Raw Cow Milk Bacterial Consortium as Bioindicator of Circulating Anti-Microbial Resistance (AMR)

 , ,
, ,  , , ,
, , ,  ,
,  , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
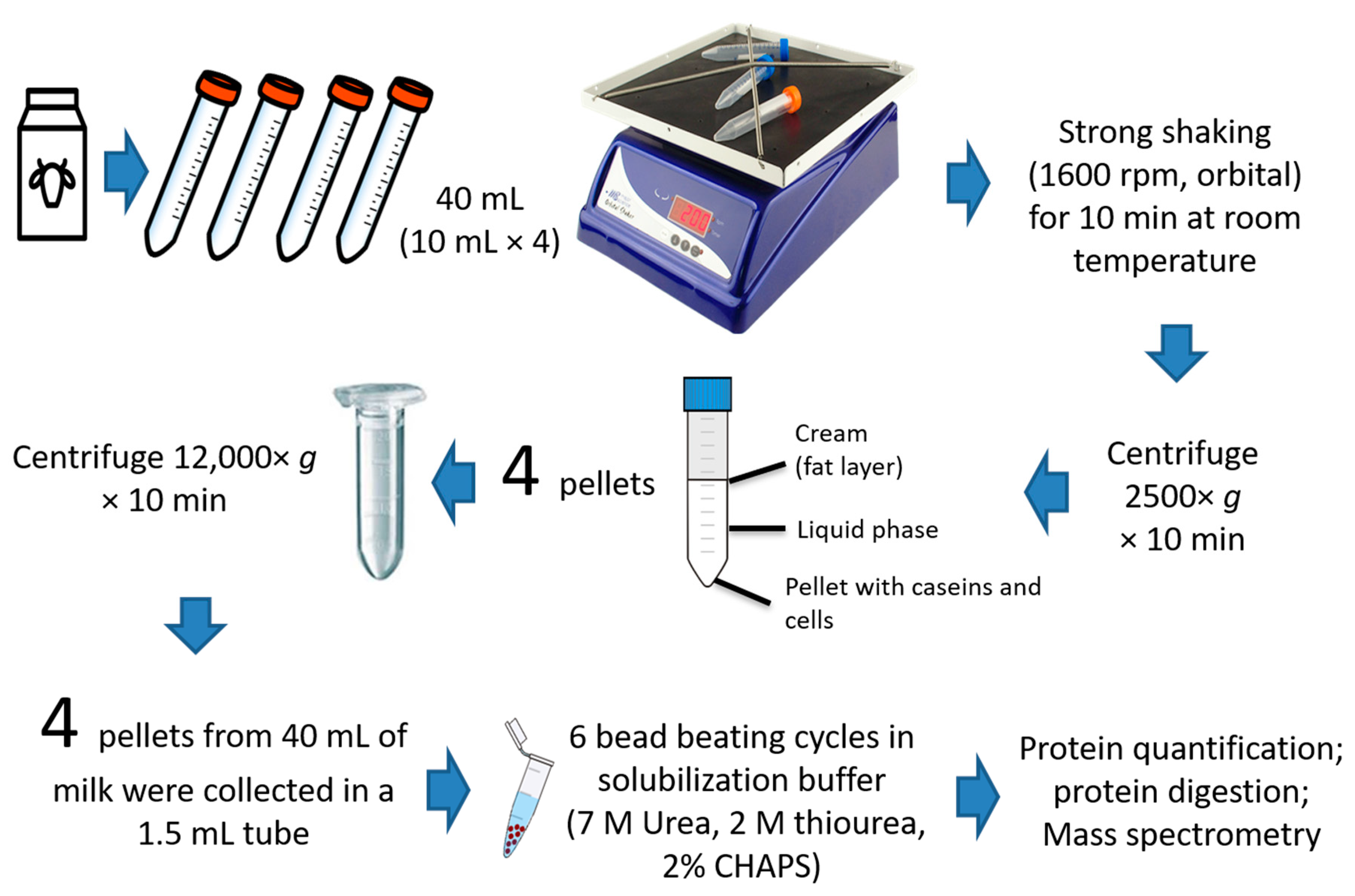
2.1. Milk Sampling
2.2. Bacterial Enrichment for Proteomics Analysis
2.3. Trypsin Digestion and Mass Spectrometry Analysis
2.4. Bioinformatics and Metaproteomics
3. Results
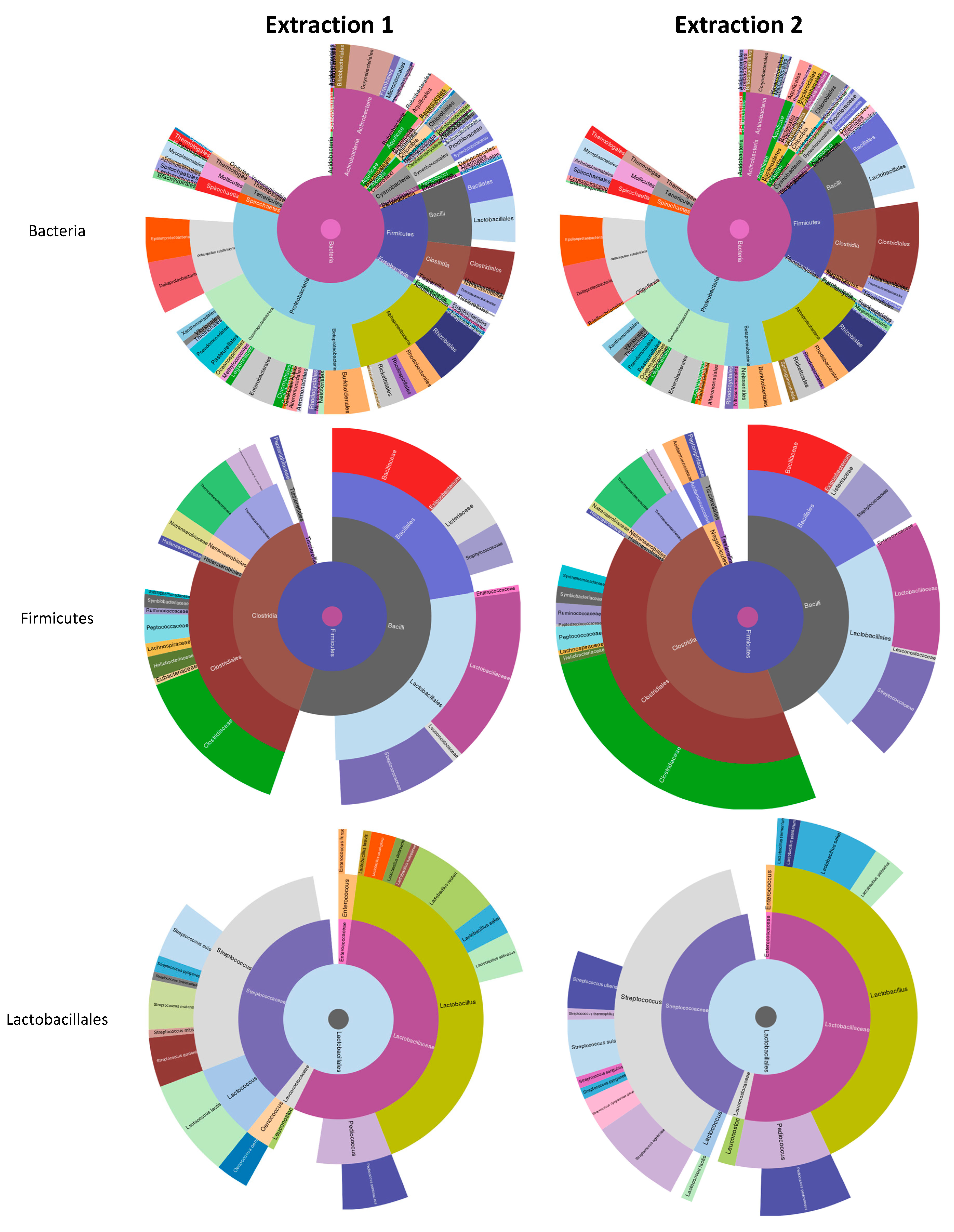
3.1. Cow Milk Microbiome Analysis
3.2. Resistome Proteins Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cox, G.; Wright, G.D. Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol. 2013, 303, 287–292. [Google Scholar] [CrossRef]
- Olivares, J.; Bernardini, A.; Egarcia-Leon, G.; Corona, F.; Sanchez, M.B.; Martínez, J. The intrinsic resistome of bacterial pathogens. Front. Microbiol. 2013, 4, 103. [Google Scholar] [CrossRef] [Green Version]
- Bhullar, K.; Waglechner, N.; Pawlowski, A.; Koteva, K.; Banks, E.D.; Johnston, M.D.; Balrton, H.A.; Wright, G.D. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS ONE 2012, 7, e34953. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Butcher, J.; Stintzi, A.; Figeys, D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome 2019, 7, 154. [Google Scholar] [CrossRef]
- Soggiu, A.; Piras, C.; Mortera, S.L.; Alloggio, I.; Urbani, A.; Bonizzi, L.; Roncada, P. Unravelling the effect of clostridia spores and lysozyme on microbiota dynamics in Grana Padano cheese: A metaproteomics approach. J. Proteom. 2016, 147, 21–27. [Google Scholar] [CrossRef]
