The MHC Class Ia Genes in Chenfu’s Treefrog (Zhangixalus chenfui) Evolved via Gene Duplication, Recombination, and Selection

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and RNA Isolation
2.2. cDNA Amplification and Cloning
2.3. Analysis of ZcMHC Class Ia
2.4. Sequence Alignment and Allele Identification
2.5. Statistical Study
2.5.1. Recombination Detection
2.5.2. Phylogenetic Analyses
2.5.3. Positive Selection Identification
3. Results
3.1. Identification and Sequencing of ZcMHC Class Ia cDNA
3.2. Allele Characterization of ZcMHC Class Ia
3.3. Number of Loci and Genetic Diversity
3.4. Recombination Detection
3.5. Selection Detection
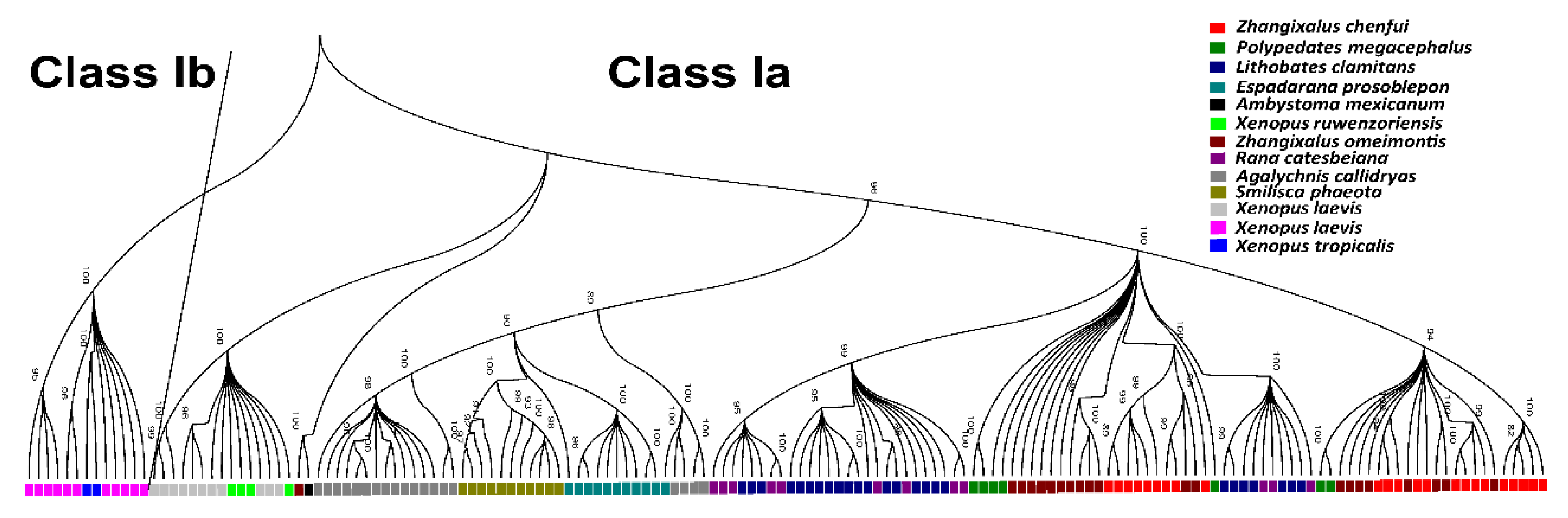
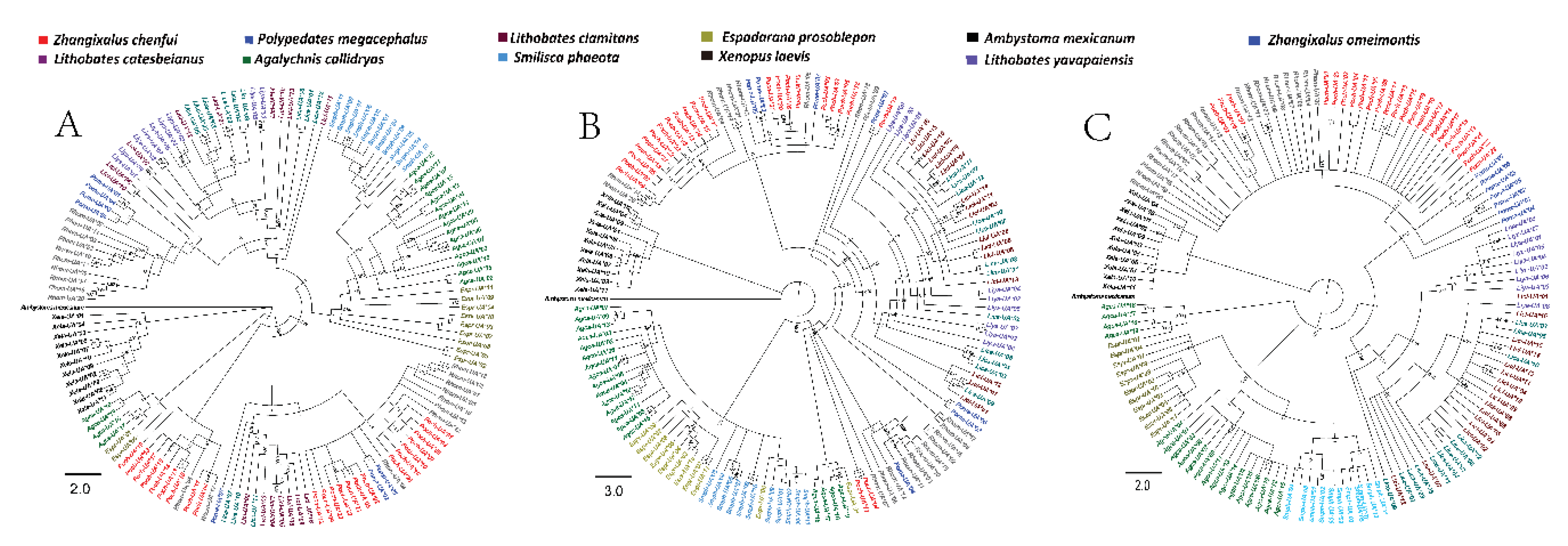
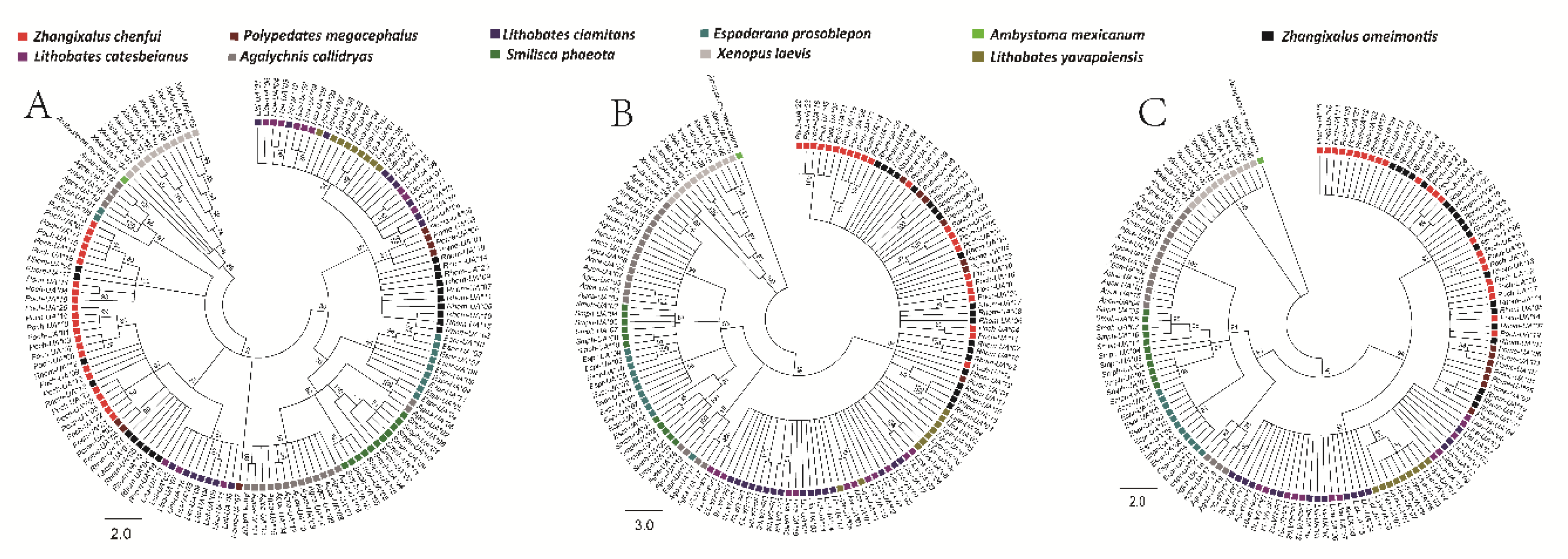
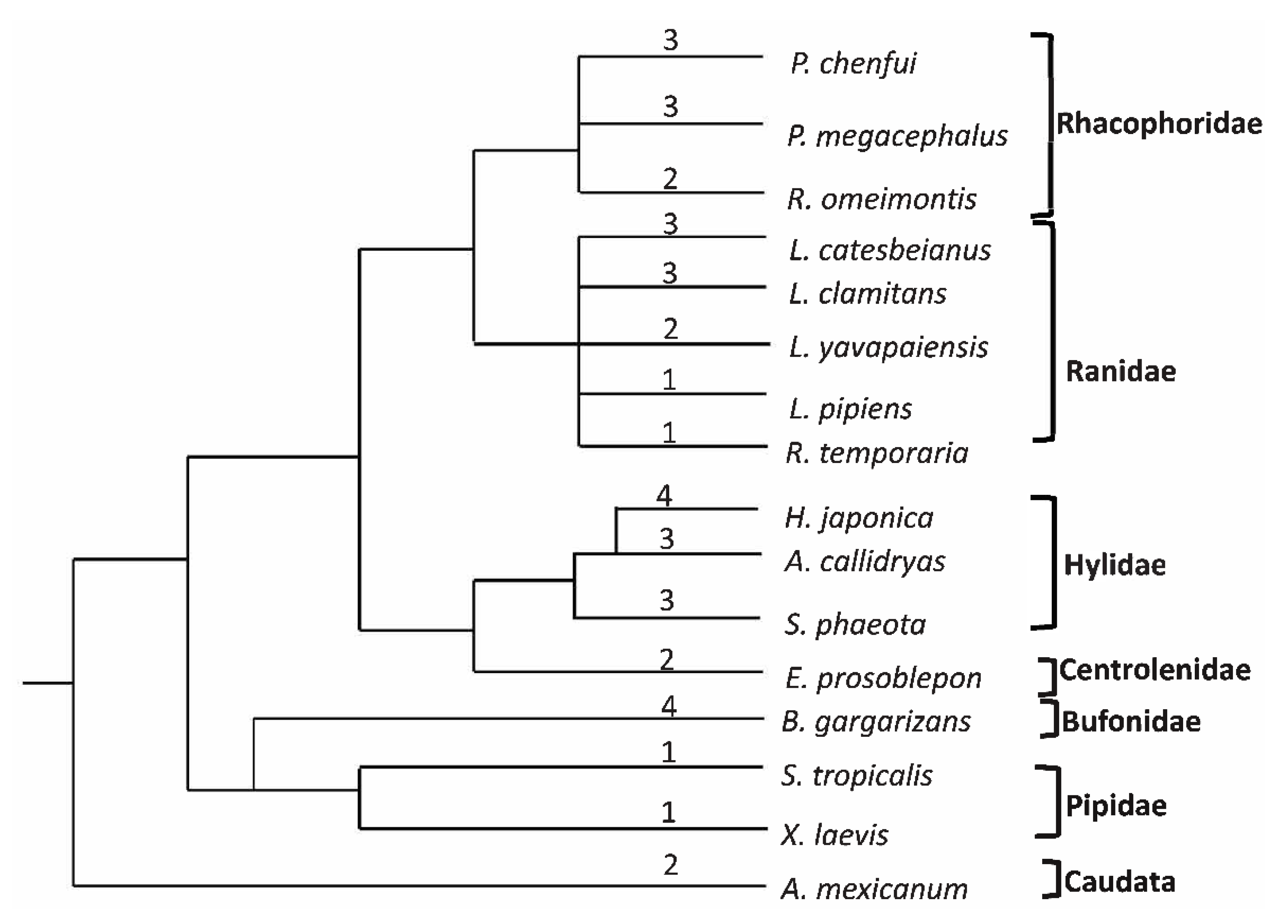
3.6. Phylogenetic Analyses
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klein, J. Natural History of the Major Histocompatibility Complex; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Leclaire, S.; Strandh, M.; Dell’Ariccia, G.; Gabirot, M.; Westerdahl, H.; Bonadonna, F. Plumage microbiota covaries with the major histocompatibility complex in blue petrels. Mol. Ecol. 2019, 28, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major histocompatibility complex (MHC) class I and MHC class II proteins: Conformational plasticity in antigen presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, A.M. Natural selection at major histocompatibility complex loci of vertebrates. Annu. Rev. Genet. 1998, 32, 415–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkman, P.J.; Saper, M.; Samraoui, B.; Bennett, W.S.; Strominger, J.L.; Wiley, D. Structure of the human class I histocompatibility antigen, HLA-A 2. Nature 1987, 329, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Piertney, S.; Oliver, M. The evolutionary ecology of the major histocompatibility complex. Heredity 2006, 96, 7–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsen, I.B.; Wilson, S.R.; Easteal, S. Patterns of reticulate evolution for the classical class I and II HLA loci. Immunogenetics 1998, 48, 312–323. [Google Scholar] [CrossRef]
