Transport to the Slaughterhouse Affects the Salmonella Shedding and Modifies the Fecal Microbiota of Finishing Pigs

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Design
2.2. Microbiological Culture
2.3. Fecal DNA Extraction and 16S rRNA Gene Sequencing
2.4. Biostatistical Analyses
3. Results
3.1. Microbiological Culture
3.2. Fecal Microbiota Composition
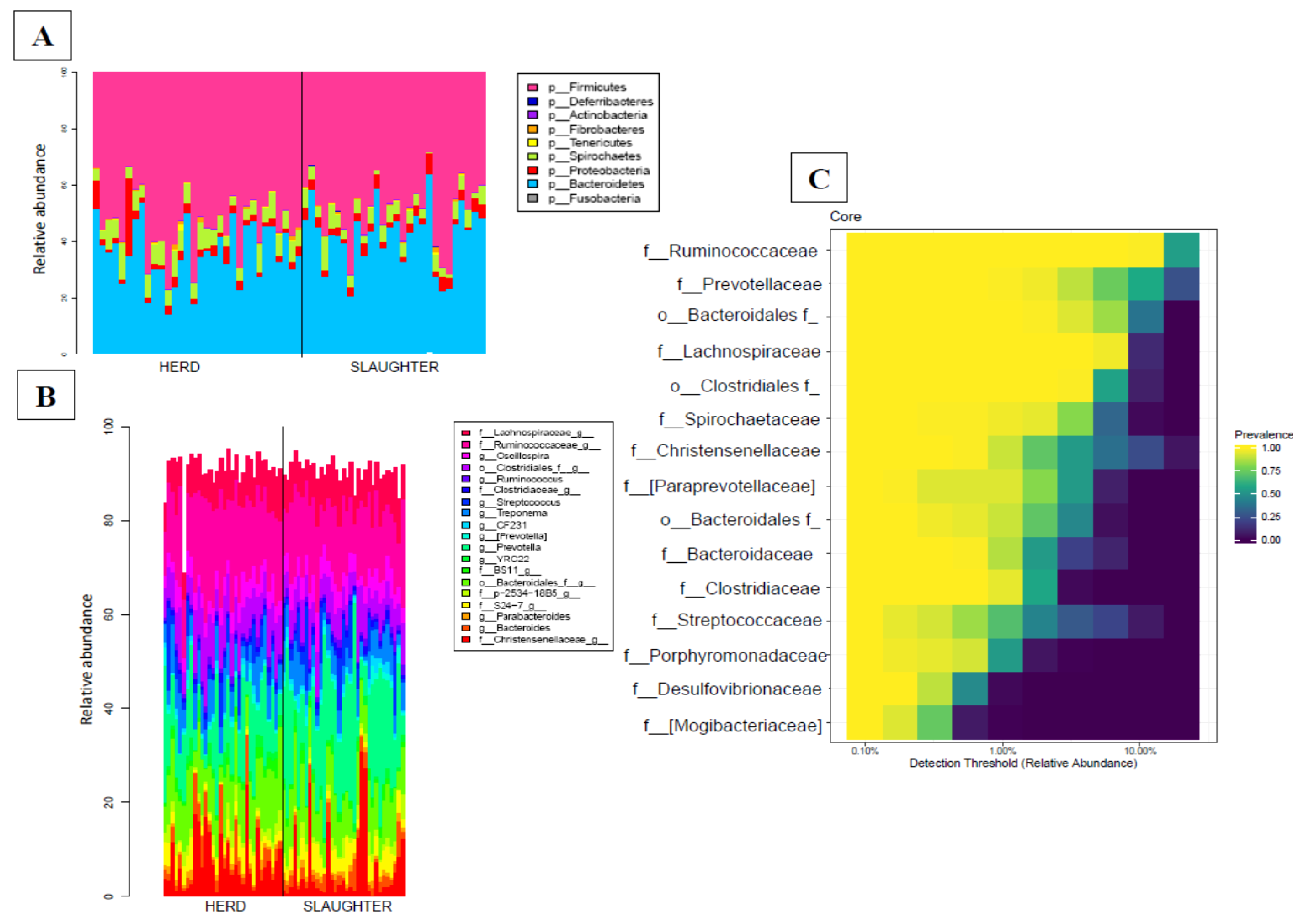
3.2.1. Fecal Microbiota Sequencing, OTU Identification and Annotation
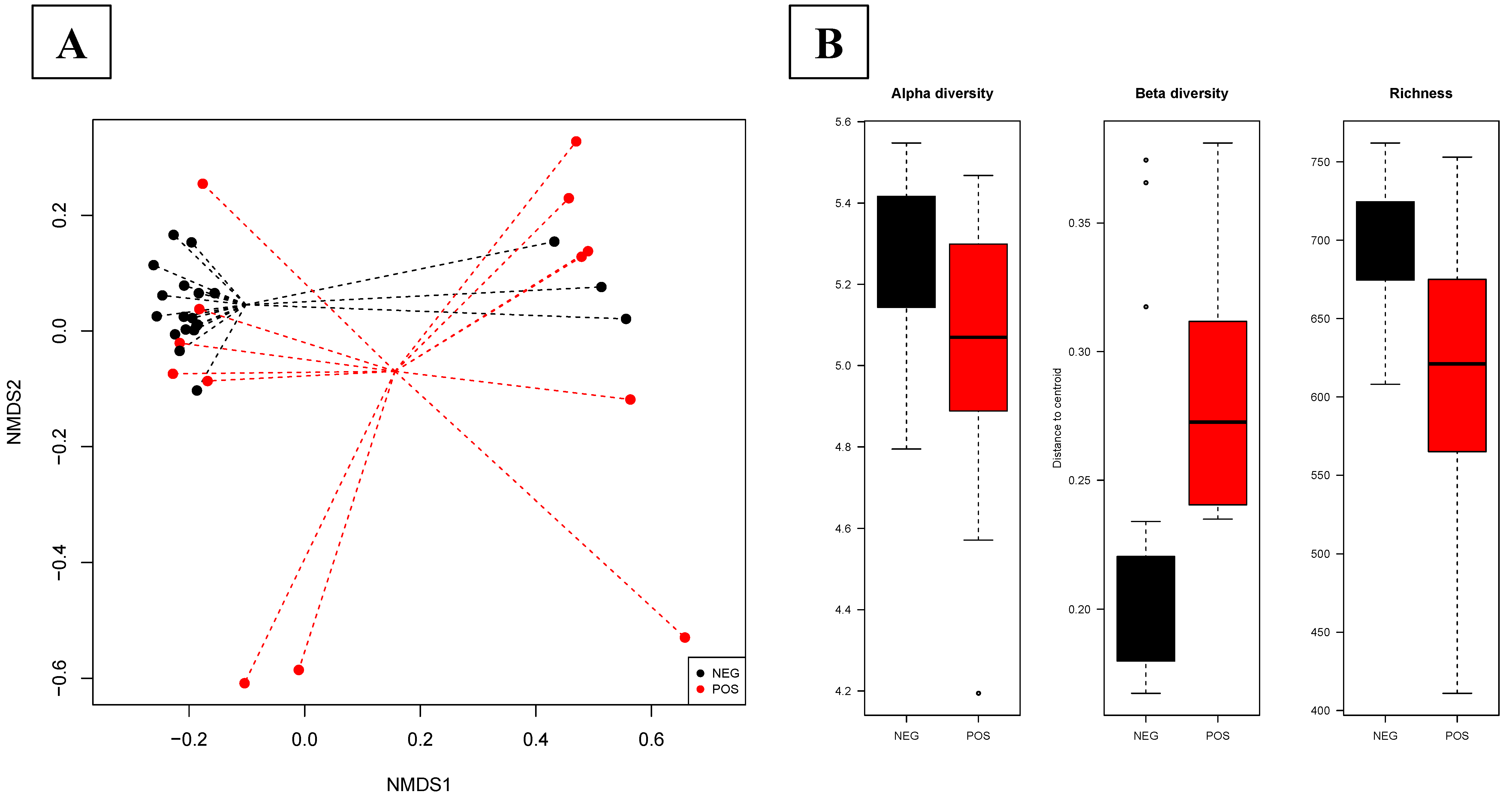
3.2.2. The Salmonella Status Is Associated with a Different Gut Microbiota Composition in Pigs at the Slaughterhouse
3.2.3. Salmonella Infection during Transport Affects the Fecal Microbiota in Pigs
3.2.4. Transport Has an Impact on the Faecal Microbiota Composition
3.2.5. The Fecal Microbial Composition on the Farm Does not Predict the ‘Will Be’ Salmonella Status at Slaughter
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- EFSA. Scientific report on the European Union one health 2018 zoonoses report. EFSA J. 2019, 17, 5926. [Google Scholar]
- Arguello, H.; Álvarez-Ordoñez, A.; Carvajal, A.; Rubio, P.; Prieto, M. Role of slaughtering in Salmonella spreading and control in pork production. J. Food Prot. 2013, 76, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Hu, X.M.; Zhang, T.; Wei, H.K.; Zhou, Y.F.; Zhou, Z.X.; Peng, J. Effects of dietary oregano essential oil and vitamin E supplementation on meat quality, stress response and intestinal morphology in pigs following transport stress. J. Vet. Med. Sci. 2017, 79, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Berends, B.R.; Van Knapen, F.; Snijders, J.M.A.; Mossel, D.A.A. Identification and quantification of risk factors regarding Salmonella spp. On pork carcasses. Int. J. Food Microbiol. 1997, 36, 199–206. [Google Scholar] [CrossRef]
- Magistrali, C.; Dionisi, A.M.; De Curtis, P.; Cucco, L.; Vischi, O.; Scuota, S.; Zicavo, A.; Pezzotti, G. Contamination of Salmonella spp. in a pig finishing herd, from the arrival of the animals to the slaughterhouse. Res. Vet. Sci. 2008, 85, 204–207. [Google Scholar] [CrossRef]