- Mortera, S.L.; Soggiu, A.; Vernocchi, P.; Del Chierico, F.; Piras, C.; Carsetti, R.; Marzano, V.; Britti, D.; Urbani, A.; Roncada, P.; et al. Metaproteomic investigation to assess gut microbiota shaping in newborn mice: A combined taxonomic, functional and quantitative approach. J. Proteom. 2019, 203, 103378. [Google Scholar] [CrossRef]
- Soggiu, A.; Piras, C.; Gaiarsa, S.; Bendixen, E.; Panitz, F.; Bendixen, C.; Sassera, D.; Brasca, M.; Bonizzi, L.; Roncada, P. Draft genome sequence of Clostridium tyrobutyricum strain DIVETGP, isolated from cow’s milk for Grana Padano production. Genome Announc. 2016, 3, 2164. [Google Scholar] [CrossRef] [Green Version]
- Hogan, J.S.; National Mastitis Council. Laboratory Handbook on Bovine Mastitis; National Mastitis Council: Madison, WI, USA, 1999. [Google Scholar]
- Brewster, J.D.; Paul, M. Short communication: Improved method for centrifugal recovery of bacteria from raw milk applied to sensitive real-time quantitative PCR detection of Salmonella spp. J. Dairy Sci. 2016, 99, 3375–3379. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Distler, U.; Kuharev, J.; Navarro, P.; Tenzer, S. Label-free quantification in ion mobility–enhanced data-independent acquisition proteomics. Nat. Protoc. 2016, 11, 795–812. [Google Scholar] [CrossRef]
- Marini, F.; Carregari, V.C.; Greco, V.; Ronci, M.; Iavarone, F.; Persichilli, S.; Castagnola, M.; Urbani, A.; Pieroni, L. Exploring the HeLa Dark Mitochondrial Proteome. Front. Cell Dev. Biol. 2020, 8, 137. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Singh, R.G.; Tanca, A.; Palomba, A.; Van Der Jeugt, F.; Verschaffelt, P.; Uzzau, S.; Martens, L.; Dawyndt, P.; Mesuere, B. Unipept 4.0: Functional Analysis of Metaproteome Data. J. Proteome Res. 2019, 18, 606–615. [Google Scholar] [CrossRef]
