
Characterization of Microbiome on Feces, Blood and Milk in Dairy Cows with Different Milk Leucocyte Pattern



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Housing
2.2. Collection of Samples
2.3. DNA Extraction, Library Preparation, Sequencing and Taxonomic Annotation
2.4. Quantitative Real-Time PCR (qPCR)
2.5. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Günther, J.; Koczan, D.; Yang, W.; Nürnberg, G.; Repsilber, D.; Schuberth, H.-J.; Park, Z.; Maqbool, N.; Molenaar, A.; Seyfert, H.-M. Assessment of the Immune Capacity of Mammary Epithelial Cells: Comparison with Mammary Tissue after Challenge with Escherichia Coli. Vet. Res. 2009, 40, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzilli, M.; Zecconi, A. Assessment of Epithelial Cells’ Immune and Inflammatory Response to Staphylococcus Aureus When Exposed to a Macrolide. J. Dairy Res. 2010, 77, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M. The Origin of Human Milk Bacteria: Is There a Bacterial Entero-Mammary Pathway during Late Pregnancy and Lactation? Adv. Nutr. 2014, 5, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, S.L.; Corl, C.M.; Sordillo, L.M. Immunopathology of Mastitis: Insights into Disease Recognition and Resolution. J. Mammary Gland Biol. Neoplasia 2011, 4, 291–304. [Google Scholar] [CrossRef]
- Damm, M.; Holm, C.; Blaabjerg, M.; Bro, M.N.; Schwarz, D. Differential Somatic Cell Count—A Novel Method for Routine Mastitis Screening in the Frame of Dairy Herd Improvement Testing Programs. J. Dairy Sci. 2017, 100, 4926–4940. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.; Lipkens, Z.; Piepers, S.; De Vliegher, S. Investigation of Differential Somatic Cell Count as a Potential New Supplementary Indicator to Somatic Cell Count for Identification of Intramammary Infection in Dairy Cows at the End of the Lactation Period. Prev. Vet. Med. 2019, 172, 104803. [Google Scholar] [CrossRef]
- Zecconi, A.; Zanini, L.; Cipolla, M.; Stefanon, B. Factors Affecting the Patterns of Total Amount and Proportions of Leukocytes in Bovine Milk. Animals 2020, 10, 992. [Google Scholar] [CrossRef]
- Bobbo, T.; Penasa, M.; Cassandro, M. Combining Total and Differential Somatic Cell Count to Better Assess the Association of Udder Health Status with Milk Yield, Composition and Coagulation Properties in Cattle. Ital. J. Anim. Sci. 2020, 19, 697–703. [Google Scholar] [CrossRef]
- Sordillo, L.M.; Raphael, W. Significance of Metabolic Stress, Lipid Mobilization, and Inflammation on Transition Cow Disorders. Vet. Clin. Food Anim. Pract. 2013, 29, 267–278. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic Cells and the Control of Immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Uhr, T. Induction of Protective IgA by Intestinal Dendritic Cells Carrying Commensal Bacteria. Science 2004, 303, 1662–1665. [Google Scholar] [CrossRef] [Green Version]
