
RNA-Seq Implies Divergent Regulation Patterns of LincRNA on Spermatogenesis and Testis Growth in Goats


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sampling
2.3. RNA Isolation, Library Construction, and Sequencing
2.4. Mapping and Assembling
2.5. Coding Potential Analysis of Transcripts
2.6. Principal Component Analysis (PCA) and Correlation Analysis of Samples
2.7. Differential Expression Analysis
2.8. Quantitative Real-Time PCR (qRT-PCR) Validation
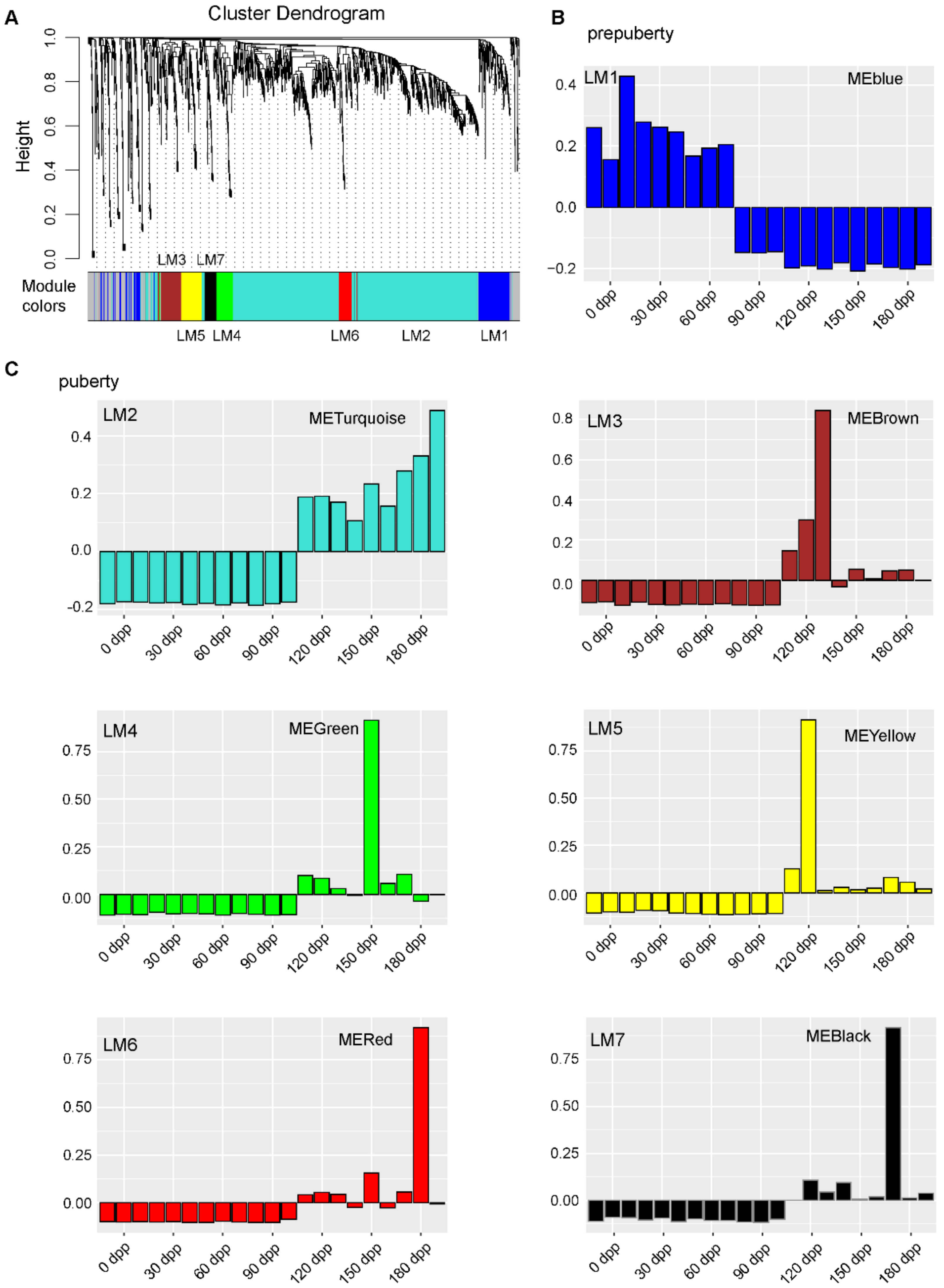
2.9. WGCNA Analysis
2.10. Functional Enrichment Analysis
2.11. The Prediction of the Target Genes of LincRNAs
3. Results
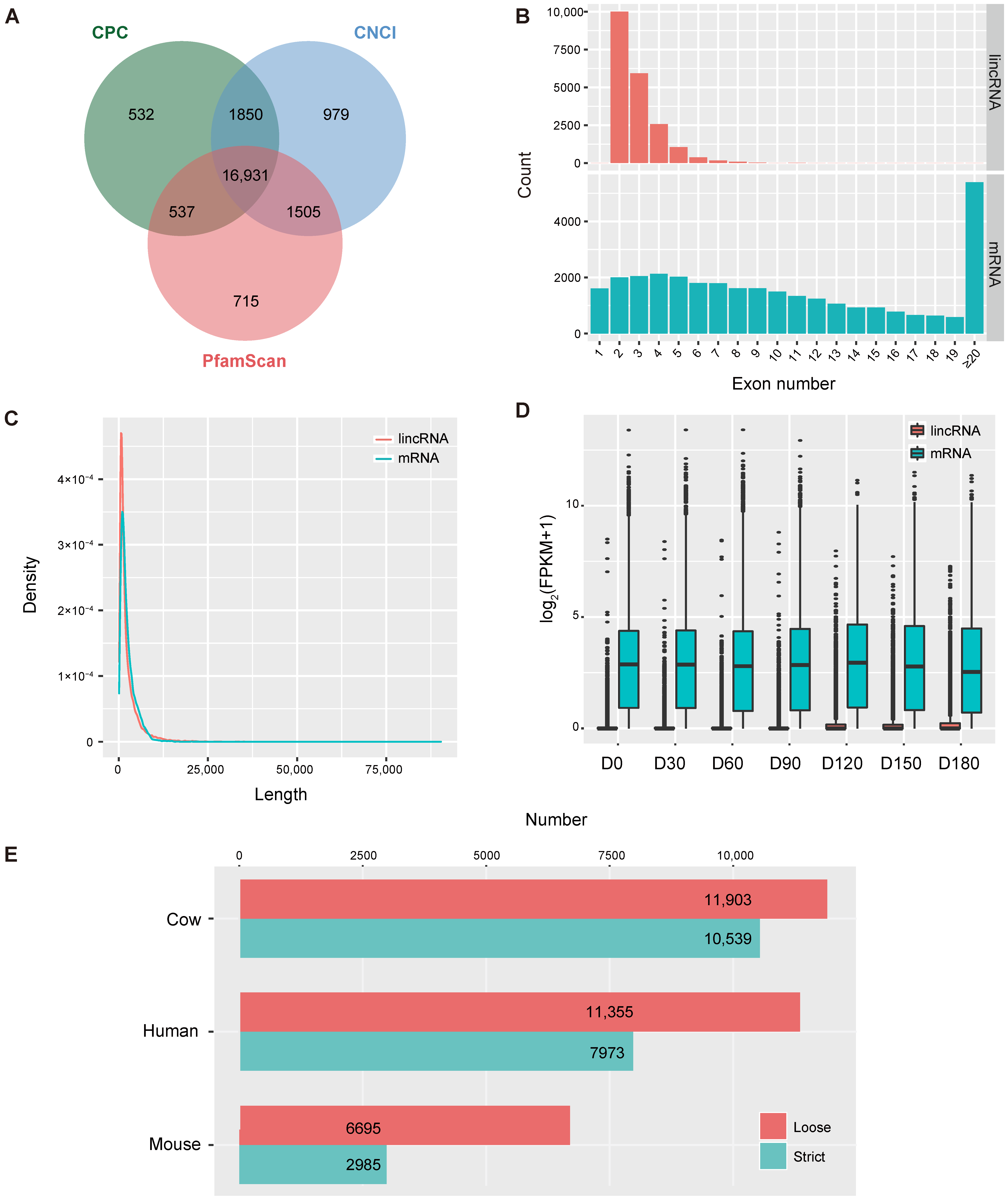
3.1. Identification of LincRNAs in the Goat Testis
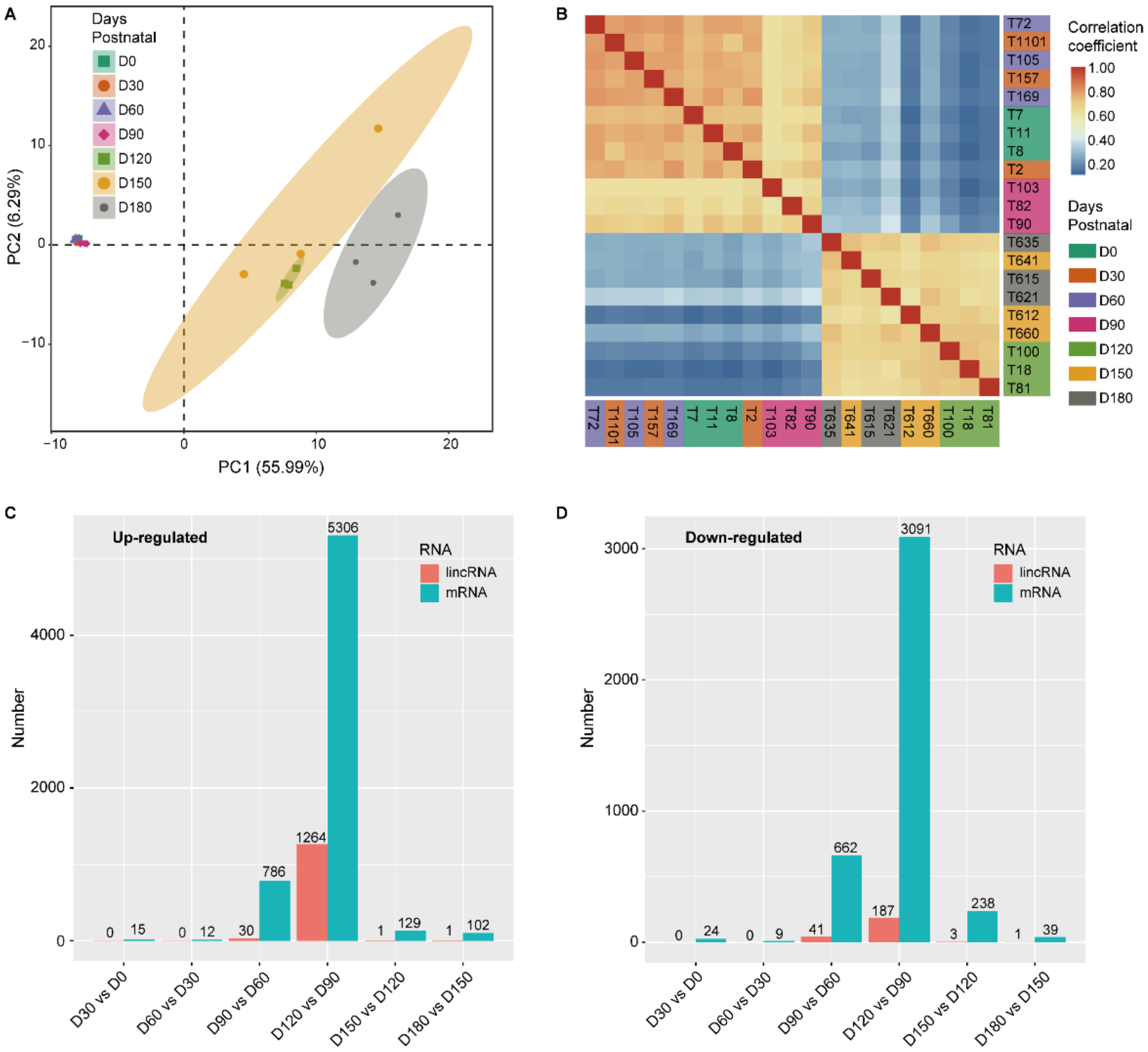
3.2. The Expression Profile of LincRNAs
3.3. The Time-Specific Expression of LincRNAs Contributed to the Transition of Testicular Development Stages
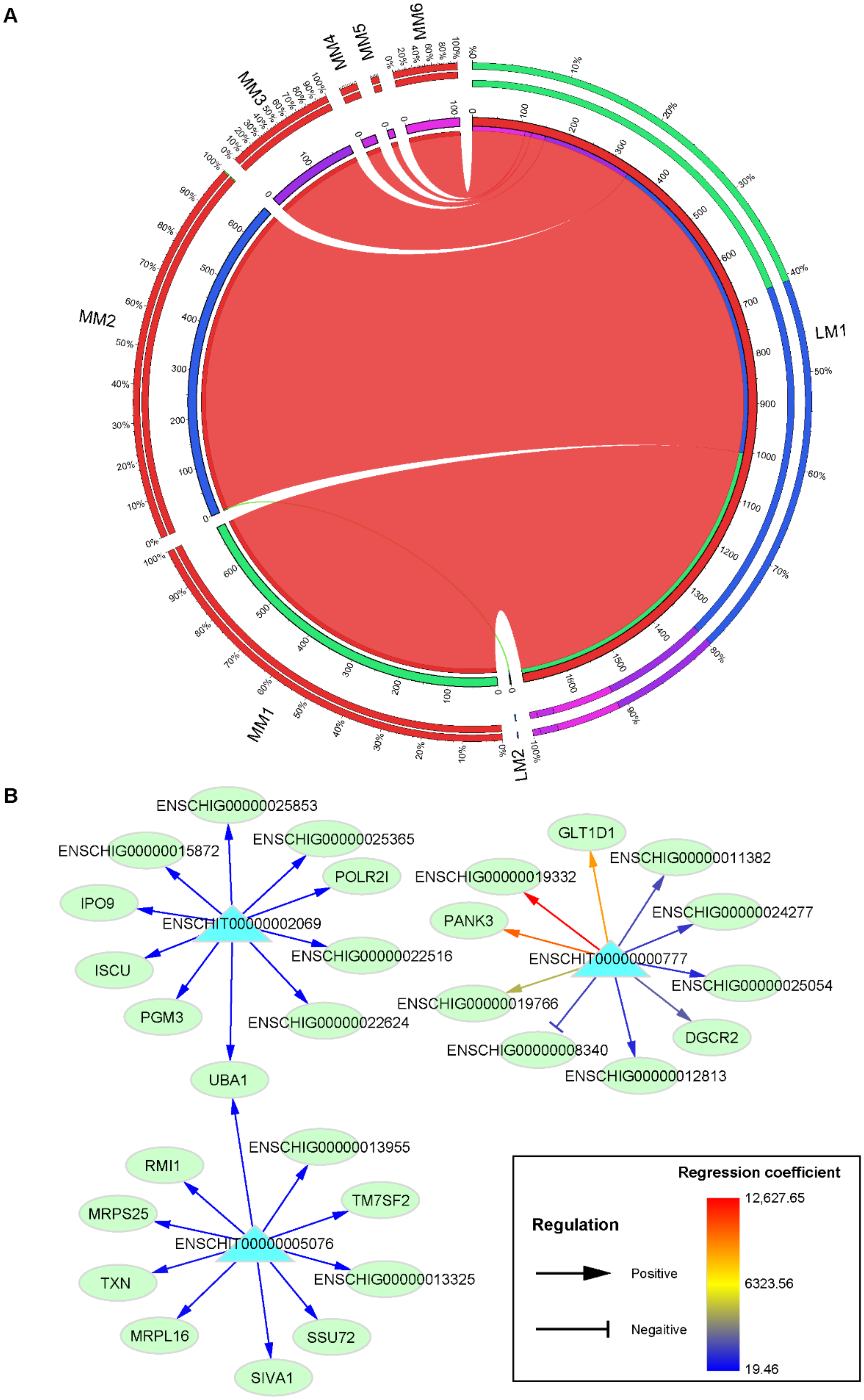
3.4. Prediction of LincRNA Target Genes
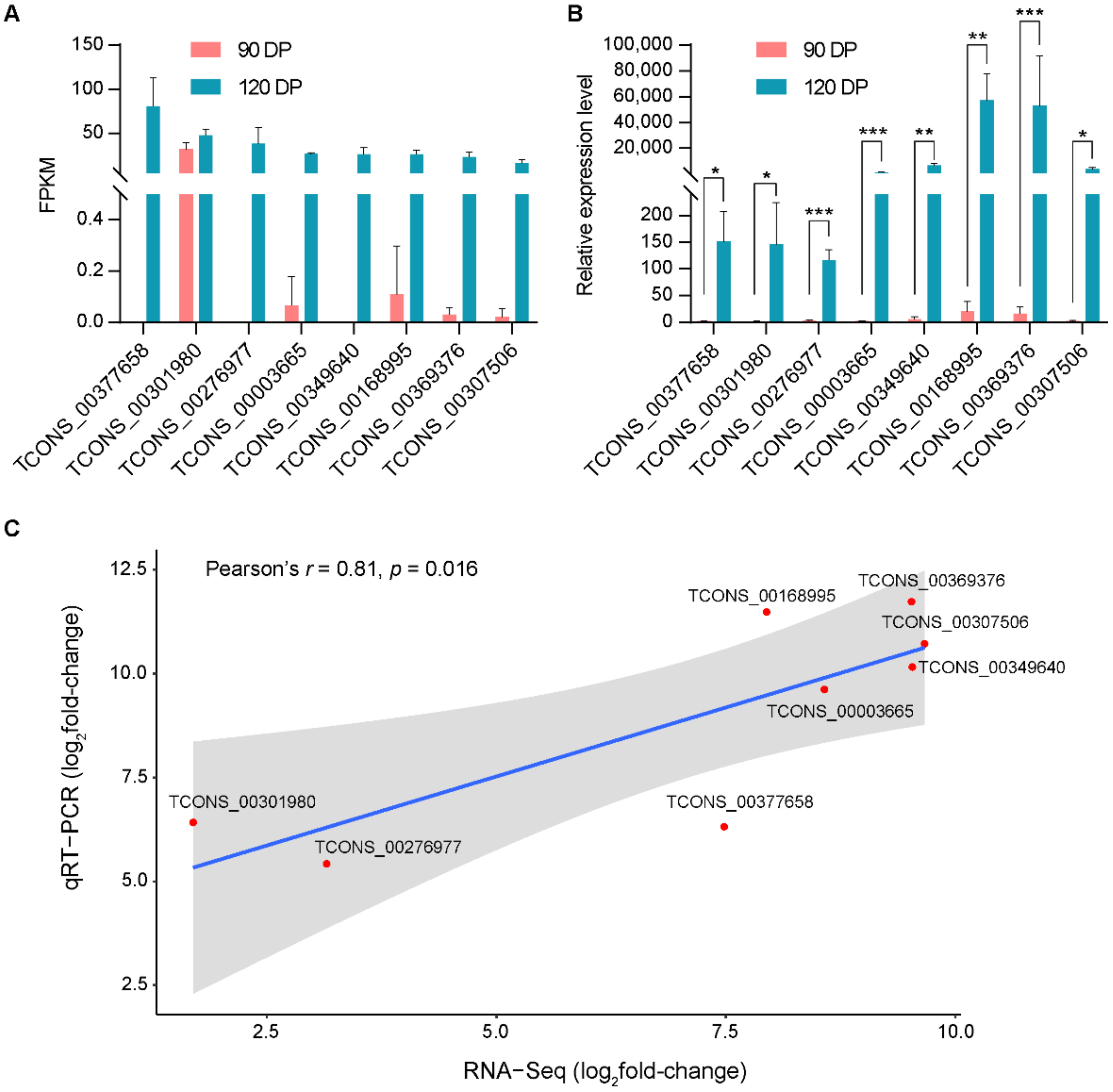
3.5. qRT-PCR Test
4. Discussion
4.1. Characteristics of LincRNAs Identified in This Study
4.2. The Specific Expression Pattern of LincRNAs
4.3. The Role of LincRNAs in Goat Testicular Development
4.3.1. The Down-Regulation of LincRNAs at 90 DP
4.3.2. The Up-Regulation of LincRNAs at 120 DP
4.3.3. The Divergent Regulation Patterns of LincRNA on Spermatogenesis and Organ Growth
4.4. Prediction of the Target Genes of LincRNAs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, K.C.; Frith, M.C.; Mattick, J.S. Rapid evolution of noncoding RNAs: Lack of conserva-tion does not mean lack of function. Trends Genet. 2006, 22, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [Green Version]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [Green Version]