- Consuegra, S.; Megens, H.J.; Schaschl, H.; Leon, K.; Stet, R.J.M.; Jordan, W.C. Rapid Evolution of the MH Class I Locus Results in Different Allelic Compositions in Recently Diverged Populations of Atlantic Salmon. Mol. Biol. Evol. 2005, 22, 1095. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhong, L.; Wei, Q.; Gan, X.; He, S. Evolution of MHC class I genes in two ancient fish, paddlefish (Polyodon spathula) and Chinese sturgeon (Acipenser sinensis). FEBS Lett. 2010, 584, 3331–3339. [Google Scholar] [CrossRef] [Green Version]
- Wutzler, R.; Foerster, K.; Kempenaers, B. MHC class I variation in a natural blue tit population (Cyanistes caeruleus). Genetica 2012, 140, 349–364. [Google Scholar] [CrossRef]
- Spurgin, L.G.; Richardson, D.S. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc. R. Soc. Lond. Ser. B 2010, 277, 979–988. [Google Scholar] [CrossRef] [Green Version]
- Garrigan, D.; Hedrick, P.W. Perspective: Detecting adaptive molecular polymorphism: Lessons from the MHC. Evolution 2003, 57, 1707. [Google Scholar] [CrossRef] [PubMed]
- Mccairns, R.J.; Bourget, S.; Bernatchez, L. Putative causes and consequences of MHC variation within and between locally adapted stickleback demes. Mol. Ecol. 2011, 20, 486–502. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, A.; Roemer, G.; Debenham, S.; Binns, M.; Garcelon, D.; Wayne, R.K. High MHC diversity maintained by balancing selection in an otherwise genetically monomorphic mammal. Proc. Natl. Acad. Sci. USA 2004, 101, 3490–3494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincek, V.; Nizetić, D.; Golubić, M.; Figueroa, F.; Nevo, E.; Klein, J. Evolutionary expansion of MHC class I loci in the mole-rat, Spalax ehrenbergi. Mol. Biol. Evol. 1987, 4, 483–491. [Google Scholar]
- Figueroa, F.; Mayer, W.E.; Sato, A.; Zaleska-Rutczynska, Z.; Hess, B.; Tichy, H.; Klein, J. MHC class I genes of swordtail fishes, Xiphophorus: Variation in the number of loci and existence of ancient gene families. Immunogenetics 2001, 53, 695–708. [Google Scholar] [CrossRef]
- Gaudieri, S.; Dawkins, R.L.; Habara, K.; Kulski, J.K.; Gojobori, T. SNP profile within the human major histocompatibility complex reveals an extreme and interrupted level of nucleotide diversity. Genome Res. 2000, 10, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Meyer, D.; Thomson, G. How selection shapes variation of the human major histocompatibility complex: A review. Ann. Hum. Genet. 2001, 65, 1–26. [Google Scholar] [CrossRef]