- Gabanyi, I.; Muller, P.A.; Feighery, L.; Oliveira, T.Y.; Costa-Pinto, F.A.; Mucida, D. Neuro-Immune Interactions Drive Tissue Programming in Intestinal Macrophages. Cell 2016, 164, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Rimoldi, M.; Chieppa, M.; Salucci, V.; Avogadri, F.; Sonzogni, A.; Sampietro, G.M.; Nespoli, A.; Viale, G.; Allavena, P.; Rescigno, M. Intestinal Immune Homeostasis Is Regulated by the Crosstalk between Epithelial Cells and Dendritic Cells. Nat. Immunol. 2005, 6, 507–514. [Google Scholar] [CrossRef]
- Capaldo, C.T.; Powell, D.N.; Kalman, D. Layered Defense: How Mucus and Tight Junctions Seal the Intestinal Barrier. J. Mol. Med. 2017, 95, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Kvidera, S.K.; Dickson, M.J.; Abuajamieh, M.; Snider, D.B.; Fernandez, M.V.S.; Johnson, J.S.; Keating, A.F.; Gorden, P.J.; Green, H.B.; Schoenberg, K.M.; et al. Intentionally Induced Intestinal Barrier Dysfunction Causes Inflammation, Affects Metabolism, and Reduces Productivity in Lactating Holstein Cows. J. Dairy Sci. 2017, 100, 4113–4127. [Google Scholar] [CrossRef] [Green Version]
- Nakagaki, B.N.; Vieira, A.T.; Rezende, R.M.; David, B.A.; Menezes, G.B. Tissue Macrophages as Mediators of a Healthy Relationship with Gut Commensal Microbiota. Cell. Immunol. 2018, 330, 16–26. [Google Scholar] [CrossRef]
- Young, W.; Hine, B.C.; Wallace, O.A.M.; Callaghan, M.; Bibiloni, R. Transfer of Intestinal Bacterial Components to Mammary Secretions in the Cow. PeerJ 2015, 3, e888. [Google Scholar] [CrossRef]
- Mandal, R.K.; Jiang, T.; Al-Rubaye, A.A.; Rhoads, D.D.; Wideman, R.F.; Zhao, J.; Pevzner, I.; Kwon, Y.M. An Investigation into Blood Microbiota and Its Potential Association with Bacterial Chondronecrosis with Osteomyelitis (BCO) in Broilers. Sci. Rep. 2016, 6, 25882. [Google Scholar] [CrossRef] [Green Version]
- Vientós-Plotts, A.I.; Ericsson, A.C.; Rindt, H.; Grobman, M.E.; Graham, A.; Bishop, K.; Cohn, L.A.; Reinero, C.R. Dynamic Changes of the Respiratory Microbiota and Its Relationship to Fecal and Blood Microbiota in Healthy Young Cats. PLoS ONE 2017, 12, e0173818. [Google Scholar] [CrossRef]
- Li, Q.; Wang, C.; Tang, C.; Zhao, X.; He, Q.; Li, J. Identification and Characterization of Blood and Neutrophil-Associated Microbiomes in Patients with Severe Acute Pancreatitis Using Next-Generation Sequencing. Front. Cell. Infect. Microbiol. 2018, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Zhou, H.; Jing, Y.; Dong, C. Association between Blood Microbiome and Type 2 Diabetes Mellitus: A Nested Case-Control Study. J. Clin. Lab. Anal. 2019, 33, e22842. [Google Scholar] [CrossRef] [Green Version]
- Whittle, E.; Leonard, M.O.; Harrison, R.; Gant, T.W.; Tonge, D.P. Multi-Method Characterization of the Human Circulating Microbiome. Front. Microbiol. 2019, 9, 3266. [Google Scholar] [CrossRef] [Green Version]
- Païssé, S.; Valle, C.; Servant, F.; Courtney, M.; Burcelin, R.; Amar, J.; Lelouvier, B. Comprehensive Description of Blood Microbiome from Healthy Donors Assessed by 16S Targeted Metagenomic Sequencing. Transfusion 2016, 56, 1138–1147. [Google Scholar] [CrossRef]