- World Health Organization. Antimicrobial Resistance Global Report on Surveillance; WHO Press, World Health Organization: Geneva, Switzerland, June 2014. [Google Scholar]
- Noyes, N.R.; Yang, X.; Linke, L.M.; Magnuson, R.J.; Dettenwanger, A.; Cook, S.; Geornaras, I.; Woerner, E.D.; Gow, S.P.; McAllister, A.T.; et al. Resistome diversity in cattle and the environment decreases during beef production. Elife 2016, 5, e13195. [Google Scholar] [CrossRef]
- Liu, J.; Taft, D.H.; Maldonado-Gomez, M.X.; Johnson, D.; Treiber, M.L.; Lemay, D.G.; Depeters, E.J.; Mills, D.A. The fecal resistome of dairy cattle is associated with diet during nursing. Nat. Commun. 2019, 10, 4406. [Google Scholar] [CrossRef] [Green Version]
- Noyes, N.R.; Yang, X.; Linke, L.M.; Magnuson, R.J.; Cook, S.R.; Zaheer, R.; Yang, H.; Woerner, D.R.; Geornaras, I.; McArt, J.A.; et al. Characterization of the resistome in manure, soil and wastewater from dairy and beef production systems. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Michael, G.B.; Freitag, C.; Wendlandt, S.; Eidam, C.; Feßler, A.T.; Lopes, G.V.; Kadlec, K.; Schwarz, S. Emerging issues in antimicrobial resistance of bacteria from food-producing animals. Future Microbiol. 2015, 10, 427–443. [Google Scholar] [CrossRef]
- Rovira, P.; McAllister, T.; Lakin, S.M.; Cook, S.R.; Doster, E.; Noyes, N.R.; Weinroth, M.D.; Yang, X.; Parker, J.K.; Boucher, C.; et al. Characterization of the microbial resistome in conventional and “raised without antibiotics” beef and dairy production systems. Front. Microbiol. 2019, 10, 1980. [Google Scholar] [CrossRef] [Green Version]