- Pavelko, K.D.; Mendez-Fernandez, Y.; Bell, M.P.; Hansen, M.J.; Johnson, A.J.; David, C.S.; Rodriguez, M.; Pease, L.R. Nonequivalence of classical MHC class I loci in ability to direct effective antiviral immunity. PLoS Pathog. 2012, 8, e1002541. [Google Scholar] [CrossRef]
- Kelley, J.; Walter, L.; Trowsdale, J. Comparative genomics of major histocompatibility complexes. Immunogenetics 2005, 56, 683–695. [Google Scholar] [CrossRef]
- Nonaka, M.; Namikawa, C.; Kato, Y.; Sasaki, M.; Salter-Cid, L.; Flajnik, M.F. Major histocompatibility complex gene mapping in the amphibian Xenopus implies a primordial organization. Proc. Natl. Acad. Sci. USA 1997, 94, 5789–5791. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.; Garcia-Montijano, M. MHC diversity and differential exposure to pathogens in kestrels (Aves: Falconidae). Mol. Ecol. 2010, 19, 691–705. [Google Scholar]
- May, S.; Zeisset, I.; Beebee, T.J. Larval fitness and immunogenetic diversity in chytrid-infected and uninfected natterjack toad (Bufo calamita) populations. Conserv. Genet. 2011, 12, 805–811. [Google Scholar] [CrossRef]
- Sommer, S. The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2005, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernatchez, L.; Landry, C. MHC studies in nonmodel vertebrates: What have we learned about natural selection in 15 years? J. Evol. Biol. 2003, 16, 363–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roved, J.; Hansson, B.; Tarka, M.; Hasselquist, D.; Westerdahl, H. Evidence for sexual conflict over major histocompatibility complex diversity in a wild songbird. Proc. R. Soc. Lond. Ser. B 2018, 285, 20180841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammut, B.; Du Pasquier, L.; Ducoroy, P.; Laurens, V.; Marcuz, A.; Tournefier, A. Axolotl MHC architecture and polymorphism. Eur. J. Immunol 1999, 29, 2897–2907. [Google Scholar] [CrossRef]
- Bos, D.H.; Waldman, B. Evolution by recombination and transspecies polymorphism in the MHC class I gene of Xenopus laevis. Mol. Biol. Evol. 2006, 23, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Bos, D.; Waldman, B. Polymorphism, natural selection, and structural modeling of class Ia MHC in the African clawed frog (Xenopus laevis). Immunogenetics 2006, 58, 433–442. [Google Scholar] [CrossRef]