- Van Hoek, A.H.A.M.; de Jonge, R.; van Overbeek, W.M.; Bouw, E.; Pielaat, A.; Smid, J.H.; Malorny, B.; Junker, E.; Löfström, C.; Pedersen, K.; et al. A quantitative approach towards a better understanding of the dynamics of Salmonella spp. in a pork slaughter-line. Int. J. Food Microbiol. 2012, 153, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Pesciaroli, M.; Cucco, L.; De Luca, S.; Massacci, F.R.; Maresca, C.; Medici, L.; Paniccià, M.; Scoccia, E.; Staffolani, M.; Pezzotti, G.; et al. Association between pigs with high caecal Salmonella loads and carcass contamination. Int. J. Food Microbiol. 2017, 242, 82–86. [Google Scholar] [CrossRef]
- Argüello, H.; Estellé, J.; Zaldívar-lópez, S.; Jiménez-marín, Á.; Carvajal, A.; López-bascón, M.A.; Crispie, F.; Sullivan, O.O.; Cotter, P.D.; Priego-capote, F.; et al. Early Salmonella Typhimurium infection in pigs disrupts Microbiome composition and functionality principally at the ileum mucosa. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Drumo, R.; Pesciaroli, M.; Ruggeri, J.; Tarantino, M.; Chirullo, B.; Pistoia, C.; Petrucci, P.; Martinelli, N.; Moscati, L.; Manuali, E.; et al. Salmonella enterica serovar typhimurium exploits inflammation to modify swine intestinal microbiota. Front. Cell. Infect. Microbiol. 2016, 5, 1–13. [Google Scholar] [CrossRef]
- Bearson, S.M.D.; Allen, H.K.; Bearson, B.L.; Looft, T.; Brunelle, B.W.; Kich, J.D.; Tuggle, C.K.; Bayles, D.O.; Alt, D.; Levine, U.Y.; et al. Profiling the gastrointestinal microbiota in response to Salmonella: Low versus high Salmonella shedding in the natural porcine host. Infect. Genet. Evol. 2013, 16, 330–340. [Google Scholar] [CrossRef]
- Argüello, H.; Estellé, J.; Leonard, F.C.; Crispie, F.; Cotter, P.D.; O’Sullivan, O.; Lynch, H.; Walia, K.; Duffy, G.; Lawlor, P.G.; et al. Influence of the intestinal microbiota on colonization resistance to Salmonella and the shedding pattern of naturally exposed pigs. mSystems 2019, 4, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.B.; Isaacson, R.E. Salmonella in swine: Microbiota interactions. Annu. Rev. Anim. Biosci. 2017, 5, 43–63. [Google Scholar] [CrossRef] [PubMed]
- International-Standards. Microbiology of Food and Animal Feeding Stuffs—Horizontal Method for the Detection of Salmonella spp.—Amendment 1: Annex D: Detection of Salmonella spp. in Animal Feces and in Environmental Samples from the Primary Production St.; UNI EN ISO.; EN ISO 6579-2:2012/A1; International-Standards: Geneva, Switzerland, 2012. [Google Scholar]