- Mattick, J.S.; Rinn, J.L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Non–coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001, 2, 919–929. [Google Scholar] [CrossRef]
- Gabory, A.; Jammes, H.; Dandolo, L. The H19 locus: Role of an imprinted non-coding RNA in growth and development. Bioessays 2010, 32, 473–480. [Google Scholar] [CrossRef]
- Plath, K.; Mlynarczyk-Evans, S.; Nusinow, D.A.; Panning, B. Xist RNA and the mechanism of X chromosome inactivation. Annu. Rev. Genet. 2002, 36, 233–278. [Google Scholar] [CrossRef] [PubMed]
- Bouckenheimer, J.; Assou, S.; Riquier, S.; Hou, C.; Philippe, N.; Sansac, C.; Lavabre-Bertrand, T.; Commes, T.; Lemaitre, J.M.; Boureux, A.; et al. Long non-coding RNAs in human early embryonic development and their potential in ART. Hum. Reprod. Update 2016, 23, 19–40. [Google Scholar] [CrossRef]
- Matsumoto, A.; Pasut, A.; Matsumoto, M.; Yamashita, R.; Fung, J.; Monteleone, E.; Saghatelian, A.; Nakayama, K.I.; Clohessy, J.G.; Pandolfi, P.P. MTORC1 and muscle regeneration are regulated by the LINC00961-encoded SPAR polypeptide. Nature 2016, 541, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Satpathy, A.T.; Chang, H.Y. Gene regulation in the immune system by long noncoding RNAs. Nat. Immunol. 2017, 18, 962–972. [Google Scholar] [CrossRef]