- Ohta, Y.; Goetz, W.; Hossain, M.Z.; Nonaka, M.; Flajnik, M.F. Ancestral organization of the MHC revealed in the amphibian Xenopus. J. Immunol. 2006, 176, 3674–3685. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Wang, Y.; Shen, H.; Li, C.; Chen, C.; Luo, Z.; Wu, H. Evolution by selection, recombination, and gene duplication in MHC class I genes of two Rhacophoridae species. BMC Evol. Biol. 2013, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Kiemnec-Tyburczy, K.; Richmond, J.; Savage, A.; Lips, K.; Zamudio, K. Genetic diversity of MHC class I loci in six non-model frogs is shaped by positive selection and gene duplication. Heredity 2012, 109, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejun, X.L.W.G.L. Studies on the Karyotypes of Rhacoohorus Chenfui and Rana Japonica Japonica. Acta Acad. Med. Zunyi 1990, 2, 008. [Google Scholar]
- Scheffler, K.; Martin, D.P.; Seoighe, C. Robust inference of positive selection from recombining coding sequences. Bioinformatics 2006, 22, 2493–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lankat-Buttgereit, B.; Tampë, R. The transporter associated with antigen processing TAP: Structure and function. FEBS Lett. 1999, 464, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Dijkstra, J.M. Major Histocompatibility Complex (MHC) Genes and Disease Resistance in Fish. Cells 2019, 8, 378. [Google Scholar] [CrossRef] [Green Version]
- Miller, H.C.; Belov, K.; Daugherty, C.H. MHC class I genes in the tuatara (Sphenodon spp.): Evolution of the MHC in an ancient reptilian order. Mol. Biol. Evol. 2006, 23, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Lukacs, M.F.; Harstad, H.; Bakke, H.G.; Beetz-Sargent, M.; McKinnel, L.; Lubieniecki, K.P.; Koop, B.F.; Grimholt, U. Comprehensive analysis of MHC class I genes from the U-, S-, and Z-lineages in Atlantic salmon. BMC Genom. 2010, 11, 154. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Klein, D.; Sültmann, H.; Figueroa, F.; O’hUigin, C.; Klein, J. Class I Mhc genes of cichlid fishes: Identification, expression, and polymorphism. Immunogenetics 1997, 46, 63–72. [Google Scholar] [CrossRef]