- WHO. White-Kauffman-Le Minor Scheme “Antigenic Formulae of the Salmonella Serovars—WHO Collaborating Centre for Reference and Research on Salmonella”, WHO 9th ed.; WHO: Geneva, Switzerland, 2007. [Google Scholar]
- Dore, J.; Ehrlich, S.D.; Levenez, F.; Pelletier, E.; Alberti, A.; Bertrand, L.; Bork, P.; Costea, P.I.; Sunagawa, S.; Guarner, F.; et al. IHMS_SOP 06 V1: Standard operating procedure for fecal samples DNA extraction, protocol Q. International Human Microbiome Standards. 2015. Available online: http://www.microbiome-standards.org (accessed on 15 January 2019).
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Rideout, J.R.; He, Y.; Navas-Molina, J.A.; Walters, W.A.; Ursell, L.K.; Gibbons, S.M.; Chase, J.; McDonald, D.; Gonzalez, A.; Robbins-Pianka, A.; et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2014, 2, e545. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Navas-Molina, J.A.; Peralta-Sánchez, J.M.; González, A.; McMurdie, P.J.; Vázquez-Baeza, Y.; Ursell, L.K.; Lauber, C.; Zhou, H.; Song, S.J.; Huntley, J.; et al. Advancing our understanding of the human microbiome using QIIME. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2013; Volume 531, pp. 371–444. [Google Scholar]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2012, 10, 57–59. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, B.R.; Simpson, G.L.; Solymos, P.; et al. vegan: Community Ecology Package. R Package Version 2.5-5. 2019. Available online: https://cran.r-project.org/package=vegan/ (accessed on 15 January 2019).
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013. [Google Scholar] [CrossRef] [Green Version]
- Lahti, L.; Shetty, S. Tools for Microbiome Analysis in R. Version 1.9.95. 2017. Available online: https://github.com/microbiome/microbiome/ (accessed on 15 January 2019).
- Käsbohrer, A.; Protz, D.; Helmuth, R.; Nöckler, K.; Blaha, T.; Conraths, F.J.; Geue, L. Salmonella in slaughter pigs of German origin: An epidemiological study. Eur. J. Epidemiol. 2000, 16, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Rajkowski, K.T.; Eblen, S.; Laubauch, C. Efficacy of washing and sanitizing trailers used for swine transport in reduction of Salmonella and Escherichia coli. J. Food Prot. 1998, 61, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Mannion, C.; Leonard, F.C.; Lynch, P.B.; Egan, J. Efficacy of cleaning and disinfection on pig farms in Ireland. Vet. Rec. 2007, 161, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, R.E.; Firkins, L.D.; Weigel, R.M.; Zuckermann, F.A.; DiPietro, J.A. Effect of transportation and feed withdrawal on shedding of Salmonella Typhimurium among experimentally infected pigs. Am. J. Vet. Res. 1999, 60, 1155–1158. [Google Scholar]
