
Identification and Characterization of lncRNA and mRNA in Testes of Landrace and Hezuo Boars

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Testis Sample Collection
2.3. Hematoxylin and Eosin Staining
2.4. RNA Extraction and Qualification
2.5. Library Preparation and Sequencing
2.6. Transcriptome Assemble
2.7. Gene Expression Quantification
2.8. Identification of Potential lncRNA Candidates
2.9. Target Gene Prediction and Functional Enrichment Analysis
2.10. LncRNA–mRNA Network Construction
2.11. Validation of Differentially Expressed Genes by qRT-PCR
3. Results
3.1. Morphology of Testicular Tissues
3.2. Quality Analysis of RNA-Seq Data
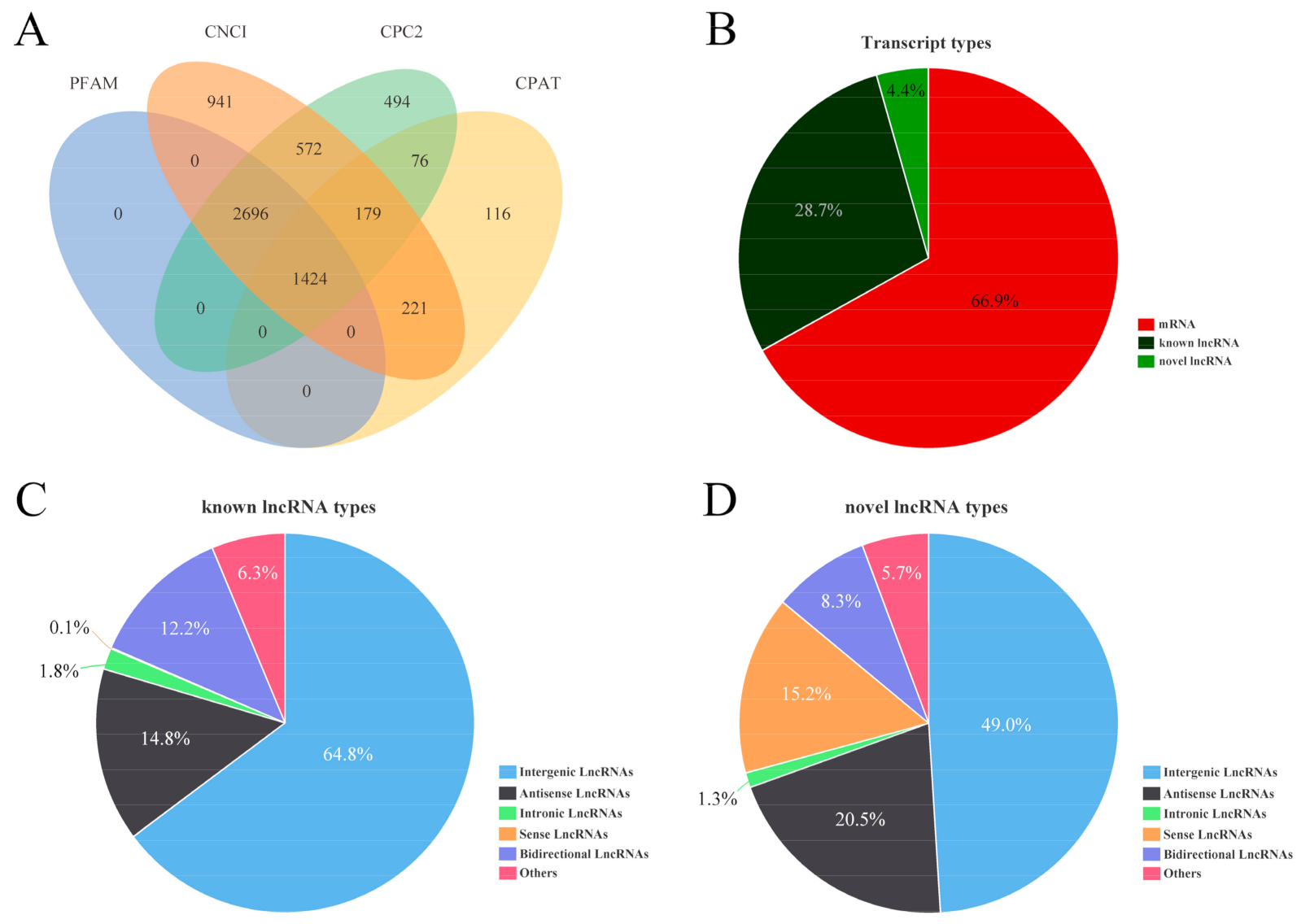
3.3. Identification and Characteristics of lncRNA and mRNA
3.4. Differential Expression Analyses of lncRNA and mRNA
3.5. GO Terms and KEGG Pathways Analysis of Differentially Expressed mRNA
3.6. GO Terms and KEGG Pathways Enrichment Analyses of Target Genes of Differentially Expressed lncRNA
3.7. GO Terms and KEGG Pathways Enrichment Analysis of Genes Related to Precocious Puberty
3.8. LncRNA–mRNA Interaction Network Analysis
3.9. Validation of Expression Levels of lncRNA and mRNA Detected in RNA-Seq
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cesario, S.K.; Hughes, L.A. Precocious Puberty: A Comprehensive Review of Literature. J. Obstet. Gynecol. Neonatal Nurs. 2007, 36, 263–274. [Google Scholar] [CrossRef] [PubMed]
- De Camargo, G.M.F.; Costa, R.B.; de Albuquerque, L.G.; Regitano, L.C.D.; Baldi, F.; Tonhati, H. Association between JY-1 gene polymorphisms and reproductive traits in beef cattle. Gene 2014, 533, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Zhao, Y.Z.; Chu, M.X.; Zhang, Y.J.; Fang, L.; Di, R.; Cao, G.L.; Li, N. Association Between Sexual Precocity and Alleles ofKISS-1andGPR54Genes in Goats. Anim. Biotechnol. 2009, 20, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Guo, X.; Wang, Y.; Wang, Y.; Cao, G.; Jiang, Y. Genome-Wide Analysis on the Landscape of Transcriptomes and Their Relationship With DNA Methylomes in the Hypothalamus Reveals Genes Related to Sexual Precocity in Jining Gray Goats. Front. Endocrinol. 2018, 9, 501. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.; Zhang, Y.; Zhang, N.; Zang, L.; Wang, M.; Cui, X.; Jiang, Y. Identification of differentially expressed genes in ovaries of chicken attaining sexual maturity at different ages. Mol. Biol. Rep. 2011, 39, 3037–3045. [Google Scholar] [CrossRef]
- Ding, H.; Luo, Y.; Liu, M.; Huang, J.; Xu, D. Histological and transcriptome analyses of testes from Duroc and Meishan boars. Sci. Rep. 2016, 6, 20758. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Struhl, K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat. Struct. Mol. Biol. 2007, 14, 103–105. [Google Scholar] [CrossRef]
