
Genotyping Strategies Using ddRAD Sequencing in Farmed Arctic Charr (Salvelinus alpinus)

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Background and Phenotypes
2.2. DNA Extraction and Quantification
2.3. ddRAD Library Preparation and Sequencing
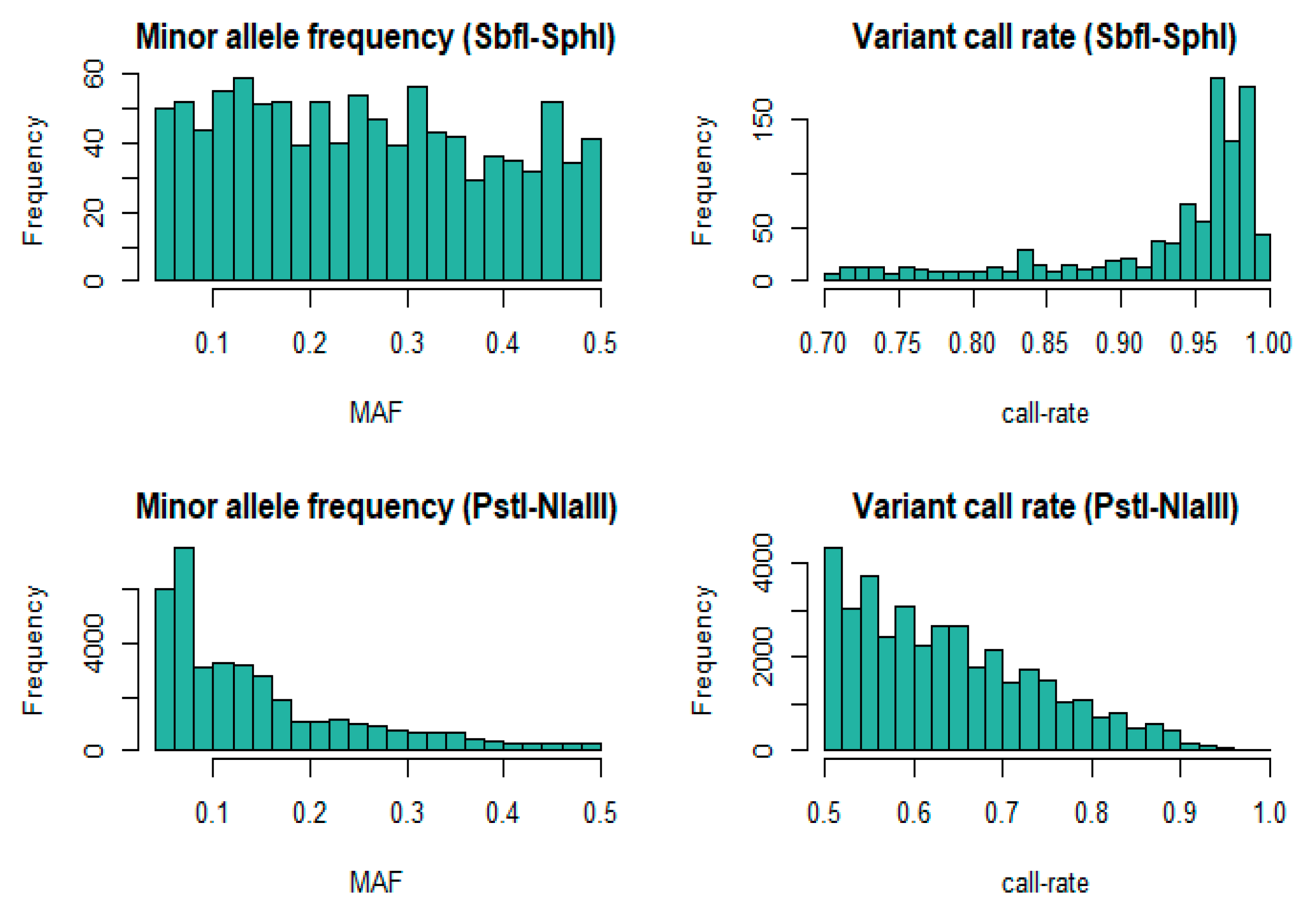
2.4. Sequence Data Analysis and SNP Genotyping
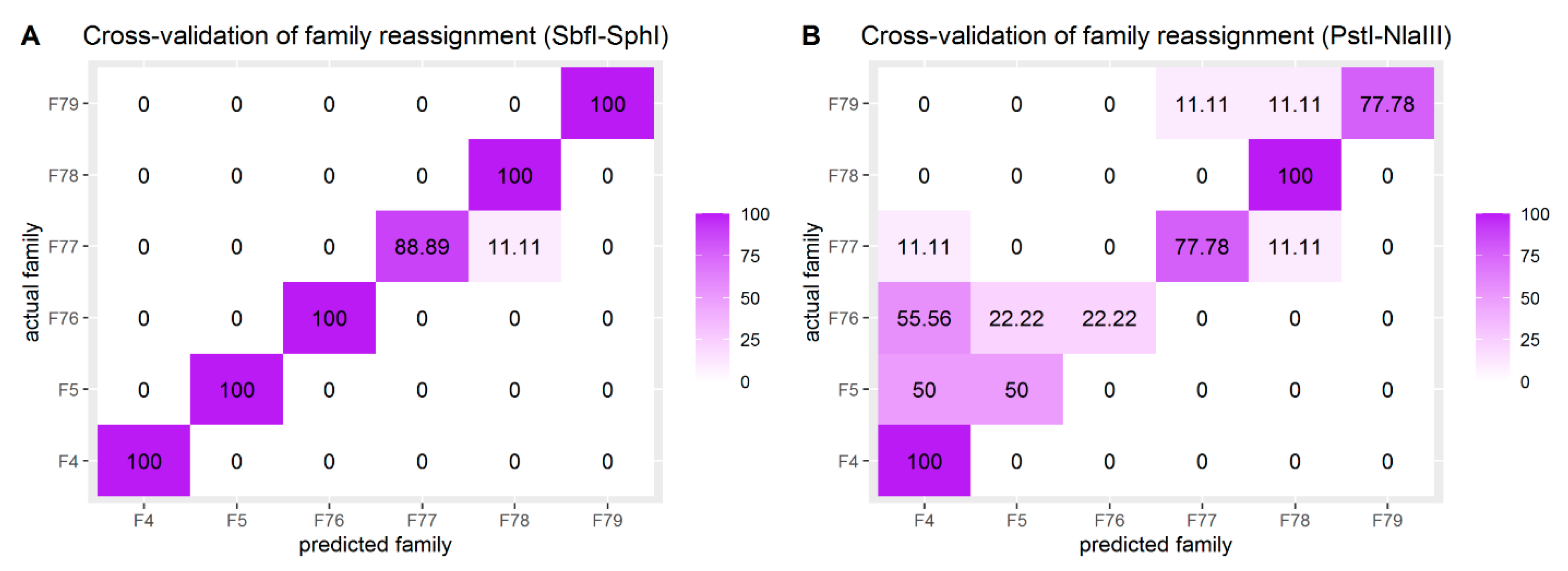
2.5. Genetic Diversity and Kinship
2.6. Association with Phenotypic Traits
3. Results
3.1. Genotypic Information
3.2. Descriptive Statistics of Phenotypic Traits
3.3. Genetic Diversity
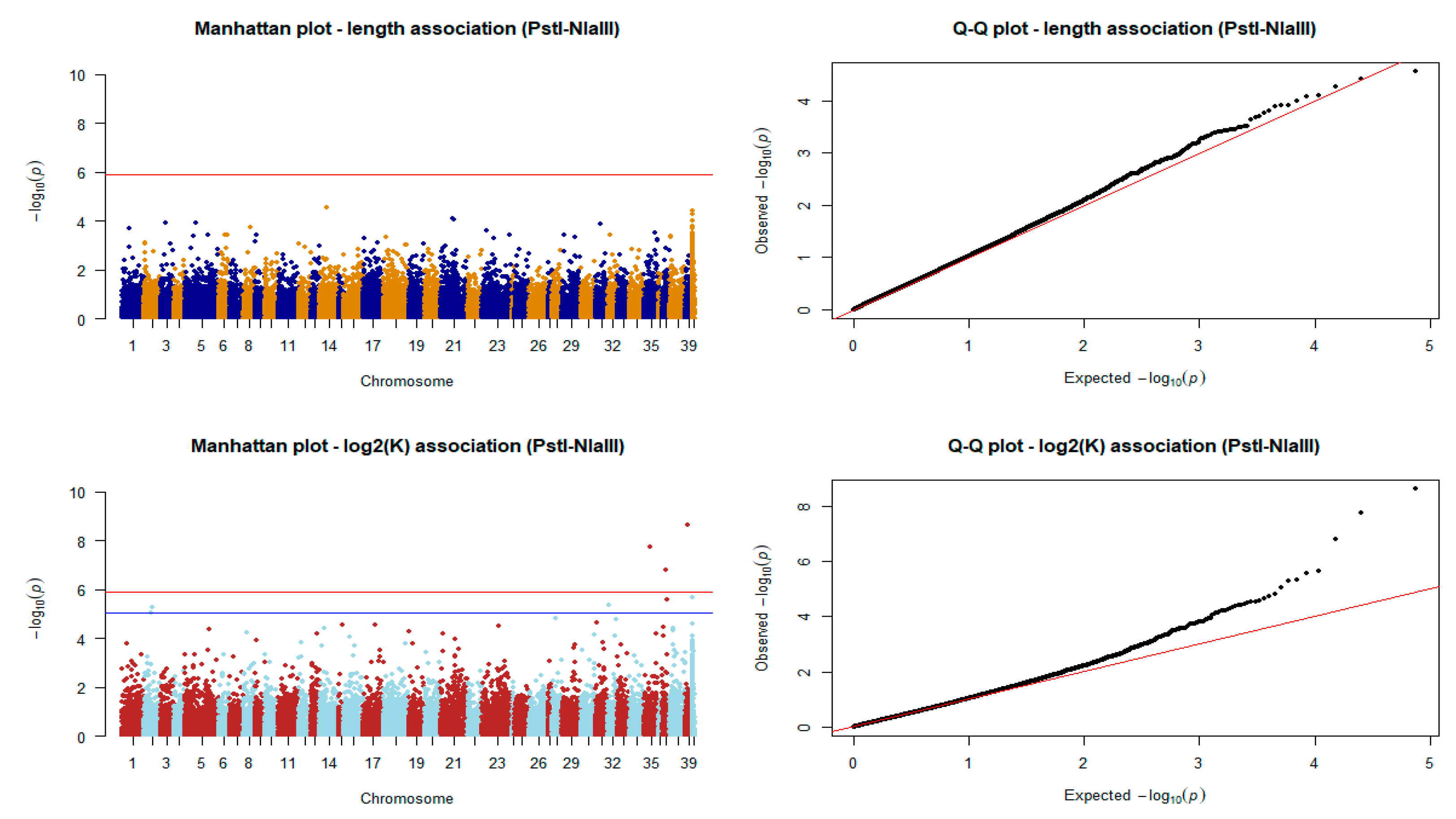
3.4. Association Scans for Phenotypic Traits
4. Discussion
4.1. Genetic Diversity
4.2. Kinship Investigation
4.3. Association Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Houston, R.D.; Bean, T.P.; Macqueen, D.J.; Gundappa, M.K.; Jin, Y.H.; Jenkins, T.L.; Selly, S.L.C.; Martin, S.A.M.; Stevens, J.R.; Santos, E.M.; et al. Harnessing Genomics to Fast-Track Genetic Improvement in Aquaculture. Nat. Rev. Genet. 2020, 21, 389–409. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Shan, X.; Shi, Q. Research Advances in the Genomics and Applications for Molecular Breeding of Aquaculture Animals. Aquaculture 2020, 526, 735357. [Google Scholar] [CrossRef]
- Robledo, D.; Palaiokostas, C.; Bargelloni, L.; Martínez, P.; Houston, R. Applications of Genotyping by Sequencing in Aquaculture Breeding and Genetics. Rev. Aquac. 2017, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the Power of RADseq for Ecological and Evolutionary Genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; Anderson, R.M.; van Stijn, T.C.; Kristjánsson, T.; Clarke, S.M. Construction of Relatedness Matrices Using Genotyping-by-Sequencing Data. BMC Genom. 