- Kaufman, J.; Milne, S.; Göbel, T.W.; Walker, B.A.; Jacob, J.P.; Auffray, C.; Zoorob, R.; Beck, S. The chicken B locus is a minimal essential major histocompatibility complex. Nature 1999, 401, 923–925. [Google Scholar] [CrossRef]
- Teacher, A.G.F.; Garner, T.W.J.; Nichols, R.A. Evidence for Directional Selection at a Novel Major Histocompatibility Class I Marker in Wild Common Frogs (Rana temporaria) Exposed to a Viral Pathogen (Ranavirus). PLoS ONE 2009, 4, e4616. [Google Scholar] [CrossRef] [Green Version]
- Flajnik, M.F.; Ohta, Y.; Greenberg, A.S.; Salter-Cid, L.; Carrizosa, A.; Du Pasquier, L.; Kasahara, M. Two ancient allelic lineages at the single classical class I locus in the Xenopus MHC. J. Immunol. 1999, 163, 3826–3833. [Google Scholar] [PubMed]
- Flajnik, M.; Ohta, Y.; Namikawa-Yomada, C.; Nonaka, M. Insight into the primordial MHC from studies in ectothermic vertebrates. Immunol. Rev. 1999, 167, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Goyos, A.; Sowa, J.; Ohta, Y.; Robert, J. Remarkable conservation of distinct nonclassical MHC class I lineages in divergent amphibian species. J. Immunol. 2011, 186, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didinger, C.; Eimes, J.A.; Lillie, M.; Waldman, B. Multiple major histocompatibility complex class I genes in Asian anurans: Ontogeny and phylogeny. Dev. Comp. Immunol. 2017, 70, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Roelants, K.; Gower, D.J.; Wilkinson, M.; Loader, S.P.; Biju, S.; Guillaume, K.; Moriau, L.; Bossuyt, F. Global patterns of diversification in the history of modern amphibians. Proc. Natl. Acad. Sci. USA 2007, 104, 887–892. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Gu, X.; Sitnikova, T. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc. Natl. Acad. Sci. USA 1997, 94, 7799–7806. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Ren, W.; Zhou, X.; Zhou, K.; Yang, G. Sequence polymorphism and geographical variation at a positively selected MHC-DRB gene in the finless porpoise (Neophocaena phocaenoides): Implication for recent differentiation of the Yangtze Finless porpoise? J. Mol. Evol. 2010, 71, 6–22. [Google Scholar] [CrossRef]