- Cuny, C.; Arnold, P.; Hermes, J.; Eckmanns, T.; Mehraj, J.; Schoenfelder, S.; Ziebuhr, W.; Zhao, Q.; Wang, Y.; Feßler, A.T.; et al. Occurrence of cfr-mediated multiresistance in staphylococci from veal calves and pigs, from humans at the corresponding farms, and from veterinarians and their family members. Vet. Microbiol. 2017, 200, 88–94. [Google Scholar] [CrossRef]
- Tolle, A. The Microflora of the Udder. In: Factors Influencing the Bacteriological Quality of Raw Milk. Int. Dairy J. 1980, 120, 4. [Google Scholar]
- Derakhshani, H.; Fehr, K.B.; Sepehri, S.; Francoz, D.; De Buck, J.; Barkema, H.W.; Plaizier, J.C.; Khafipour, E. Invited review: Microbiota of the bovine udder: Contributing factors and potential implications for udder health and mastitis susceptibility. J. Dairy Sci. 2018, 101, 10605–10625. [Google Scholar] [CrossRef] [Green Version]
- Hale, O.J.; Morris, M.; Jones, B.; Reynolds, C.K.; Cramer, R. Liquid Atmospheric Pressure Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Adds Enhanced Functionalities to MALDI MS Profiling for Disease Diagnostics. ACS Omega 2019, 4, 12759–12765. [Google Scholar] [CrossRef] [Green Version]
- Piras, C.; Ceniti, C.; Hartmane, E.; Costanzo, N.; Morittu, V.M.; Roncada, P.; Britti, D.; Cramer, R. Rapid liquid AP-MALDI MS profiling of lipids and proteins from goat and sheep milk for speciation and colostrum analysis. Proteomes 2020, 8, 20. [Google Scholar] [CrossRef]
- Oikonomou, G.; Machado, V.S.; Santisteban, C.; Schukken, Y.H.; Bicalho, R.C. Microbial diversity of bovine mastitic milk as described by pyrosequencing of metagenomic 16s rDNA. PLoS ONE 2012, 7, e47671. [Google Scholar] [CrossRef] [Green Version]
- Kuehn, J.S.; Gorden, P.J.; Munro, D.; Rong, R.; Dong, Q.; Plummer, P.J.; Wang, C.; Phillips, G.J. Bacterial community profiling of milk samples as a means to understand culture-negative bovine clinical mastitis. PLoS ONE 2013, 8, e61959. [Google Scholar] [CrossRef] [Green Version]