2015, 16, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilton, T.P.; McEwan, J.C.; Clarke, S.M.; Brauning, R.; van Stijn, T.C.; Rowe, S.J.; Dodds, K.G. Linkage Disequilibrium Estimation in Low Coverage High-Throughput Sequencing Data. Genetics 2018, 209, 389–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilton, T.P.; Schofield, M.R.; Black, M.A.; Chagné, D.; Wilcox, P.L.; Dodds, K.G. Accounting for Errors in Low Coverage High-Throughput Sequencing Data When Constructing Genetic Maps Using Biparental Outcrossed Populations. Genetics 2018, 209, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilton, T.P.; Chappell, A.J.; Clarke, S.M.; Brauning, R.; Dodds, K.G.; McEwan, J.C.; Rowe, S.J. Using Genotyping-by-sequencing to Predict Gender in Animals. Anim. Genet. 2019, 50, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; van Stijn, T.C.; Rowe, S.J.; McEwan, K.M.; Clarke, S.M. Exclusion and Genomic Relatedness Methods for Assignment of Parentage Using Genotyping-by-Sequencing Data. G3 Genes Genomes Genet. 2019, 9, 3239–3247. [Google Scholar] [CrossRef] [Green Version]
- Faville, M.J.; Ganesh, S.; Cao, M.; Jahufer, M.Z.Z.; Bilton, T.P.; Easton, H.S.; Ryan, D.L.; Trethewey, J.A.K.; Rolston, M.P.; Griffiths, A.G.; et al. Predictive Ability of Genomic Selection Models in a Multi-Population Perennial Ryegrass Training Set Using Genotyping-by-Sequencing. Theor. Appl. Genet. 2018, 131, 703–720. [Google Scholar] [CrossRef] [Green Version]
- Palaiokostas, C.; Clarke, S.M.; Jeuthe, H.; Brauning, R.; Bilton, T.P.; Dodds, K.G.; McEwan, J.C.; De Koning, D.-J. Application of Low Coverage Genotyping by Sequencing in Selectively Bred Arctic Charr ( Salvelinus alpinus). G3 Genes|Genomes|Genetics 2020. [Google Scholar] [CrossRef] [Green Version]
- Carlberg, H.; Nilsson, J.; Brännäs, E.; Alanärä, A. An Evaluation of 30years of Selective Breeding in the Arctic Charr (Salvelinus alpinus L.) and Its Implications for Feeding Management. Aquaculture 2018, 495, 428–434. [Google Scholar] [CrossRef]
- Eriksson, L.O.; Alanärä, A.; Nilsson, J.; Brännäs, E. The Arctic Charr Story: Development of Subarctic Freshwater Fish Farming in Sweden. Hydrobiologia 2010, 650, 265–274. [Google Scholar] [CrossRef]