- Zhao, W.; Mu, Y.; Ma, L.; Wang, C.; Tang, Z.; Yang, S.; Zhou, R.; Meng-Hua, L.; Li, M.-H.; Li, K. Systematic identification and characterization of long intergenic non-coding RNAs in fetal porcine skeletal muscle development. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Shibayama, Y.; Fanucchi, S.; Magagula, L.; Mhlanga, M.M. lncRNA and gene looping: What’s the connection? Transcription 2014, 5, e28658. [Google Scholar] [CrossRef] [Green Version]
- Bischoff, S.R.; Tsai, S.Q.; Hardison, N.E.; Motsinger-Reif, A.A.; Freking, B.A.; Nonneman, D.J.; Rohrer, G.A.; Piedrahita, J.A. Differences in X-Chromosome Transcriptional Activity and Cholesterol Metabolism between Placentae from Swine Breeds from Asian and Western Origins. PLoS ONE 2013, 8, e55345. [Google Scholar] [CrossRef]
- Roberts, T.C.; Morris, K.; Weinberg, M.S. Perspectives on the mechanism of transcriptional regulation by long non-coding RNAs. Epigenetics 2013, 9, 13–20. [Google Scholar] [CrossRef]
- Sleutels, F.; Zwart, R.; Barlow, D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nat. Cell Biol. 2002, 415, 810–813. [Google Scholar] [CrossRef]
- Cui, W.; Qian, Y.; Zhou, X.; Lin, Y.; Jiang, J.; Chen, J.; Zhao, Z.; Shen, B. Discovery and characterization of long intergenic non-coding RNAs (lincRNA) module biomarkers in prostate cancer: An integrative analysis of RNA-Seq data. BMC Genom. 2015, 16, S3. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Jameson, J.L. Minireview: Transcriptional Regulation of Gonadal Development and Differentiation. Endocrinology 2005, 146, 1035–1042. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Li, W.; Tian, H.; Hu, T.; Wang, L.; Lin, Y.; Li, Y.; Huang, H.; Sun, F. Sequential expression of long noncoding RNA as mRNA gene expression in specific stages of mouse spermatogenesis. Sci. Rep. 2014, 4, 5966. [Google Scholar] [CrossRef] [Green Version]
- Chalmel, F.; Lardenois, A.; Evrard, B.; Rolland, A.D.; Sallou, O.; Dumargne, M.-C.; Coiffec, I.; Collin, O.; Primig, M.; Jegou, B. High-Resolution Profiling of Novel Transcribed Regions During Rat Spermatogenesis1. Biol. Reprod. 2014, 91, 5. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, P.; Song, D.; Xiong, S.; Zhang, H.; Fu, J.; Gao, F.; Chen, H.; Zeng, X. Expression profiles and characteristics of human lncRNA in normal and asthenozoospermia sperm. Biol. Reprod. 2019, 100, 982–993. [Google Scholar] [CrossRef]
- Ran, M.; Chen, B.; Li, Z.; Wu, M.; Liu, X.; He, C.; Zhang, S.; Li, Z. Systematic Identification of Long Noncoding RNAs in Immature and Mature Porcine Testes1. Biol. Reprod. 2016, 94, 77. [Google Scholar] [CrossRef]