- Pradas-Juni, M.; Hansmeier, N.R.; Link, J.C.; Schmidt, E.; Larsen, B.D.; Klemm, P.; Meola, N.; Topel, H.; Loureiro, R.; Dhaouadi, I.; et al. A MAFG-lncRNA axis links systemic nutrient abundance to hepatic glucose metabolism. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, K.; Yang, L.; Xiong, T.; Di, C.; Ma, D.; Wu, M.; Xue, Z.; Zhang, X.; Long, L.; Zhang, W.; et al. Critical roles of long noncoding RNAs in Drosophila spermatogenesis. Genome Res. 2016, 26, 1233–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long in-tergenic non-coding RNA. Nat. Rev. Mol. Cell. Biol. 2018, 19, 143. [Google Scholar] [CrossRef]
- Salemi, M.; Cannarella, R.; Condorelli, R.A.; Cimino, L.; Ridolfo, F.; Giurato, G.; Romano, C.; La Vignera, S.; Calogero, A.E. Evidence for long noncoding RNA GAS5 up-regulationin patients with Klinefelter syndrome. BMC Med. Genet. 2019, 20, 4. [Google Scholar] [CrossRef]
- Liu, M.X.; Chen, X.; Chen, G.; Cui, Q.H.; Yan, G.Y. A computational framework to infer human disease-associated long noncoding RNAs. PLoS ONE 2014, 9, e84408. [Google Scholar] [CrossRef]
- Stamou, M.; Ng, S.Y.; Brand, H.; Wang, H.; Plummer, L.; Best, L.; Havlicek, S.; Hibberd, M.; Khor, C.C.; Gusella, J.; et al. A Balanced Translocation in Kallmann Syndrome Implicates a Long Noncoding RNA, RMST, as a GnRH Neuronal Regulator. J. Clin. Endocrinol. Metab. 2020, 105, e231–e244. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, B.B.; Olcese, U.; Cabrera, J.R.; Horabin, J.I. An interactive network of long non-coding RNAs facilitates the Drosophila sex determination decision. Biochim. Biophys. Acta 2014, 1839, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Roeszler, K.N.; Itman, C.; Sinclair, A.H.; Smith, C.A. The long non-coding RNA, MHM, plays a role in chicken embryonic development, including gonadogenesis. Dev. Biol. 2012, 366, 317–326. [Google Scholar] [CrossRef]