- Kajungiro, R.A.; Palaiokostas, C.; Pinto, F.A.L.; Mmochi, A.J.; Mtolera, M.; Houston, R.D.; de Koning, D.J. Population Structure and Genetic Diversity of Nile Tilapia (Oreochromis niloticus) Strains Cultured in Tanzania. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Moses, M.; Mtolera, M.S.P.; Chauka, L.J.; Lopes, F.A.; de Koning, D.J.; Houston, R.D.; Palaiokostas, C. Characterizing the Genetic Structure of Introduced Nile Tilapia (Oreochromis niloticus) Strains in Tanzania Using Double Digest RAD Sequencing. Aquac. Int. 2019. [Google Scholar] [CrossRef] [Green Version]
- Torati, L.S.; Taggart, J.B.; Varela, E.S.; Araripe, J.; Wehner, S.; Migaud, H. Genetic Diversity and Structure in Arapaima Gigas Populations from Amazon and Araguaia-Tocantins River Basins. BMC Genet. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, S.; Kikuchi, K.; Nagashima, H.; Onodera, J.; Sugimoto, K.; Satoh, K.; Matsuzaki, K.; Yasugi, M.; Nagano, A.J.; Kumagayi, A.; et al. Assessment of Genetic Diversity in Coho Salmon (Oncorhynchus kisutch) Populations with No Family Records Using DdRAD-Seq. BMC Research Notes 2018, 11, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai, W.; Nomura, K.; Fujiwara, A.; Nakamura, Y.; Yasuike, M.; Ojima, N.; Masaoka, T.; Ozaki, A.; Kazeto, Y.; Gen, K.; et al. A DdRAD-Based Genetic Map and Its Integration with the Genome Assembly of Japanese Eel (Anguilla japonica) Provides Insights into Genome Evolution after the Teleost-Specific Genome Duplication. BMC Genom. 2014, 15, 233. [Google Scholar] [CrossRef] [Green Version]
- Oral, M.; Colléter, J.; Bekaert, M.; Taggart, J.B.; Palaiokostas, C.; McAndrew, B.J.; Vandeputte, M.; Chatain, B.; Kuhl, H.; Reinhardt, R.; et al. Gene-Centromere Mapping in Meiotic Gynogenetic European Seabass. BMC Genom. 