- EFSA. Report of the task force on zoonoses data collection on the analysis of the baseline survey on the prevalence of Salmonella in slaughter pigs, in the EU, 2006–2007—Part A: Salmonella prevalence estimates. EFSA J. 2008, 6, 135. [Google Scholar] [CrossRef]
- Arguello, H.; Carvajal, A.; Collazos, J.A.; García-Feliz, C.; Rubio, P. Prevalence and serovars of Salmonella enterica on pig carcasses, slaughtered pigs and the environment of four Spanish slaughterhouses. Food Res. Int. 2012, 45, 905–912. [Google Scholar] [CrossRef]
- Bonardi, S.; Bruini, I.; Alpigiani, I.; Vismarra, A.; Barilli, E.; Brindani, F.; Morganti, M.; Bellotti, P.; Bolzoni, L.; Pongolini, S. Influence of pigskin on Salmonella contamination of pig carcasses and cutting lines in an italian slaughterhouse. Ital. J. Food Saf. 2016, 5, 65–68. [Google Scholar] [CrossRef] [Green Version]
- Snary, E.L.; Swart, A.N.; Simons, R.R.L.; Domingues, A.R.C.; Vigre, H.; Evers, E.G.; Hald, T.; Hill, A.A. A quantitative microbiological risk assessment for Salmonella in pigs for the European Union. Risk Anal. 2016, 36, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Hurd, H.S.; Gailey, J.K.; McKean, J.D.; Rostagno, M.H. Rapid infection in market-weight swine following exposure to a Salmonella Typhimurium-contaminated environment. Am. J. Vet. Res. 2001, 62, 1194–1197. [Google Scholar] [CrossRef]
- Boughton, C.; Egan, J.; Kelly, G.; Markey, B.; Leonard, N. Rapid infection of pigs following exposure to environments contaminated with different levels of Salmonella Typhimurium. Foodborne Pathog. Dis. 2007, 4, 33–40. [Google Scholar] [CrossRef]
- McCormack, U.M.; Curiao, T.; Buzoianu, S.G.; Prieto, M.L.; Ryan, T.; Varley, P.; Crispie, F.; Magowan, E.; Metzler-Zebeli, B.U.; Berry, D.; et al. Exploring a possible link between the intestinal microbiota and feed efficiency in pigs. Appl. Environ. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massacci, F.R.; Tofani, S.; Forte, C.; Bertocchi, M.; Lovito, C.; Orsini, S.; Tentellini, M.; Marchi, L.; Lemonnier, G.; Luise, D.; et al. Host genotype and amoxicillin administration affect the incidence of diarrhoea and faecal microbiota of weaned piglets during a natural multiresistant ETEC infection. J. Anim. Breed. Genet. 2020, 137, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Massacci, F.R.; Berri, M.; Lemonnier, G.; Guettier, E.; Blanc, F.; Jardet, D.; Rossignol, M.N.; Mercat, M.J.; Doré, J.; Lepage, P.; et al. Late weaning is associated with increased microbial diversity and higher F. prausnitzii abundance in piglet’s gut microbiota. Anim. Microbiome 2020, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Holman, D.B.; Brunelle, B.W.; Trachsel, J.; Allen, K. Meta-analysis to define a core microbiota in the swine gut. mSystems 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Megahed, A.; Zeineldin, M.; Evans, K.; Maradiaga, N.; Blair, B.; Aldridge, B.; Lowe, J. Impacts of environmental complexity on respiratory and gut microbiome community structure and diversity in growing pigs. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Crespo-Piazuelo, D.; Estellé, J.; Revilla, M.; Criado-Mesas, L.; Ramayo-caldas, Y.; Óvilo, C.; Fernández, A.I.; Ballester, M.; Folch, J.M. Characterization of bacterial microbiota compositions along the intestinal tract in pigs and their interactions and functions. Sci. Rep. 2018, 8, 12727. [Google Scholar] [CrossRef]
- Borewicz, K.A.; Kim, H.B.; Singer, R.S.; Gebhart, C.J.; Sreevatsan, S.; Johnson, T.; Isaacson, R.E. Changes in the porcine intestinal microbiome in response to infection with Salmonella enterica and Lawsonia intracellularis. PLoS ONE 2015, 10, e0139106. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ID | Herd | Slaughterhouse | ||||
|---|---|---|---|---|---|---|
| Feces | MPN/g | Serotypes | Feces | MPN/g | Serotypes | |
| 1 | N | 0 | N | 0 | ||
| 2 | N | 0 | P | 32 | S. Rissen | |
| 3 | N | 0 | N | 0 | ||
| 4 | N | 0 | N | 0 | ||
| 5 | N | 0 | N | 0 | ||
| 6 | N | 0 | N | 0 | ||
| 8 | N | 0 | N | 0 | ||
| 9 | N | 0 | N | 0 | ||
| 10 | N | 0 | N | 0 | ||
| 11 | N | 0 | N | 0 | ||
| 12 | N | 0 | P | 78 | S. 4,12:i:- | |
| 13 | N | 0 | N | 0 | ||
| 14 | N | 0 | N | 0 | ||
| 15 | N | 0 | N | 0 | ||
| 16 | N | 0 | N | 0 | ||
| 17 | N | 0 | N | 0 | ||
| 18 | N | 0 | N | 0 | ||
| 19 | N | 0 | P | 77 | S. 4,12:i:- | |
| 20 | N | 0 | N | 0 | ||
| 21 | N | 0 | P | 74 | S. 4,12:i:- | |