- Weng, B.; Ran, M.; Chen, B.; He, C.; Dong, L.; Peng, F. Genome-wide analysis of long non-coding RNAs and their role in postnatal porcine testis development. Genomics 2017, 109, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Wichman, L.; Somasundaram, S.; Breindel, C.; Valerio, D.M.; McCarrey, J.R.; Hodges, C.A.; Khalil, A.M. Dynamic expression of long noncoding RNAs reveals their potential roles in spermatogenesis and fertility. Biol. Reprod. 2017, 97, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Bo, D.; Jiang, X.; Liu, G.; Xu, F.; Hu, R.; Wassie, T.; Chong, Y.; Ahmed, S.; Liu, C.; Girmay, S. Multipathway synergy promotes testicular transition from growth to spermatogenesis in early-puberty goats. BMC Genom. 2020, 21, 372. [Google Scholar] [CrossRef]
- Wang, Y.; Song, F.; Zhu, J.; Zhang, S.; Yang, Y.; Chen, T.; Tang, B.; Dong, L.; Ding, N.; Zhang, Q.; et al. GSA: Genome Sequence Archive. Genom. Proteom. Bioinform. 2017, 15, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Members, B.I.G.D.C. Database Resources of the BIG Data Center in 2019. Nucleic Acids Res. 2019, 47, D8–D14. [Google Scholar] [CrossRef] [PubMed]
- FastQC—A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 3 January 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35 (Suppl. 2), W345–W349. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, P.; Longden, L.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, K.; Taniguchi, H.; Yoda, K.; Shimizu, M.; Sakiyama, S. Nucleotide sequence of a full-length cDNA for mouse cytoskeletal beta-actin mRNA. Nucleic Acids Res. 1986, 14, 2829. [Google Scholar] [CrossRef]