- Cogen, A.L.; Nizet, V.; Gallo, R.L. Skin Microbiota: A Source of Disease or Defence? Br. J. Dermatol. 2008, 158, 442–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potgieter, M.; Bester, J.; Kell, D.B.; Pretorius, E. The Dormant Blood Microbiome in Chronic, Inflammatory Diseases. FEMS Microbiol. Rev. 2015, 39, 567–591. [Google Scholar] [CrossRef] [Green Version]
- Mercier, R.; Kawai, Y.; Errington, J. General Principles for the Formation and Proliferation of a Wall-Free (L-Form) State in Bacteria. eLife 2014, 3, e04629. [Google Scholar] [CrossRef]
- Zecconi, A.; Vairani, D.; Cipolla, M.; Rizzi, N.; Zanini, L. Assessment of Subclinical Mastitis Diagnostic Accuracy by Differential Cell Count in Individual Cow Milk. Ital. J. Anim. Sci. 2019, 18, 460–465. [Google Scholar] [CrossRef]
- Zecconi, A.; dell’Orco, F.; Rizzi, N.; Vairani, D.; Cipolla, M.; Pozzi, P.; Zanini, L. Cross-Sectional Study on the Prevalence of Contagious Pathogens in Bulk Tank Milk and Their Effects on Somatic Cell Counts and Milk Yield. Ital. J. Anim. Sci. 2020, 19, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Schukken, Y.H.; Wilson, D.J.; Welcome, F.; Garrison-Tikofsky, L.; Gonzalez, R.N. Monitoring Udder Health and Milk Quality Using Somatic Cell Counts. Vet. Res. 2003, 34, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zecconi, A.; Sesana, G.; Vairani, D.; Cipolla, M.; Rizzi, N.; Zanini, L. Somatic Cell Count as a Decision Tool for Selective Dry Cow Therapy in Italy. Ital. J. Anim. Sci. 2019, 18, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Lima, S.F.; Bicalho, M.L.D.S.; Bicalho, R.C. Evaluation of Milk Sample Fractions for Characterization of Milk Microbiota from Healthy and Clinical Mastitis Cows. PLoS ONE 2018, 13, e0193671. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- AlShawaqfeh, M.K.; Wajid, B.; Minamoto, Y.; Markel, M.; Lidbury, J.A.; Steiner, J.M.; Serpedin, E.; Suchodolski, J.S. A Dysbiosis Index to Assess Microbial Changes in Fecal Samples of Dogs with Chronic Inflammatory Enteropathy. FEMS Microbiol. Ecol. 2017, 93, fix136. [Google Scholar] [CrossRef] [Green Version]
- Scarsella, E.; Sandri, M.; Monego, S.D.; Licastro, D.; Stefanon, B. Blood Microbiome: A New Marker of Gut Microbial Population in Dogs? Vet. Sci. 2020, 7, 198. [Google Scholar] [CrossRef]
- Addinsoft. XLSTAT Statistical and Data Analysis Solution; Addinsoft: Boston, MA, USA, 2020. [Google Scholar]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Tolle, A. The Microflora of the Udder. In: Factors Influencing the Bacteriological Quality of Raw Milk. Int. Dairy Federation Bull. 1980, 120, 4. [Google Scholar]
- Smith, K.L.; Todhunter, D.A.; Schoenberger, P.S. Environmental Mastitis: Cause, Prevalence, Prevention1, 2. J. Dairy Sci. 1985, 68, 1531–1553. [Google Scholar] [CrossRef]
- Paulrud, C.O. Basic Concepts of the Bovine Teat Canal. Vet. Res. Commun. 2005, 29, 215–245. [Google Scholar] [CrossRef]
- Kim, M.; Wells, J.E. A Meta-Analysis of Bacterial Diversity in the Feces of Cattle. Curr. Microbiol. 2016, 72, 145–151. [Google Scholar] [CrossRef]