- Gao, Y.; Li, S.; Lai, Z.; Zhou, Z.; Wu, F.; Huang, Y.; Lan, X.; Lei, C.; Chen, H.; Dang, R. Analysis of Long Non-Coding RNA and mRNA Expression Profiling in Immature and Mature Bovine (Bos taurus) Testes. Front. Genet. 2019, 10, 646. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, F.; Li, F.; Ren, C.; Pang, J.; Wan, Y.; Wang, Z.; Feng, X.; Zhang, Y. Comprehensive analysis of long noncoding RNA and mRNA expression patterns in sheep testicular maturation†. Biol. Reprod. 2018, 99, 650–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Gun, S. Research on Hezuo pig germplasm resources. Swine Ind. Sci. 2013, 30, 124–127. (In Chinese) [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.E.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Mistry, J.; Bateman, A.; Finn, R.D. Predicting active site residue annotations in the Pfam database. BMC Bioinform. 2007, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Guil, S.; Esteller, M. Cis-acting noncoding RNAs: Friends and foes. Nat. Struct. Mol. Biol. 2012, 19, 1068–1075. [Google Scholar] [CrossRef]
- Liao, Q.; Liu, C.; Yuan, X.; Kang, S.; Miao, R.; Xiao, H.; Zhao, G.; Luo, H.; Bu, D.; Zhao, H.; et al. Large-scale prediction of long non-coding RNA functions in a coding–non-coding gene co-expression network. Nucleic Acids Res. 2011, 39, 3864–3878. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhou, P.-H.; Hu, W.; Zhang, X.-B.; Wang, W.; Zhang, L.-J. Protective Effect of Adrenomedullin on Rat Leydig Cells from Lipopolysaccharide-Induced Inflammation and Apoptosis via the PI3K/Akt Signaling Pathway ADM on Rat Leydig Cells from Inflammation and Apoptosis. Mediat. Inflamm. 2016, 2016, 1–16. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, W.; Luo, H.; Wang, X.; Chen, Z.; Zhang, J.J.; Wang, Y.; Li, X. Thyroid hormone inhibits the proliferation of piglet Sertoli cell via PI3K signaling pathway. Theriogenology 2015, 83, 86–94. [Google Scholar] [CrossRef]
- Amoyel, M.; Anderson, J.; Suisse, A.; Glasner, J.; Bach, E.A. Socs36E Controls Niche Competition by Repressing MAPK Signaling in the Drosophila Testis. PLoS Genet. 2016, 12, e1005815. [Google Scholar] [CrossRef] [Green Version]
- Qi, S.; Fu, W.; Wang, C.; Liu, C.; Quan, C.; Kourouma, A.; Yan, M.; Yu, T.; Duan, P.; Yang, K. BPA-induced apoptosis of rat Sertoli cells through Fas/FasL and JNKs/p38 MAPK pathways. Reprod. Toxicol. 2014, 50, 108–116. [Google Scholar] [CrossRef]
- Lai, M.-S.; Cheng, Y.-S.; Chen, P.-R.; Tsai, S.-J.; Huang, B.-M. Fibroblast Growth Factor 9 Activates Akt and MAPK Pathways to Stimulate Steroidogenesis in Mouse Leydig Cells. PLoS ONE 2014, 9, e90243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Luo, B.; Li, J.; Dai, J. Perfluorooctanoic acid disrupts the blood-testis barrier and activates the TNFα/p38 MAPK signal-ing pathway in vivo and in vitro. Arch. Toxicol. 2016, 90, 971–983. [Google Scholar] [CrossRef]
