rainfall and low aboveground biomass [
78].
Besides the aforementioned
Citrus virga-like virus and
Cowpea Mild Mottle virus, the other 12 species sharing similarity with our discoveries are plant-infecting viruses. The most prominent of those species, as seen in
Figure 5E, is
Sisal-associated Closterovirus A, the only representative species of the family
Closteroviridae, which is the most represented viral family in our samples, sharing similarity with
Pistachio ampelovirus A, first described by Al Rwahnih et al. (2018) [
79]. This species is present in all the samples and plant taxa (
Figure 5E and
Figure 6) but is especially highly expressed in
A. sisalana (
Figure 5E). This +ssRNA family has been described by Rubio et al. (2013) [
80] as transmitted through mealybugs, aphids, or the whitefly, which is also likely responsible for the presence of CPMMV in our samples. The occurrence of mealybugs is described by da Silva et al. in the state of Paraíba, affecting cultivation of cotton (2013) [
81] and peanuts (2018) [
82], reinforcing the role of this insect in the transmission of viral infections in this region. Our results also revealed six viral species sharing similarities with viruses initially described in grapevines, three of which belonging to the family
Betaflexiviridae (the third most represented family in the stems, and fourth most represented in leaves and roots);
Sisal-associated Betaflexivirus C, sharing similarity with
Grapevine Pinot gris virus, as seen in Giampetruzzi et al. (2012) [
51] and
Sisal-associated Betaflexivirus E, sharing similarity with
Grapevine virus H, as seen in Candresse et al. (2018) [
50], both of which are unique to
A. fourcroydes, with higher expression in the leaves.
Sisal-associated Betaflexivirus D, sharing similarity with
Grapevine virus G as seen in Blouin et al. (2018) [
83], which was absent in the organs of A. hybrid 11648, and
Sisal-associated Ribovirus A, sharing similarity with
Grapevine virga-like virus (unpublished), which is unique to the stems and roots of
A. sisalana and A. hybrid 11648. The family Betaflexiviridae, which also includes CMMV, affects exclusively plants [
84], and also includes the Sisal-
associated Betaflexivirus A (sharing similarity with
Apple stem pitting virus), not expressed in
A. sisalana, and
Sisal-associated Betaflexivirus B (sharing similarity with
Diuris virus A), not expressed in
A. hybrid 11648. Taking together, our findings indicate that there is no organ tropism for the family Betaflexiviridae in our samples, even though some species seem to favor the leaves or roots. Other two species (
Sisal-associated Botourmiavirus A and
Sisal-associated Botourmiavirus B) share similarities with the oomycete-infecting viral species,
Plasmopara viticola associated ourmia-like virus 29 and
Plasmopara viticola associated ourmia-like virus 6, respectively, both of which were described by Chiapello et al. (2020) [
85]. Since the production of grapes in the state of Paraíba [
86] and the
caatinga biome [
87] is limited, with a local study by Medeiros et al. (2017) [
88] considering the climate conditions unsuitable for the cultivation of vines, the similarity of some of our new viral species to grapevine-infecting viruses suggests that these species have a broader host range than just the species from which they were originally isolated.
The family Alphaflexiviridae includes three species in our samples, sharing similarities on BLASTx with plant-infecting species in this same family;
Alternanthera mosaic virus [
89],
Cassia mild mosaic virus [
90], and
Nerine virus X [
91]. As well as Betaflexiviridae, this family does not display an expression pattern to the family level.
Sisal-associated Alphaflexivirus A is expressed only in the stems and roots of
A. sisalana and the hybrid, and in the leaves of
A. sisalana while
Sisal-associated Alphaflexivirus B is exclusive of the roots of
A. fourcroydes and
A. sisalana, and
Sisal-associated Alphaflexivirus C is unique to the roots of
A. fourcroydes. Sisal-associated Unclassified virus A is also unique to
A. fourcroydes but to leaves instead of roots.
Finally, a curious result is the detection of
Sisal-associated Unclassified virus E, which shares similarity on BLASTx with
Halhan virus 3, described by Rosani et al. (2019) [
92]. This viral species was infecting the bivalve
Haliotis discus, a sea snail species. By contrast with the aforementioned fungal and plant species, the occurrence of sea snails in the Brazilian
caatinga is virtually impossible. Thus, considering the expression of
Sisal-associated Unclassified virus E only in the stems and roots of
A. fourcroydes, we hypothesize that other
Gastropoda species inhabiting the roots of this plant, as described by Pearce and Örstan (2006) [
93] and Pratt (1971) [
94], might have left viable viral RNA on the organs where it was detected, and that this group of species occurs in a broader range of environments than previously thought.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}