- Oikonomou, G.; Bicalho, M.L.; Meira, E.; Rossi, R.E.; Foditsch, C.; Machado, V.S.; Teixeira, A.G.V.; Santisteban, C.; Schukken, Y.H.; Bicalho, R.C. Microbiota of cow’s milk; distinguishing healthy, sub-clinically and clinically diseased quarters. PLoS ONE 2014, 9, e85904. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, N.; Kulak, N.A.; Cox, J.; Neuhauser, N.; Mayr, K.; Hoerning, O.; Vorm, O.; Mann, M. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top orbitrap. Mol. Cell. Proteom. 2012, 11, 1–284. [Google Scholar] [CrossRef] [Green Version]
- Chavez, M.V.; Caicedo, L.D.; Castillo, J.E. Occurrence of β-Lactamase-Producing Gram-Negative Bacterial Isolates in Water Sources in Cali City, Colombia. Int. J. Microbiol. 2019, 2019, 1375060. [Google Scholar]
- Adelowo, O.O.; Ikhimiukor, O.O.; Knecht, C.; Vollmers, J.; Bhatia, M.; Kaster, A.-K.; Müller, J.A. A survey of extended-spectrum beta-lactamase-producing Enterobacteriaceae in urban wetlands in southwestern Nigeria as a step towards generating prevalence maps of antimicrobial resistance. PLoS ONE 2020, 15, e0229451. [Google Scholar] [CrossRef]
- Adesoji, A.T.; Ogunjobi, A.A. Detection of extended spectrum beta-lactamases resistance genes among bacteria isolated from selected drinking water distribution channels in southwestern Nigeria. BioMed Res. Int. 2016, 2016, 7149295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesa, R.J.; Blanc, V.; Blanch, A.R.; Cortés, P.; Gonzalez, J.J.; Lavilla, S.; Miró, E.; Muniesa, M.; Saco, M.; Tórtola, M.T.; et al. Extended-spectrum β-lactamase-producing Enterobacteriaceae in different environments (humans, food, animal farms and sewage). J. Antimicrob. Chemother. 