| 22 | P | 6 | S. Rissen | N | 0 | |
| 23 | N | 0 | P | 5900 | S. Rissen | |
| 24 | N | 0 | N | 0 | ||
| 25 | P | 32 | S. Rissen | P | 4000 | S. 4,12:i:- |
| 26 | N | 0 | P | 120 | S. 4,12:i:- | |
| 27 | N | 0 | N | 0 | ||
| 28 | N | 0 | P | 51 | S. 4,12:i:- | |
| 29 | N | 0 | N | 0 | ||
| 30 | N | 0 | N | 0 | ||
| 31 | N | 0 | N | 0 | ||
| 32 | N | 0 | N | 0 | ||
| 33 | N | 0 | N | 0 | ||
| 34 | N | 0 | N | 0 | ||
| 35 | N | 0 | N | 0 | ||
| 36 | N | 0 | P | 33 | S. Rissen | |
| 38 | N | 0 | N | 51 | ||
| 40 | N | 0 | N | 0 | ||
| 42 | N | 0 | N | 0 | ||
| 43 | N | 0 | N | 0 | ||
| 44 | P | 100 | S. Rissen | N | 0 | |
| 46 | P | 3100 | S. 4,12:i:- | P | 25000000 | S. 4,12:i:- |
| 47 | P | 420 | S. 4,12:i:- | N | 0 | |
| 48 | N | 0 | P | 160 | S. 4,12:i:- | |
| 49 | N | 0 | N | 0 | ||
| 53 | N | 0 | N | 0 | ||
| 54 | N | 0 | N | 0 | ||
| 55 | N | 0 | P | 84 | S. 4,12:i:- | |
| 57 | N | 0 | P | 1100 | S. 4,12:i:- | |
| 59 | N | 6 | N | 0 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massacci, F.R.; Morelli, A.; Cucco, L.; Castinel, A.; Ortenzi, R.; Tofani, S.; Pezzotti, G.; Estellé, J.; Paniccià, M.; Magistrali, C.F. Transport to the Slaughterhouse Affects the Salmonella Shedding and Modifies the Fecal Microbiota of Finishing Pigs. Animals 2020, 10, 676. https://doi.org/10.3390/ani10040676
Massacci FR, Morelli A, Cucco L, Castinel A, Ortenzi R, Tofani S, Pezzotti G, Estellé J, Paniccià M, Magistrali CF. Transport to the Slaughterhouse Affects the Salmonella Shedding and Modifies the Fecal Microbiota of Finishing Pigs. Animals. 2020; 10(4):676. https://doi.org/10.3390/ani10040676
Chicago/Turabian StyleMassacci, Francesca Romana, Alessandra Morelli, Lucilla Cucco, Adrien Castinel, Roberta Ortenzi, Silvia Tofani, Giovanni Pezzotti, Jordi Estellé, Marta Paniccià, and Chiara Francesca Magistrali. 2020. "Transport to the Slaughterhouse Affects the Salmonella Shedding and Modifies the Fecal Microbiota of Finishing Pigs" Animals 10, no. 4: 676. https://doi.org/10.3390/ani10040676
APA StyleMassacci, F. R., Morelli, A., Cucco, L., Castinel, A., Ortenzi, R., Tofani, S., Pezzotti, G., Estellé, J., Paniccià, M., & Magistrali, C. F. (2020). Transport to the Slaughterhouse Affects the Salmonella Shedding and Modifies the Fecal Microbiota of Finishing Pigs. Animals, 10(4), 676. https://doi.org/10.3390/ani10040676