- Lau, Q.; Igawa, T.; Komaki, S.; Satta, Y. Characterisation of major histocompatibility complex class I genes in Japanese Ranidae frogs. Immunogenetics 2016, 68, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Shum, B.P.; Guethlein, L.; Flodin, L.R.; Adkison, M.A.; Hedrick, R.P.; Nehring, R.B.; Stet, R.J.; Secombes, C.; Parham, P. Modes of salmonid MHC class I and II evolution differ from the primate paradigm. J. Immunol. 2001, 166, 3297–3308. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.L.; Hughes, M.K.; Watkins, D.I. Contrasting roles of interallelic recombination at the HLA-A and HLA-B loci. Genetics 1993, 133, 669–680. [Google Scholar]
- Adams, E.J.; Cooper, S.; Thomson, G.; Parham, P.; Adams, E. Common chimpanzees have greater diversity than humans at two of the three highly polymorphic MHC class I genes. Immunogenetics 2000, 51, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, M.I.; Aizawa, K.; Mitani, H.; Bannai, H.P.; Nonaka, M. Retained orthologous relationships of the MHC class I genes during euteleost evolution. Mol. Biol. Evol. 2011, 28, 3099–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, J.; Sato, A.; Nagl, S.; O’hUigín, C. Molecular trans-species polymorphism. Annu. Rev. Ecol. Syst. 1998, 29, 1–21. [Google Scholar] [CrossRef]
- Roelants, K.; Haas, A.; Bossuyt, F. Anuran radiations and the evolution of tadpole morphospace. Proc. Natl. Acad. Sci. USA 2011, 108, 8731–8736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piontkivska, H.; Nei, M. Birth-and-death evolution in primate MHC class I genes: Divergence time estimates. Mol. Biol. Evol. 2003, 20, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5-3′) | Applications |
|---|---|---|
| ZcMHC-F | 5′CTGCGSWAYTATKABACWGCAGTCTC 3′ | ZcMHC cloning |
| ZcMHC-R | 5′TYCAGRCTGCTGTGSTCCACAT 3′ | ZcMHC cloning |
| ZcMHC-F1 | 5′CCGTCAGTCCGATGGTGCGA 3′ | ZcMHC 3′RACE outer |
| ZcMHC-F2 | 5′GAAGTGCCAACAACCGAAGGAGAC 3′ | ZcMHC 3′RACE inner |
| ZcMHC-R1 | 5′GAGCCTCGTTCATGGTGGGGATA 3′ | ZcMHC 5′RACE outer |
| ZcMHC-R2 | 5′GAGTCACAGGACGAGCCTGATGA T3′ | ZcMHC 5′RACE inner |
| ZcMHC-F3 | 5′GAGGTGACACACAGGAGGAGATGGA 3′ | ZcMHC coding region PCR |
| ZcMHC-R3 | 5′CGGCATRGTGTACGCTCAGGCTKT 3′ | ZcMHC coding region PCR |
| Amino Acid. | K2P Nucleotide Distance | Standard Error | Poisson-Corrected Amino Acid Distance | Standard Error |
|---|---|---|---|---|
| complete sequence | 0.075 | 0.005 | 0.130 | 0.013 |
| leading peptide | 0.014 | 0.007 | 0.042 | 0.021 |