2017, 18, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manousaki, T.; Tsakogiannis, A.; Taggart, J.B.; Palaiokostas, C.; Tsaparis, D.; Lagnel, J.; Chatziplis, D.; Magoulas, A.; Papandroulakis, N.; Mylonas, C.C.; et al. Exploring a Nonmodel Teleost Genome Through RAD Sequencing—Linkage Mapping in Common Pandora, Pagellus erythrinus and Comparative Genomic Analysis. G3 Genes|Genomes|Genetics 2016, 6, 509–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Zhou, Z.; Liu, B.; Kong, S.; Chen, B.; Bai, H.; Li, L.; Pu, F.; Xu, P. Construction of a High-Density Genetic Linkage Map and QTL Mapping for Growth-Related Traits in Takifugu Bimaculatus. Mar. Biotechnol. 2020, 22, 130–144. [Google Scholar] [CrossRef]
- Barría, A.; Christensen, K.A.; Yoshida, G.M.; Correa, K.; Jedlicki, A.; Lhorente, J.P.; Davidson, W.S.; Yáñez, J.M. Genomic Predictions and Genome-Wide Association Study of Resistance Against Piscirickettsia Salmonis in Coho Salmon (Oncorhynchus kisutch) Using ddRAD Sequencing. G3 Genes Genomes Genet. 2018, 8, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.K.; Taggart, J.B.; Bekaert, M.; Wehner, S.; Palaiokostas, C.; Setiawan, A.N.; Symonds, J.E.; Penman, D.J. Mapping the Sex Determination Locus in the Hāpuku (Polyprion oxygeneios) Using ddRAD Sequencing. BMC Genom. 2016, 17, 448. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.L.; Gu, X.H.; Li, B.J.; Zhu, Z.X.; Qin, H.; ning Meng, Z.; Lin, H.R.; Xia, J.H. Identifying a Long QTL Cluster Across ChrLG18 Associated with Salt Tolerance in Tilapia Using GWAS and QTL-Seq. Mar. Biotechnol. 2019, 21, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, D.; Kanterakis, A.; Manousaki, T.; Tsakogiannis, A.; Tsagris, M.; Tsamardinos, I.; Papaharisis, L.; Chatziplis, D.; Potamias, G.; Tsigenopoulos, C.S. Scanning of Genetic Variants and Genetic Mapping of Phenotypic Traits in Gilthead Sea Bream Through ddRAD Sequencing. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Taslima, K.; Wehner, S.; Taggart, J.B.; de Verdal, H.; Benzie, J.A.H.; Bekaert, M.; McAndrew, B.J.; Penman, D.J. Sex Determination in the GIFT Strain of Tilapia Is Controlled by a Locus in Linkage Group 23. BMC Genet. 2020, 21, 49. [Google Scholar] [CrossRef]