- Cendron, F.; Niero, G.; Carlino, G.; Penasa, M.; Cassandro, M. Characterizing the Fecal Bacteria and Archaea Community of Heifers and Lactating Cows through 16S RRNA Next-Generation Sequencing. J. Appl. Genet. 2020, 61, 593–605. [Google Scholar] [CrossRef]
- Derakhshani, H.; Fehr, K.B.; Sepehri, S.; Francoz, D.; De Buck, J.; Barkema, H.W.; Plaizier, J.C.; Khafipour, E. Invited Review: Microbiota of the Bovine Udder: Contributing Factors and Potential Implications for Udder Health and Mastitis Susceptibility. J. Dairy Sci. 2018, 101, 10605–10625. [Google Scholar] [CrossRef] [Green Version]
- Verdier-Metz, I.; Gagne, G.; Bornes, S.; Monsallier, F.; Veisseire, P.; Delbès-Paus, C.; Montel, M.-C. Cow Teat Skin, a Potential Source of Diverse Microbial Populations for Cheese Production. Appl. Environ. Microbiol. 2012, 78, 326–333. [Google Scholar] [CrossRef] [Green Version]
- Taponen, S.; McGuinness, D.; Hiitiö, H.; Simojoki, H.; Zadoks, R.; Pyörälä, S. Bovine Milk Microbiome: A More Complex Issue than Expected. Vet. Res. 2019, 50, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the Bovine Rumen Bacterial Community from Birth to Adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef] [Green Version]
- Plaizier, J.C.; Li, S.; Danscher, A.M.; Derakshani, H.; Andersen, P.H.; Khafipour, E. Changes in Microbiota in Rumen Digesta and Feces Due to a Grain-Based Subacute Ruminal Acidosis (SARA) Challenge. Microb. Ecol. 2017, 74, 485–495. [Google Scholar] [CrossRef]
- Khafipour, E.; Li, S.; Tun, H.; Derakhshani, H.; Moossavi, S.; Plaizier, K. Effects of Grain Feeding on Microbiota in the Digestive Tract of Cattle. Anim. Front. 2016, 6, 13–19. [Google Scholar] [CrossRef]
- Kell, D.B.; Kaprelyants, A.S.; Weichart, D.H.; Harwood, C.R.; Barer, M.R. Viability and Activity in Readily Culturable Bacteria: A Review and Discussion of the Practical Issues. Antonie Van Leeuwenhoek 1998, 73, 169–187. [Google Scholar] [CrossRef]
- Kell, D.B.; Pretorius, E. On the Translocation of Bacteria and Their Lipopolysaccharides between Blood and Peripheral Locations in Chronic, Inflammatory Diseases: The Central Roles of LPS and LPS-Induced Cell Death. Integr. Biol. 2015, 7, 1339–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, L.K. Mycoplasma Mastitis: Causes, Transmission, and Control. Vet. Clin. Food Anim. Pract. 2012, 28, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.L. Resistance in Gram-Negative Bacteria: Enterobacteriaceae. Am. J. Infect. Control 2006, 34, S20–S28. [Google Scholar] [CrossRef]
- Bertazzoni Minelli, E.; Benini, A.; Marzotto, M.; Sbarbati, A.; Ruzzenente, O.; Ferrario, R.; Hendriks, H.; Dellaglio, F. Assessment of Novel Probiotic Lactobacillus Casei Strains for the Production of Functional Dairy Foods. Int. Dairy J. 2004, 14, 723–736. [Google Scholar] [CrossRef]
- Butler, M.I.; Bastiaanssen, T.F.S.; Long-Smith, C.; Berding, K.; Morkl, S.; Cusack, A.-M.; Strain, C.; Busca, K.; Porteous-Allen, P.; Claesson, M.J.; et al. Recipe for a Healthy Gut: Intake of Unpasteurised Milk Is Associated with Increased Lactobacillus Abundance in the Human Gut Microbiome. Nutrients 2020, 12, 1468. [Google Scholar] [CrossRef]