2006, 58, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenover, F.C.; Filpula, D.; Phillips, K.L.; Plorde, J.J. Cloning and sequencing of a gene encoding an aminoglycoside 6′-N-acetyltransferase from an R factor of Citrobacter diversus. J. Bacteriol. 1988, 170, 471–473. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | GO Term | Name |
|---|---|---|
| 4001 | GO:0005524 | ATP binding |
| 1985 | GO:0003677 | DNA binding |
| 1595 | GO:0046872 | metal ion binding |
| 1164 | GO:0000287 | magnesium ion binding |
| 1061 | GO:0008270 | zinc ion binding |
| 620 | GO:0003899 | DNA-directed 5′-3′ RNA polymerase activity |
| 514 | GO:0016787 | hydrolase activity |
| 511 | GO:0000049 | tRNA binding |
| 481 | GO:0005525 | GTP binding |
| 473 | GO:0046933 | proton-transporting ATP synthase activity, rotational mechanism |
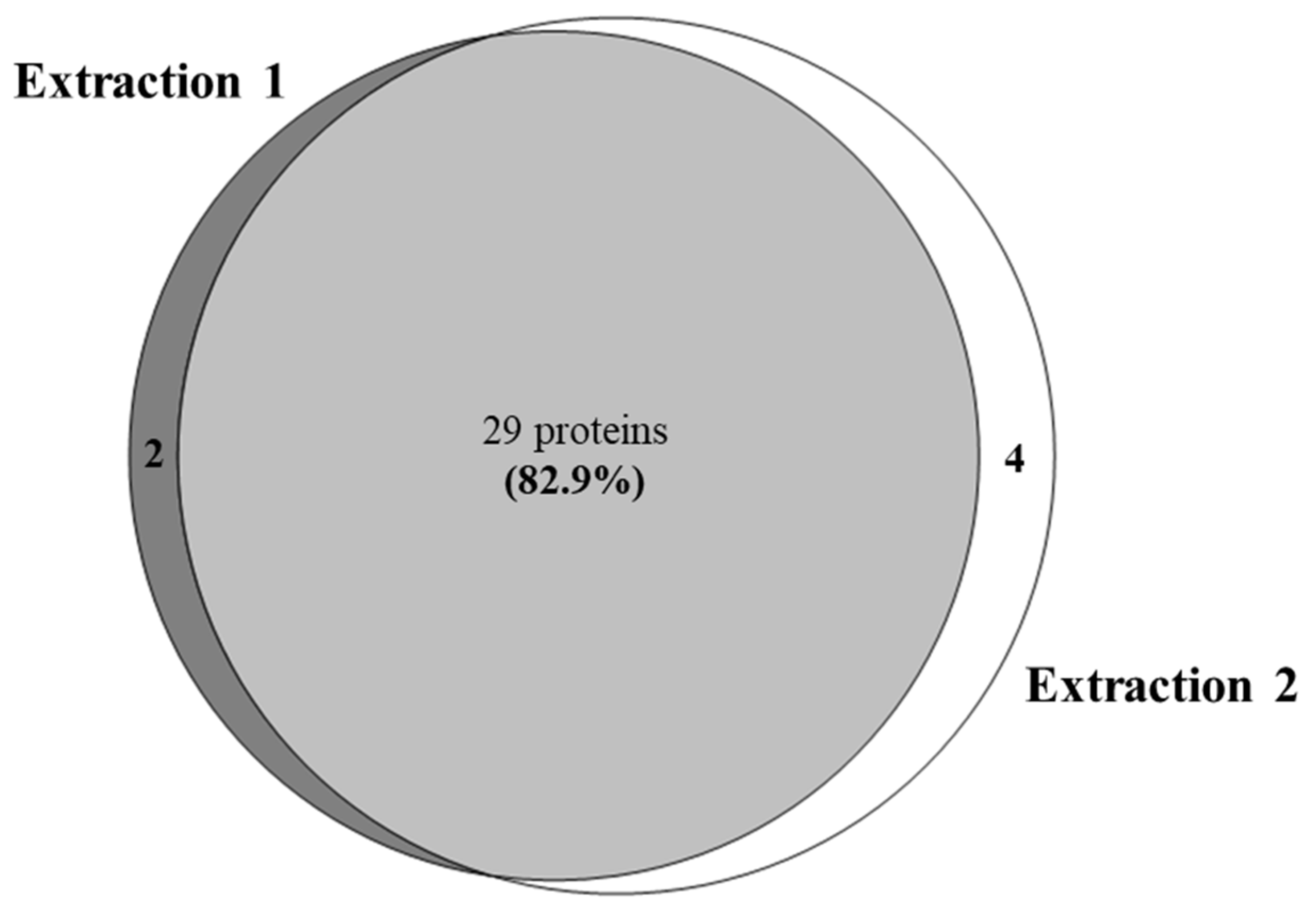
| 29 Common Elements in “Extraction 1” and “Extraction 2”: | |||
|---|---|---|---|
| Protein.Entry | Protein.Accession | Protein.Description | Uniprot |
| ARO:3001066 | AAA87176.1 | SHV-7 [Escherichia coli] | Q46759 |
| ARO:3001077 | AAF34333.1 | SHV-19 [Klebsiella pneumoniae] | Q9LAR9 |
| ARO:3001078 | AAF34334.1 | SHV-20 [Klebsiella pneumoniae] | Q9LAR8 |
| ARO:3001079 | AAF34335.1 | SHV-21 [Klebsiella pneumoniae] | Q9LAR7 |
| ARO:3001076 | AAF64386.1 | SHV-18 [Klebsiella pneumoniae] | Q9LAJ9 |
| ARO:3001073 | AAG17550.1 | SHV-14 [Klebsiella pneumoniae] | Q9F918 |
| ARO:3001087 | AAG49894.1 | SHV-29 [Klebsiella pneumoniae] | Q9AHN9 |
| ARO:3001092 | AAK64187.1 | SHV-34 [Escherichia coli] | Q93LM8 |