- Swift, M.L. GraphPad prism, data analysis, and scientific graphing. J. Chem. Inf. Comput. Sci. 1997, 37, 411–412. [Google Scholar] [CrossRef]
- ggpubr: ’ggplot2’ Based Publication Ready Plots. Available online: https://cran.r-project.org/web/packages/ggpubr (accessed on 3 August 2020).
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A. Ensembl BioMarts: A hub for data retrieval across taxonomic space. In Database; 2011; Volume 2011, p. bar30. [Google Scholar] [CrossRef]
- Carbon, S.; Douglass, E.; Dunn, N.; Good, B.; Harris, N.L.; Lewis, S.E.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; et al. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.Y.; Muruganujan, A.; Ebert, D.; Huang, X.S.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Li, H.; Fang, S.S.; Kang, Y.; Wu, W.; Hao, Y.J.; Li, Z.Y.; Bu, D.C.; Sun, N.H.; Zhang, M.Q.; et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016, 44, D203–D208. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local regu-lation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef]
- Kornienko, A.E.; Guenzl, P.M.; Barlow, D.P.; Pauler, F.M. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.T. Epigenetic regulation by long noncoding RNAs. Science 2012, 338, 1435–1439. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Fu, L.; Koganti, P.P.; Wang, L.; Hand, J.M.; Ma, H.; Yao, J. Identification and Functional Prediction of Large Inter-genic Noncoding RNAs (lincRNAs) in Rainbow Trout (Oncorhynchus mykiss). Mar. Biotechnol. 