- Niu, Z.; Zheng, L.; Wu, S.; Mu, H.; Ma, F.; Song, W.; Zhu, H.; Wu, J.; He, X.; Hua, J. Ras/ERK1/2 pathway regulates the self-renewal of dairy goat spermatogonia stem cells. Reproduction 2015, 149, 445–452. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Cheng, C.Y. TGF-β3 regulates anchoring junction dynamics in the seminiferous epithelium of the rat testis via the Ras/ERK signaling pathway: An in vivo study. Dev. Biol. 2005, 280, 321–343. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Xu, Y.; Zuo, W.-L.; Yang, H.-X.; Liao, Q.-P.; Xin, Z.-C.; Guo, Y.-L. Effect of Ras signal regulating Cox7a2 expression and its sub-cellular location in testicular interstitial cell. J. Peking Univ. Health Sci. 2012, 44, 507–510. [Google Scholar]
- Duan, P.; Hu, C.; Quan, C.; Yu, T.; Huang, W.; Chen, W.; Tang, S.; Shi, Y.; Martin, F.L.; Yang, K. 4-Nonylphenol induces autophagy and attenuates mTOR-p70S6K/4EBP1 signaling by modulating AMPK activation in Sertoli cells. Toxicol. Lett. 2017, 267, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.-S.; Li, T.-P.; Ton, H.; Mao, X.-D.; Chen, Y.-J. Advances of Long Noncoding RNAs-mediated Regulation in Reproduction. Chin. Med. J. 2018, 131, 226–234. [Google Scholar] [CrossRef]
- Paixão, G.; Esteves, A.; Carolino, N.; Pires, M.D.A.; Payan-Carreira, R. Evaluation of gonadal macroscopic and microscopic morphometry reveals precocious puberty in Bísaro pig. Reprod. Domest. Anim. 2020, 55, 1706–1713. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, C.; Yang, S.; Tian, R.; Wang, J.; Yuan, Q.; Dong, H.; He, Z.; Wang, S.; Li, Z. Dynamics of the Transcriptome during Human Spermatogenesis: Predicting the Potential Key Genes Regulating Male Gametes Generation. Sci. Rep. 2016, 6, srep19069. [Google Scholar] [CrossRef] [Green Version]
- Mulvey, B.B.; Olcese, U.; Cabrera, J.R.; Horabin, J.I. An interactive network of long non-coding RNAs facilitates the Drosophila sex determination decision. Biochim. Biophys. Acta Bioenerg. 2014, 1839, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, R.; Kuroda, K.; Ikemoto, Y.; Ochiai, A.; Matsumoto, A.; Kumakiri, J.; Kitade, M.; Itakura, A.; Muter, J.; Brosens, J.; et al. Reprogramming of the retinoic acid pathway in decidualizing human endometrial stromal cells. PLoS ONE 2017, 12, e0173035. [Google Scholar] [CrossRef] [PubMed]
- Paquin, J.; Danalache, B.A.; Jankowski, M.; McCann, S.M.; Gutkowska, J. Oxytocin induces differentiation of P19 embryonic stem cells to cardiomyocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 9550–9555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pares, X.; Farrés, J.; Kedishvili, N.; Duester, G. Medium- and short-chain dehydrogenase/reductase gene and protein families. Cell. Mol. Life Sci. 2008, 65, 3936–3949. [Google Scholar] [CrossRef] [Green Version]