| ARO:3001093 | AAL68926.1 | SHV-35 [Klebsiella pneumoniae] | Q8VP57 |
| ARO:3001088 | AAT75225.1 | SHV-30 [Enterobacter cloacae] | Q6DLX7 |
| ARO:3001146 | ABN49111.1 | SHV-94 [Klebsiella pneumoniae] | A3FFR3 |
| ARO:3001148 | ABN49112.1 | SHV-96 [Acinetobacter baumannii] | A3FFR4 |
| ARO:3001182 | AEK80394.1 | SHV-140 [Klebsiella pneumoniae] | G1EC65 |
| ARO:3001183 | AFC60795.1 | SHV-141 [Klebsiella pneumoniae] | H9CTU8 |
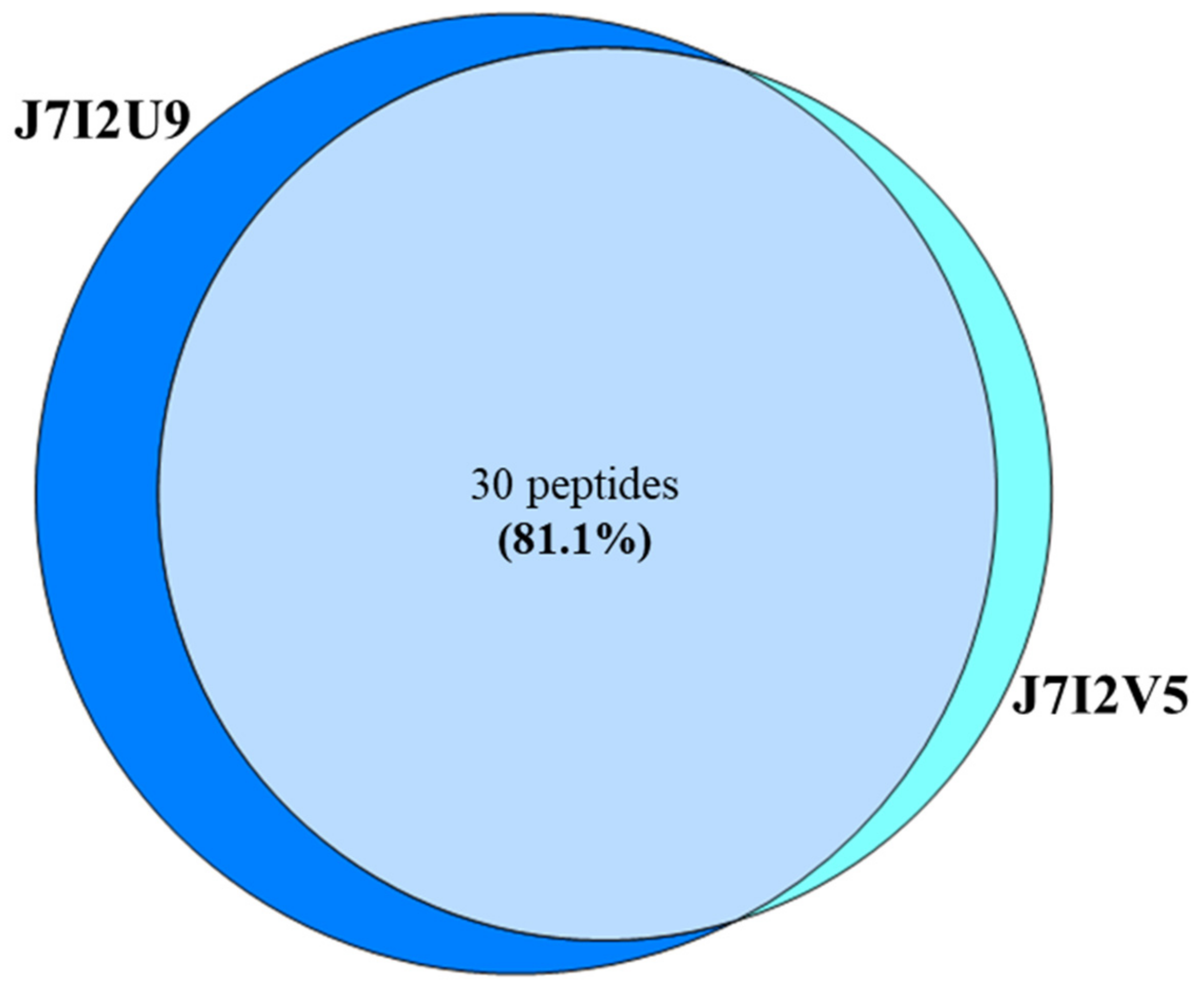
| ARO:3001188 | AFQ23955.1 | SHV-149 [Klebsiella pneumoniae] | J7I2U9 |
| ARO:3001190 | AFQ23957.1 | SHV-151 [Klebsiella pneumoniae] | J7I6M7 |
| ARO:3001193 | AFQ23960.1 | SHV-154 [Klebsiella pneumoniae] | J7I2V5 |
| ARO:3001195 | AFQ23962.1 | SHV-156 [Klebsiella pneumoniae] | J7I6N3 |
| ARO:3001197 | AFQ23964.1 | SHV-158 [Klebsiella pneumoniae] | J7I616 |
| ARO:3001198 | AFQ23965.1 | SHV-159 [Klebsiella pneumoniae] | J7I2W1 |
| ARO:3001200 | AFQ23967.1 | SHV-161 [Klebsiella pneumoniae] | J7I616 |
| ARO:3001202 | AFQ23969.1 | SHV-163 [Klebsiella pneumoniae] | J7I622 |
| ARO:3001357 | AHA80959.1 | SHV-173 [Klebsiella pneumoniae] | V5N2H6 |
| ARO:3001364 | AJO16042.1 | SHV-182 [Klebsiella pneumoniae] | A0A0C5C1Y7 |
| ARO:3003156 | AJO16047.1 | SHV-189 [Klebsiella pneumoniae] | A0A0C5C1Z0 |
| ARO:3001204 | BAM28879.1 | SHV-167 [Klebsiella pneumoniae] | I7GSH3 |
| ARO:3002589 | CAE50925.1 | AAC(6′)-Iid [Enterococcus hirae] | Q70E72 |
| ARO:3001337 | CAQ03504.1 | SHV-99 [Klebsiella pneumoniae] | B7FDD8 |
| ARO:3003155 | CEA29751.1 | SHV-188 [Klebsiella pneumoniae] | A0A0A1ISX2 |
| Protein.Entry | Protein Description | Uniprot Accession | Peptides Included Exclusively | RT | |
|---|---|---|---|---|---|
| Mean | %CV | ||||