2016, 18, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cheng, T.; Liu, C.; Liu, D.; Zhang, Q.; Long, R.; Zhao, P.; Xia, Q. Systematic identification and characterization of long non-coding RNAs in the silkworm, Bombyx mori. PLoS ONE 2016, 11, e0147147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabili, M.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, Z.; Chen, D.; Zhang, B.; Tian, R.R.; Wu, J.; Zhang, Y.; Xu, K.; Yang, L.M.; Cheng, C.; et al. Annotation and cluster analysis of spatiotemporal- and sex-related lncRNA expression in rhesus macaque brain. Genome Res. 2017, 27, 1608–1620. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncod-ing RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [Green Version]
- Arenas, M.I.; Bethencourt, F.R.; Fraile, B.; Paniagua, R. Immunocytochemical and quantitative study of the tunica albuginea testis in young and ageing men. Histochem. Cell Biol. 1997, 107, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, R.M. Local-Control of Testicular Function. Q. J. Exp. Physiol. 1983, 68, 265–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Wu, J.; Liu, B.; Jiang, Y.; Chen, W.; Li, J.; He, Q.; He, Z. The roles and mechanisms of Leydig cells and myoid cells in regulating spermatogenesis. Cell Mol. Life Sci. 2019, 1–15. [Google Scholar] [CrossRef]
- Potter, S.J.; DeFalco, T. Role of the testis interstitial compartment in spermatogonial stem cell function. Reproduction 2017, 153, R151. [Google Scholar] [CrossRef] [Green Version]
- Fijak, M.; Meinhardt, A. The testis in immune privilege. Immunol. Rev. 2006, 213, 66–81. [Google Scholar] [CrossRef]