- Taslima, K.; Davie, A.; McAndrew, B.J.; Penman, D.J. DNA Sampling from Mucus in the Nile Tilapia, Oreochromis niloticus: Minimally Invasive Sampling for Aquaculture-Related Genetics Research. Aquac. Res. 2016, 47, 4032–4037. [Google Scholar] [CrossRef]
- Rivera-Colón, A.G.; Rochette, N.C.; Catchen, J.M. Simulation with RADinitio Improves RADseq Experimental Design and Sheds Light on Sources of Missing Data. Mol. Ecol. Resour. 2021, 21, 363–378. [Google Scholar] [CrossRef]
- Palaiokostas, C.; Bekaert, M.; Khan, M.G.; Taggart, J.B.; Gharbi, K.; McAndrew, B.J.; Penman, D.J. A Novel Sex-Determining QTL in Nile Tilapia (Oreochromis niloticus). BMC Genom. 2015, 16, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Rochette, N.C.; Rivera-Colón, A.G.; Catchen, J.M. Stacks 2: Analytical Methods for Paired-End Sequencing Improve RADseq-Based Population Genomics. Mol. Ecol. 2019, 28, 4737–4754. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant Analysis of Principal Components: A New Method for the Analysis of Genetically Structured Populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endelman, J.B. Ridge Regression and Other Kernels for Genomic Selection with R Package RrBLUP. Plant Genome 2011, 4. [Google Scholar] [CrossRef] [Green Version]
- Endelman, J.B.; Jannink, J.-L. Shrinkage Estimation of the Realized Relationship Matrix. G3 Genes|Genomes|Genetics 2012, 2, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.; Endelman, J.; Dawson, J.; Rutkoski, J.; Wu, S.; Manes, Y.; Dreisigacker, S.; Crossa, J.; Sánchez-Villeda, H.; Sorrells, M.; et al. Genomic Selection in Wheat Breeding Using Genotyping-by-Sequencing. Plant Genome 2012, 5. [Google Scholar] [CrossRef] [Green Version]
- Perdry, H.; Dandine-Roulland, C.; Bandyopadhyay, D.; Kettner, L. Pack Package ‘Gaston’: Genetic Data Handling (QC, GRM, LD, PCA) and Linear Mixed Models. Available online: https://cran.r-project.org/web/packages/gaston/gaston.pdf.