| ARO:3001188 | SHV-149; [Klebsiella pneumoniae] | J7I2U9 | LSESRLSGSVGMIEMDLASGR | 63.23 | 1.28 |
| LSGSVGMIEMDLASGR | 72.80 | 1.14 | |||
| LSGSVGMIEMDLASGRTLTAWR | 73.01 | 1.07 | |||
| SVLPAGWFIADKTGAGER | 65.34 | 1.16 | |||
| TGAGERGAR | 79.06 | 0.95 | |||
| ARO:3001193 | SHV-154; [Klebsiella pneumoniae] | J7I2V5 | LSESQLSGSVGMIEMDLASGR | 64.68 | 1.15 |
| LSESQLSGSVGMIEMDLASGRTLTAWR | 91.27 | 0.76 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piras, C.; Greco, V.; Gugliandolo, E.; Soggiu, A.; Tilocca, B.; Bonizzi, L.; Zecconi, A.; Cramer, R.; Britti, D.; Urbani, A.; et al. Raw Cow Milk Bacterial Consortium as Bioindicator of Circulating Anti-Microbial Resistance (AMR). Animals 2020, 10, 2378. https://doi.org/10.3390/ani10122378
Piras C, Greco V, Gugliandolo E, Soggiu A, Tilocca B, Bonizzi L, Zecconi A, Cramer R, Britti D, Urbani A, et al. Raw Cow Milk Bacterial Consortium as Bioindicator of Circulating Anti-Microbial Resistance (AMR). Animals. 2020; 10(12):2378. https://doi.org/10.3390/ani10122378
Chicago/Turabian StylePiras, Cristian, Viviana Greco, Enrico Gugliandolo, Alessio Soggiu, Bruno Tilocca, Luigi Bonizzi, Alfonso Zecconi, Rainer Cramer, Domenico Britti, Andrea Urbani, and et al. 2020. "Raw Cow Milk Bacterial Consortium as Bioindicator of Circulating Anti-Microbial Resistance (AMR)" Animals 10, no. 12: 2378. https://doi.org/10.3390/ani10122378
APA StylePiras, C., Greco, V., Gugliandolo, E., Soggiu, A., Tilocca, B., Bonizzi, L., Zecconi, A., Cramer, R., Britti, D., Urbani, A., & Roncada, P. (2020). Raw Cow Milk Bacterial Consortium as Bioindicator of Circulating Anti-Microbial Resistance (AMR). Animals, 10(12), 2378. https://doi.org/10.3390/ani10122378