- Umbreit, N.T.; Zhang, C.-Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368. [Google Scholar] [CrossRef]
- Brandt, M.P.; Gust, K.M.; Bon, D.; Tsaur, I.; Bartsch, G. Trend analysis and regional tumor incidence in Germany for testicu-lar cancer between 2003 and 2014. Andrology 2019, 7, 408–414. [Google Scholar] [CrossRef]
- Lin, Z.; Tong, M.-H. m6A mRNA modification regulates mammalian spermatogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 403–411. [Google Scholar] [CrossRef]
- Kotaja, N. MicroRNAs and spermatogenesis. Fertil. Steril. 2014, 101, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.H.; Zhao, Y.M. The regulatory functions of piRNA/PIWI in spermatogenesis. Yi Chuan 2017, 39, 683–691. [Google Scholar] [CrossRef] [PubMed]
- St. Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Elling, R.; Robinson, E.K.; Shapleigh, B.; Liapis, S.C.; Covarrubias, S.; Katzman, S.; Groff, A.F.; Jiang, Z.; Agarwal, S.; Motwani, M. Genetic models reveal cis and trans immune-regulatory activities for lincRNA-Cox2. Cell Rep. 2018, 25, 1511–1524. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Ye, J.; Yang, C.; Zhang, K.; Li, X.; Luo, L.; Ding, J.; Li, Y.; Cao, H.; Ling, Y.; et al. Screening and evaluating of long noncoding RNAs in the puberty of goats. BMC Genom. 2017, 18, 164. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Song, X.; Wang, J.; Zhou, M. Genome-Wide Identification and Characterization of Fusarium gramine-arum-Responsive lncRNAs in Triticum aestivum. Genes 2020, 11, 1135. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| ACTB * | GTCACCAACTGGGACGACAT | CATCTTCTCACGGTTGGCCT |