- Peñaloza, C.; Manousaki, T.; Franch, R.; Tsakogiannis, A.; Sonesson, A.; Aslam, M.L.; Allal, F.; Bargelloni, L.; Houston, R.D.; Tsigenopoulos, C.S. Development and Validation of a Combined Species SNP Array for the European Seabass (Dicentrarchus labrax) and Gilthead Seabream (Sparus aurata). bioRxiv 2020, 2020.12.17.423305. [Google Scholar] [CrossRef]
- Gorjanc, G.; Cleveland, M.A.; Houston, R.D.; Hickey, J.M. Potential of Genotyping-by-Sequencing for Genomic Selection in Livestock Populations. Genet. Sel. Evol. 2015, 47, 12. [Google Scholar] [CrossRef]
- Nilsson, J.; Brännäs, E.; Eriksson, L.-O. The Swedish Arctic Charr Breeding Programme. Hydrobiologia 2010, 650, 275–282. [Google Scholar] [CrossRef]
- Nyinondi, C.S.; Mtolera, M.S.P.; Mmochi, A.J.; Pinto, F.A.L.; Houston, R.D.; de Koning, D.J.; Palaiokostas, C. Assessing the Genetic Diversity of Farmed and Wild Rufiji Tilapia (Oreochromis urolepis urolepis) Populations Using DdRAD Sequencing. Ecol. Evol. 2020, 10, 10044–10056. [Google Scholar] [CrossRef]
- Linck, E.; Battey, C.J. Minor Allele Frequency Thresholds Strongly Affect Population Structure Inference with Genomic Data Sets. Mol. Ecol. Resour. 2019, 19, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Selechnik, D.; Richardson, M.F.; Hess, M.K.; Hess, A.S.; Dodds, K.G.; Martin, M.; Chan, T.C.; Cardilini, A.P.A.; Sherman, C.D.H.; Shine, R.; et al. Inherent Population Structure Determines the Importance of Filtering Parameters for Reduced Representation Sequencing Analyses. bioRxiv 2020, 2020.11.14.383240. [Google Scholar] [CrossRef]
- Lourenco, D.A.L.; Fragomeni, B.O.; Tsuruta, S.; Aguilar, I.; Zumbach, B.; Hawken, R.J.; Legarra, A.; Misztal, I. Accuracy of Estimated Breeding Values with Genomic Information on Males, Females, or Both: An Example on Broiler Chicken. Genet. Sel. Evol. 2015, 47, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Tobasei, R.; Ali, A.; Garcia, A.L.S.; Lourenco, D.; Leeds, T.; Salem, M. Genomic Predictions for Fillet Yield and Firmness in Rainbow Trout Using Reduced-Density SNP Panels. BMC Genom. 2021, 22, 92. [Google Scholar] [CrossRef] [PubMed]
- Kriaridou, C.; Tsairidou, S.; Houston, R.D.; Robledo, D. Genomic Prediction Using Low Density Marker Panels in Aquaculture: Performance Across Species, Traits, and Genotyping Platforms. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, Y.; Yuan, J.; Zhang, X.; Huang, H.; Li, F.; Xiang, J. Effects of Marker Density and Population Structure on the Genomic Prediction Accuracy for Growth Trait in Pacific White Shrimp Litopenaeus Vannamei. BMC Genet. 2017, 18, 45. [Google Scholar] [CrossRef]
- Christoffersen, C.; Federspiel, C.K.; Borup, A.; Christensen, P.M.; Madsen, A.N.; Heine, M.; Nielsen, C.H.; Kjaer, A.; Holst, B.; Heeren, J.; et al. The Apolipoprotein M/S1P Axis Controls Triglyceride Metabolism and Brown Fat Activity. Cell Rep. 2018, 22, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, T.; Zhao, S.; Zhang, S. Expression of Apolipoprotein M and Its Association with Adiponectin in an Obese Mouse Model. Exp. Ther. Med. 2019, 18, 1685–1692. [Google Scholar] [CrossRef]