- Loriè, E.P.; Li, H.; Vahlquist, A.; Törmä, H. The involvement of cytochrome p450 (CYP) 26 in the retinoic acid metabolism of human epidermal keratinocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2009, 1791, 220–228. [Google Scholar] [CrossRef]
- Arun, G.; Akhade, V.S.; Donakonda, S.; Rao, M.R.S. mrhl RNA, a Long Noncoding RNA, Negatively Regulates Wnt Signaling through Its Protein Partner Ddx5/p68 in Mouse Spermatogonial Cells. Mol. Cell. Biol. 2012, 32, 3140–3152. [Google Scholar] [CrossRef] [Green Version]
- Anguera, M.C.; Ma, W.; Clift, D.; Namekawa, S.; Iii, R.J.K.; Lee, J.T. Tsx Produces a Long Noncoding RNA and Has General Functions in the Germline, Stem Cells, and Brain. PLoS Genet. 2011, 7, e1002248. [Google Scholar] [CrossRef] [Green Version]
- Agbor, V.A.; Tao, S.; Lei, N.; Heckert, L.L. A Wt1-Dmrt1 Transgene Restores DMRT1 to Sertoli Cells of Dmrt1−/− Testes: A Novel Model of DMRT1-Deficient Germ Cells. Biol. Reprod. 2013, 88, 51. [Google Scholar] [CrossRef] [Green Version]
- Itman, C.; Wong, C.; Whiley, P.A.; Fernando, D.; Loveland, K.L. TGFβ superfamily signalling regulators are differentially expressed in the developing and adult mouse testis. Spermatogenesis 2011, 1, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Golestaneh, N.; Beauchamp, E.; Fallen, S.; Kokkinaki, M.; Üren, A.; Dym, M. Wnt signaling promotes proliferation and stemness regulation of spermatogonial stem/progenitor cells. Reproduction 2009, 138, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Lopez, P.; Yaman, R.; Lopez-Fernandez, L.A.; Vidal, F.; Puel, D.; Clertant, P.; Cuzin, F.; Rassoulzadegan, M. Withdrawal: A novel germ line-specific gene of the phosducin-like protein (PhLP) family: A meiotic function conserved from yeast to mice. J. Biol. Chem. 2019, 294, 13832. [Google Scholar] [CrossRef] [Green Version]
- Lukacik, P.; Keller, B.; Bunkoczi, G.; Kavanagh, K.L.; Lee, W.H.; Adamski, J.; Oppermann, U. Structural and biochemical characterization of human orphan DHRS10 reveals a novel cytosolic enzyme with steroid dehydrogenase activity. Biochem. J. 2007, 402, 419–427. [Google Scholar] [CrossRef]
- Rasiah, K.K.; Gardiner-Garden, M.; Padilla, E.J.; Möller, G.; Kench, J.G.; Alles, M.C.; Eggleton, S.A.; Stricker, P.D.; Adamski, J.; Sutherland, R.L.; et al. HSD17B4 overexpression, an independent biomarker of poor patient outcome in prostate cancer. Mol. Cell. Endocrinol. 2009, 301, 89–96. [Google Scholar] [CrossRef]