| TCONS_00003665 | GCTTTGAGAAACCCTGGGGAA | CTCCCTTTCCACAAGCCAAA |
| TCONS_00068785 | GTCTCCCTGCACTGCTGTAT | ATGGGACTGCTGTCGATTCA |
| TCONS_00168995 | AGGCCTGCCTTGTAGAGGAT | TCGTCACATGGAGAAGCAGA |
| TCONS_00276977 | CGCTTCCTGAGGGTTTAGCA | CACACTGCCTGGTACATGGT |
| TCONS_00301980 | AATAGGGGGAAAGCCGGTTG | CGGAGGCCATTTGACTCCTT |
| TCONS_00307506 | CATTCTCTGAGACATTTTATC TCCC | TCAAGGTTGTATTTCTGATTG TGTG |
| TCONS_00349640 | ATGATGTCAGTTGAGACTCA GAA | TGTGTTGCTGTTGACTTGCAG |
| TCONS_00360520 | GAGCAGTTCTCCTGGGTTCC | GGGTCCCTTCATGATTGCCA |
| TCONS_00369376 | AAGCTCTGTAGCCAGTGTTCC | TTTAGCCATGCTCTGAGGTCC |
| TCONS_00377658 | ACAGCCCTGAGCGAGAAGA | AGCCTGTCCAGACGAAAGGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bo, D.; Jiang, X.; Liu, G.; Hu, R.; Chong, Y. RNA-Seq Implies Divergent Regulation Patterns of LincRNA on Spermatogenesis and Testis Growth in Goats. Animals 2021, 11, 625. https://doi.org/10.3390/ani11030625
Bo D, Jiang X, Liu G, Hu R, Chong Y. RNA-Seq Implies Divergent Regulation Patterns of LincRNA on Spermatogenesis and Testis Growth in Goats. Animals. 2021; 11(3):625. https://doi.org/10.3390/ani11030625
Chicago/Turabian StyleBo, Dongdong, Xunping Jiang, Guiqiong Liu, Ruixue Hu, and Yuqing Chong. 2021. "RNA-Seq Implies Divergent Regulation Patterns of LincRNA on Spermatogenesis and Testis Growth in Goats" Animals 11, no. 3: 625. https://doi.org/10.3390/ani11030625
APA StyleBo, D., Jiang, X., Liu, G., Hu, R., & Chong, Y. (2021). RNA-Seq Implies Divergent Regulation Patterns of LincRNA on Spermatogenesis and Testis Growth in Goats. Animals, 11(3), 625. https://doi.org/10.3390/ani11030625