- Liu, D.; Pan, J.-M.; Pei, X.; Li, J.-S. Interaction Between Apolipoprotein M Gene Single-Nucleotide Polymorphisms and Obesity and Its Effect on Type 2 Diabetes Mellitus Susceptibility. Sci. Rep. 2020, 10, 7859. [Google Scholar] [CrossRef]
- Sramkova, V.; Berend, S.; Siklova, M.; Caspar-Bauguil, S.; Carayol, J.; Bonnel, S.; Marques, M.; Decaunes, P.; Kolditz, C.-I.; Dahlman, I.; et al. Apolipoprotein M: A Novel Adipokine Decreasing with Obesity and Upregulated by Calorie Restriction. Am. J. Clin. Nutr. 2019, 109, 1499–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küttner, E.; Moghadam, H.K.; Skúlason, S.; Danzmann, R.G.; Ferguson, M.M. Genetic Architecture of Body Weight, Condition Factor and Age of Sexual Maturation in Icelandic Arctic Charr (Salvelinus alpinus). Mol. Genet Genom. 2011, 286, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.D.; Danzmann, R.G.; Glebe, B.; Ferguson, M.M. The Genetic Basis of Salinity Tolerance Traits in Arctic Charr (Salvelinus alpinus). BMC Genet. 2011, 12, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghadam, H.K.; Poissant, J.; Fotherby, H.; Haidle, L.; Ferguson, M.M.; Danzmann, R.G. Quantitative Trait Loci for Body Weight, Condition Factor and Age at Sexual Maturation in Arctic Charr (Salvelinus alpinus): Comparative Analysis with Rainbow Trout (Oncorhynchus mykiss) and Atlantic Salmon (Salmo salar). Mol. Genet Genom. 2007, 277, 647–661. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linkage Group | SNP Position (bp) | Unadjusted p-Value | BH Adjusted p-Value | Annotated or Predicted Genes within 20 kb on Either Side |
|---|---|---|---|---|
| NC_036876.1 | 6,904,331 | 2.317911 × 10−9 | 8.859984 × 10−5 | LOC111960292 ** |
| NC_036872.1 | 16,812,304 | 1.710431 × 10−8 | 3.268976 × 10−4 | nkx2.7, LOC111957894, LOC111957491 |
| NC_036874.1 | 10,834,663 | 1.568166 × 10−7 | 1.998052 × 10−3 | apom *, LOC111959084, LOC111958785 |
| NW_019945418.1 | 7585 | 2.161829 × 10−6 | 2.030243 × 10−2 | - |
| NC_036874.1 | 13,345,514 | 2.655718 × 10−6 | 2.030243 × 10−2 | - |
| NC_036869.1 | 2,707,853 | 4.399173 × 10−6 | 2.761334 × 10−2 | LOC111955246 *, pex1 |
| NC_036839.1 | 21,199,964 | 5.056859 × 10−6 | 2.761334 × 10−2 | klf7 ** |
| NC_036839.1 | 17,537,842 | 8.506600 × 10−6 | 4.064454 × 10−2 | LOC111972823 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pappas, F.; Palaiokostas, C. Genotyping Strategies Using ddRAD Sequencing in Farmed Arctic Charr (Salvelinus alpinus). Animals 2021, 11, 899. https://doi.org/10.3390/ani11030899
Pappas F, Palaiokostas C. Genotyping Strategies Using ddRAD Sequencing in Farmed Arctic Charr (Salvelinus alpinus). Animals. 2021; 11(3):899. https://doi.org/10.3390/ani11030899
Chicago/Turabian StylePappas, Fotis, and Christos Palaiokostas. 2021. "Genotyping Strategies Using ddRAD Sequencing in Farmed Arctic Charr (Salvelinus alpinus)" Animals 11, no. 3: 899. https://doi.org/10.3390/ani11030899
APA StylePappas, F., & Palaiokostas, C. (2021). Genotyping Strategies Using ddRAD Sequencing in Farmed Arctic Charr (Salvelinus alpinus). Animals, 11(3), 899. https://doi.org/10.3390/ani11030899