- Soggiu, A.; Bendixen, E.; Brasca, M.; Morandi, S.; Piras, C.; Bonizzi, L.; Roncada, P. Milk and Cheese Microbiome for Safety and Quality of Dairy Products. In Farm Animal Proteomics 2013, Proceedings of the 4th Management Committee Meeting and 3rd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002 Košice, Slovakia 25–26 April 2013; de Almeida, A., Eckersall, D., Bencurova, E., Dolinska, S., Mlynarcik, P., Vincova, M., Bhide, M., Eds.; Academic Publishers: Wageningen, The Netherlands, 2013; pp. 262–265. [Google Scholar]
- Parente, E.; Ricciardi, A.; Zotta, T. The Microbiota of Dairy Milk: A Review. Int. Dairy J. 2020, 107, 104714. [Google Scholar] [CrossRef]
- Ridyard, A.E.; Brown, J.K.; Rhind, S.M.; Else, R.W.; Simpson, J.W.; Miller, H.R.P. Apical Junction Complex Protein Expression in the Canine Colon: Differential Expression of Claudin-2 in the Colonic Mucosa in Dogs with Idiopathic Colitis. J. Histochem. Cytochem. 2007, 55, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Suchodolski, J.S. Diagnosis and Interpretation of Intestinal Dysbiosis in Dogs and Cats. Vet. J. 2016, 215, 30–37. [Google Scholar] [CrossRef]
- Stewart, A.S.; Pratt-Phillips, S.; Gonzalez, L.M. Alterations in Intestinal Permeability: The Role of the “Leaky Gut” in Health and Disease. J. Equine Vet. Sci. 2017, 52, 10–22. [Google Scholar] [CrossRef]
- Tizard, I.R.; Jones, S.W. The Microbiota Regulates Immunity and Immunologic Diseases in Dogs and Cats. Vet. Clin. Small Anim. Pract. 2018, 48, 307–322. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
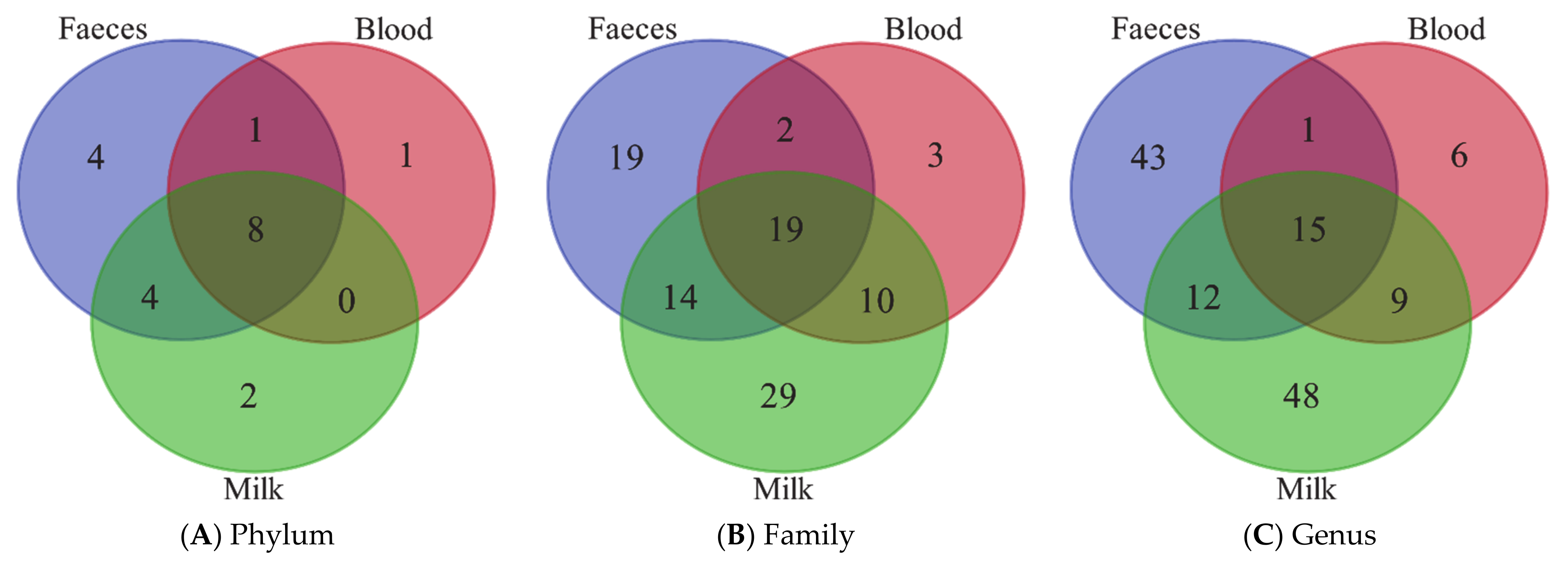{kind=link}
| Group | Number of Cows | Age (Parturitions) | DIM | ||
|---|---|---|---|---|---|
| Primiparous | Pluriparous | <70 | >70 | ||
| G | 34 | 11 | 23 | 10 | 24 |
| Y | 13 | 6 | 7 | 2 | 11 |
| R | 13 | 2 | 11 | 3 | 10 |
| Taxa | G (AA) | Y (AA) | R (AA) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | p-Value | ||||
| Family | ||||||||||
| Clostridiaceae | 21.8 | a | 10.1 | 18.6 | a | 8.0 | 31.6 | b | 11.4 | 0.002 |
| Peptostreptococcaceae | 28.7 | 15.3 | 24.8 | 13.0 | 35.9 | 14.8 | 0.077 | |||
| Ruminococcaceae | 214.8 | 56.5 | 213.9 | 59.6 | 194.5 | 60.5 | 0.077 | |||
| Turicibacteraceae | 16.2 | ab | 9.0 | 13.1 | a | 8.0 | 22.4 | b | 10.5 | 0.027 |
| Genera | ||||||||||
| Clostridium | 42.7 | a | 20.5 | 36.4 | a | 17.1 | 56.3 | b | 19.8 | 0.012 |
| Dorea | 7.2 | ab | 2.9 | 8.0 | b | 4.0 | 5.8 | a | 2.2 | 0.040 |
| Epulopiscium | 1.7 | 1.3 | 1.5 | 1.3 | 2.5 | 1.4 | 0.060 | |||
| SMB53 | 0.3 | ab | 0.2 | 0.2 | a | 0.2 | 0.4 | b | 0.3 | 0.041 |
| Turicibacter | 16.2 | ab | 9.0 | 13.1 | a | 8.0 | 22.4 | b | 10.5 | 0.027 |
| Taxa | G (AA) | Y (AA) | R (AA) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | p-Value | ||||
| Phylum | ||||||||||
| Tenericutes | 0.6 | ab | 1.2 | 1.1 | b | 1.2 | 1.4 | a | 3.8 | 0.020 |
| Genera | ||||||||||
| Corynebacterium | 0.1 | 0.2 | 0.5 | 0.8 | 0.2 | 0.2 | 0.097 | |||