| α1 domain | 0.170 | 0.020 | 0.295 | 0.040 |
| α2 domain | 0.089 | 0.012 | 0.163 | 0.030 |
| α3 domain | 0.014 | 0.003 | 0.030 | 0.009 |
| transmembrane region | 0.062 | 0.020 | 0.144 | 0.059 |
| cytoplasmic region | 0.062 | 0.018 | 0.126 | 0.045 |
| Recombinant Sequence | Nucleotide Breakpoint | Potential Parental Sequences | Methods |
|---|---|---|---|
| Poch-UA*05 | 234,764 | Poch-UA*07/Poch-UA*18 | RDP, GENECONV, BootScan, MaxChi, Chimaera, Siscan, 3Seq |
| Poch-UA*09 | 314,1046 | Unknown(Poch-UA*14)/Poch-UA*11 | GENECONV, BootScan, MaxChi, Chimaera, Siscan, 3Seq |
| Poch-UA*18 | 234,993 | Unknown(Poch-UA*07)/Poch-UA*10 | BootScan, MaxChi, Chimaera, Siscan, 3Seq |
| Method | α1 Domain | α2 Domain | α3 Domain | Transmembrane Region | Cytoplasmic Region | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 11 | 42 | 80 | 83 | 7 | 23 | 25 | 34 | 37 | 42 | 63 | 67 | 74 | 84 | 85 | 99 | 5 | 9 | 1 | 17 | 23 | |
| FELa | + | + | + | + | + | + | + | ||||||||||||||
| MEMEb | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| Codemlc | * | ** | * | ** | * | ** | ** | ** | ** | ** | * | * | |||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Huang, S.; Jiang, Y.; Han, F.; Ni, Q.; Yao, Y.; Xu, H.; Mishra, S.; Zhang, M. The MHC Class Ia Genes in Chenfu’s Treefrog (Zhangixalus chenfui) Evolved via Gene Duplication, Recombination, and Selection. Animals 2020, 10, 34. https://doi.org/10.3390/ani10010034
Chen H, Huang S, Jiang Y, Han F, Ni Q, Yao Y, Xu H, Mishra S, Zhang M. The MHC Class Ia Genes in Chenfu’s Treefrog (Zhangixalus chenfui) Evolved via Gene Duplication, Recombination, and Selection. Animals. 2020; 10(1):34. https://doi.org/10.3390/ani10010034
Chicago/Turabian StyleChen, Hu, Siqi Huang, Ye Jiang, Fuyao Han, Qingyong Ni, Yongfang Yao, Huailiang Xu, Sudhanshu Mishra, and Mingwang Zhang. 2020. "The MHC Class Ia Genes in Chenfu’s Treefrog (Zhangixalus chenfui) Evolved via Gene Duplication, Recombination, and Selection" Animals 10, no. 1: 34. https://doi.org/10.3390/ani10010034
APA StyleChen, H., Huang, S., Jiang, Y., Han, F., Ni, Q., Yao, Y., Xu, H., Mishra, S., & Zhang, M. (2020). The MHC Class Ia Genes in Chenfu’s Treefrog (Zhangixalus chenfui) Evolved via Gene Duplication, Recombination, and Selection. Animals, 10(1), 34. https://doi.org/10.3390/ani10010034