- Sun, S.-C.; Kim, N.-H. Spindle assembly checkpoint and its regulators in meiosis. Hum. Reprod. Updat. 2011, 18, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Shi, X.; Bi, Y.; Qi, L.; Guo, X.; Wang, L.; Zhou, Z.; Sha, J. SHCBP1L, a conserved protein in mammals, is predominantly expressed in male germ cells and maintains spindle stability during meiosis in testis. Mol. Hum. Reprod. 2014, 20, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Cruz, J.N.; Da Costa, K.S.; De Carvalho, T.A.A.; De Alencar, N.A.N. Measuring the structural impact of mutations on cytochrome P450 21A2, the major steroid 21-hydroxylase related to congenital adrenal hyperplasia. J. Biomol. Struct. Dyn. 2020, 38, 1425–1434. [Google Scholar] [CrossRef]
- Merke, D.P.; Bornstein, S.R.; Avila, N.A.; Chrousos, G.P. Future Directions in the Study and Management of Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency. Ann. Intern. Med. 2002, 136, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Nayak, S.; Lee, P.A.; Witchel, S.F. Variants of the Type II 3β-Hydroxysteroid Dehydrogenase Gene in Children with Premature Pubic Hair and Hyperandrogenic Adolescents. Mol. Genet. Metab. 1998, 64, 184–192. [Google Scholar] [CrossRef]
- Satterfield, M.C.; Gao, H.; Li, X.; Wu, G.; Johnson, G.A.; Spencer, T.E.; Bazer, F.W. Select Nutrients and Their Associated Transporters Are Increased in the Ovine Uterus Following Early Progesterone Administration1. Biol. Reprod. 2010, 82, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Malcher, A.; Rozwadowska, N.; Stokowy, T.; Kolanowski, T.J.; Jedrzejczak, P.; Zietkowiak, W.; Kurpisz, M. Potential biomarkers of nonobstructive azoospermia identified in microarray gene expression analysis. Fertil. Steril. 2013, 100, 1686–1694.e7. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-M.; Liu, G.; Nie, Z.-Y.; Nie, D.-S.; Deng, Y.; Lu, G.-X. Molecular cloning of a novel rat gene Tsarg1, a member of the DnaJ/HSP40 protein family. DNA Seq. 2005, 16, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Yan, Z.; Wang, P.; Yang, Q.; Huang, X.; Shi, H.; Tang, Y.; Ji, Y.; Zhang, J.; Gun, S. Identification and Characterization of lncRNA and mRNA in Testes of Landrace and Hezuo Boars. Animals 2021, 11, 2263. https://doi.org/10.3390/ani11082263
Zhang B, Yan Z, Wang P, Yang Q, Huang X, Shi H, Tang Y, Ji Y, Zhang J, Gun S. Identification and Characterization of lncRNA and mRNA in Testes of Landrace and Hezuo Boars. Animals. 2021; 11(8):2263. https://doi.org/10.3390/ani11082263
Chicago/Turabian StyleZhang, Bo, Zunqiang Yan, Pengfei Wang, Qiaoli Yang, Xiaoyu Huang, Haixia Shi, Yuran Tang, Yanan Ji, Juanli Zhang, and Shuangbao Gun. 2021. "Identification and Characterization of lncRNA and mRNA in Testes of Landrace and Hezuo Boars" Animals 11, no. 8: 2263. https://doi.org/10.3390/ani11082263
APA StyleZhang, B., Yan, Z., Wang, P., Yang, Q., Huang, X., Shi, H., Tang, Y., Ji, Y., Zhang, J., & Gun, S. (2021). Identification and Characterization of lncRNA and mRNA in Testes of Landrace and Hezuo Boars. Animals, 11(8), 2263. https://doi.org/10.3390/ani11082263