| Mycoplasma | 0.6 | b | 1.2 | 1.1 | ab | 1.2 | 1.4 | a | 3.8 | 0.02 |
| Taxa | G (AA) | Y (AA) | R (AA) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | p-Value | ||||
| Phylum | ||||||||||
| Actinobacteria | 28.6 | 18.6 | 29.5 | 15.5 | 22.1 | 21.2 | 0.099 | |||
| OD1 | 0.2 | a | 0.2 | 0.1 | a | 0.2 | 0.4 | b | 0.4 | 0.002 |
| Proteobacteria | 106.5 | ab | 63.4 | 170.6 | b | 98.8 | 90.9 | a | 54.8 | 0.033 |
| Family | ||||||||||
| Cellulomonadaceae | 0.3 | 0.5 | 1.0 | 1.7 | 0.1 | 0.2 | 0.074 | |||
| Enterobacteriaceae | 1.2 | ab | 2.5 | 5.2 | b | 13.6 | 0.3 | a | 0.5 | 0.043 |
| Microbacteriaceae | 10.7 | 8.6 | 13.2 | 8.7 | 7.5 | 8.4 | 0.091 | |||
| Moraxellaceae | 54.0 | 52.5 | 115.7 | 95.4 | 57.9 | 52.6 | 0.054 | |||
| Nocardiaceae | 1.2 | b | 1.3 | 1.7 | b | 1.9 | 0.7 | a | 1.3 | 0.017 |
| Phyllobacteriaceae | 0.5 | 1.4 | 0.6 | 0.9 | 0.0 | 0.1 | 0.092 | |||
| Propionibacteriaceae | 2.8 | 3.3 | 4.4 | 7.0 | 1.3 | 1.6 | 0.076 | |||
| Pseudomonadaceae | 5.2 | 7.4 | 7.0 | 7.0 | 2.3 | 3.9 | 0.063 | |||
| Succinivibrionaceae | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.1 | 0.085 | |||
| Williamsiaceae | 0.1 | 0.2 | 0.1 | 0.1 | 0.0 | 0.1 | 0.077 | |||
| Genera | ||||||||||
| 5-7N15 | 0.2 | 0.2 | 0.1 | 0.1 | 0.2 | 0.2 | 0.065 | |||
| Acinetobacter | 51.3 | 51.2 | 110.1 | 92.2 | 54.8 | 50.9 | 0.055 | |||
| Aminobacter | 0.5 | 1.4 | 0.6 | 0.9 | 0.0 | 0.1 | 0.092 | |||
| CF231 | 0.1 | 0.2 | 0.0 | 0.1 | 0.1 | 0.1 | 0.087 | |||
| Microbacterium | 4.8 | 4.0 | 5.3 | 3.9 | 2.4 | 2.6 | 0.052 | |||
| Olsenella | 0.0 | ab | 0.0 | 0.0 | b | 0.1 | 0.0 | a | 0.0 | 0.044 |
| Propionibacterium | 2.8 | 3.3 | 4.4 | 7.0 | 1.2 | 1.6 | 0.056 | |||
| Pseudomonas | 5.1 | 7.4 | 6.9 | 6.8 | 2.3 | 3.9 | 0.055 | |||
| Rhodococcus | 1.2 | ab | 1.3 | 1.7 | b | 1.9 | 0.7 | a | 1.3 | 0.017 |
| Ruminobacter | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.1 | 0.085 | |||
| Staphylococcus | 0.2 | 0.4 | 0.5 | 0.6 | 0.8 | 2.1 | 0.098 | |||
| Williamsia | 0.1 | 0.2 | 0.1 | 0.1 | 0.0 | 0.1 | 0.077 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarsella, E.; Zecconi, A.; Cintio, M.; Stefanon, B. Characterization of Microbiome on Feces, Blood and Milk in Dairy Cows with Different Milk Leucocyte Pattern. Animals 2021, 11, 1463. https://doi.org/10.3390/ani11051463
Scarsella E, Zecconi A, Cintio M, Stefanon B. Characterization of Microbiome on Feces, Blood and Milk in Dairy Cows with Different Milk Leucocyte Pattern. Animals. 2021; 11(5):1463. https://doi.org/10.3390/ani11051463
Chicago/Turabian StyleScarsella, Elisa, Alfonso Zecconi, Michela Cintio, and Bruno Stefanon. 2021. "Characterization of Microbiome on Feces, Blood and Milk in Dairy Cows with Different Milk Leucocyte Pattern" Animals 11, no. 5: 1463. https://doi.org/10.3390/ani11051463
APA StyleScarsella, E., Zecconi, A., Cintio, M., & Stefanon, B. (2021). Characterization of Microbiome on Feces, Blood and Milk in Dairy Cows with Different Milk Leucocyte Pattern. Animals, 11(5), 1463. https://doi.org/10.3390/ani11051463