
Changes in the Fecal Microbiota Associated with a Broad-Spectrum Antimicrobial Administration in Hospitalized Neonatal Foals with Probiotics Supplementation

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Samples Collection
- positive blood culture;
- positive culture of samples from local sites of suspected infection;
- postmortem examination. Systemic inflammatory response syndrome (SIRS) diagnosis was made as suggested by Wong and Wilkins [34].
- before the start of antimicrobial treatment (Ta, admission);
- at the end of the treatment (Te);
- the day of discharge from the hospital (Td).
2.2. Bacterial DNA Extraction and 16S rRNA Sequencing
2.3. Bioinformatics and Statistics
3. Results
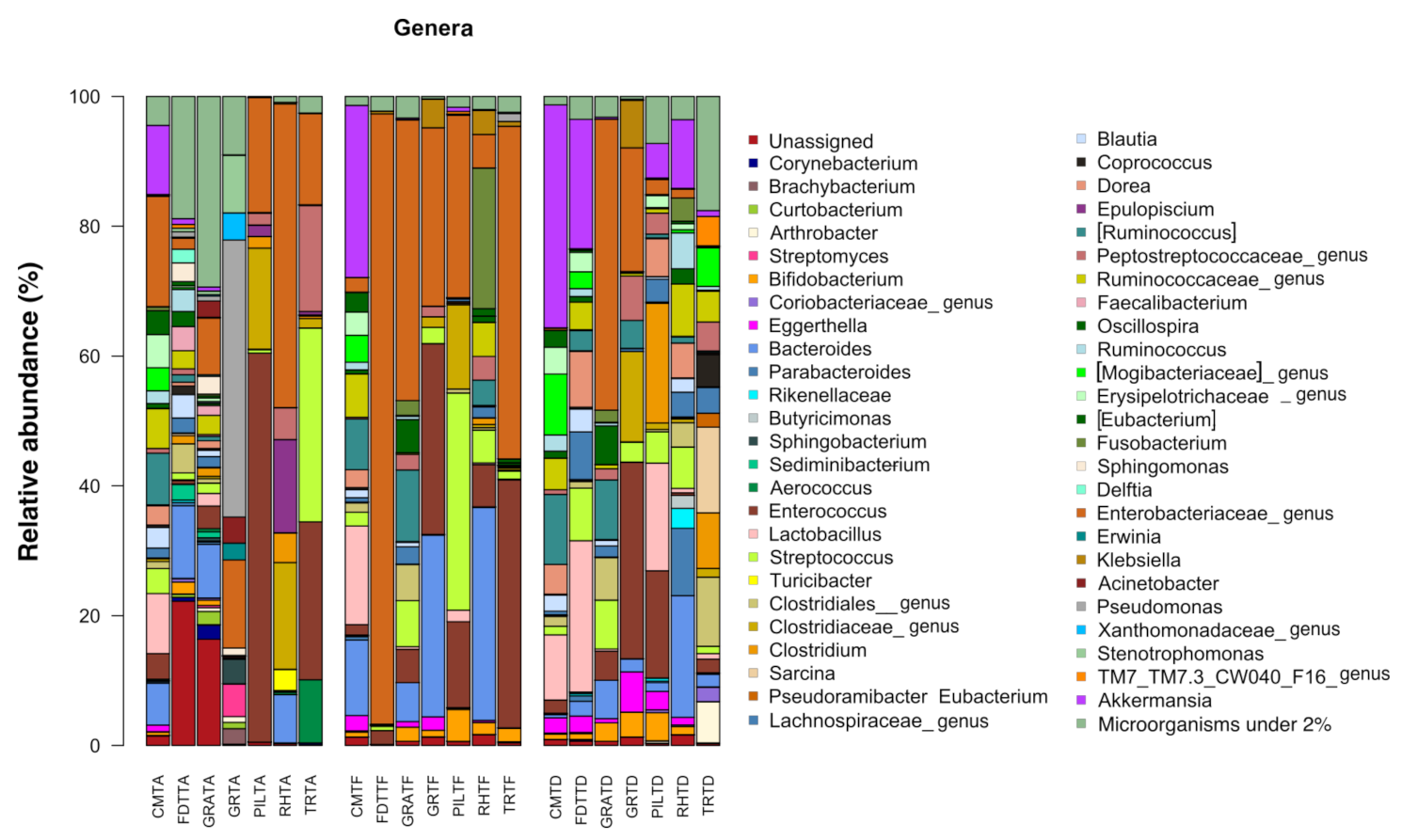
3.1. Analysis of the Composition of Gut Bacterial Microbiota in Relation to Antimicrobial Treatment
3.1.1. Characterization of Bacterial Ecosystem before Antimicrobial Treatment (Ta)
3.1.2. Characterization of Bacterial Ecosystem at the End of Antimicrobial Treatment (Te)
3.1.3. Characterization of Bacterial Ecosystem at the Time of Hospital Discharge (Td)
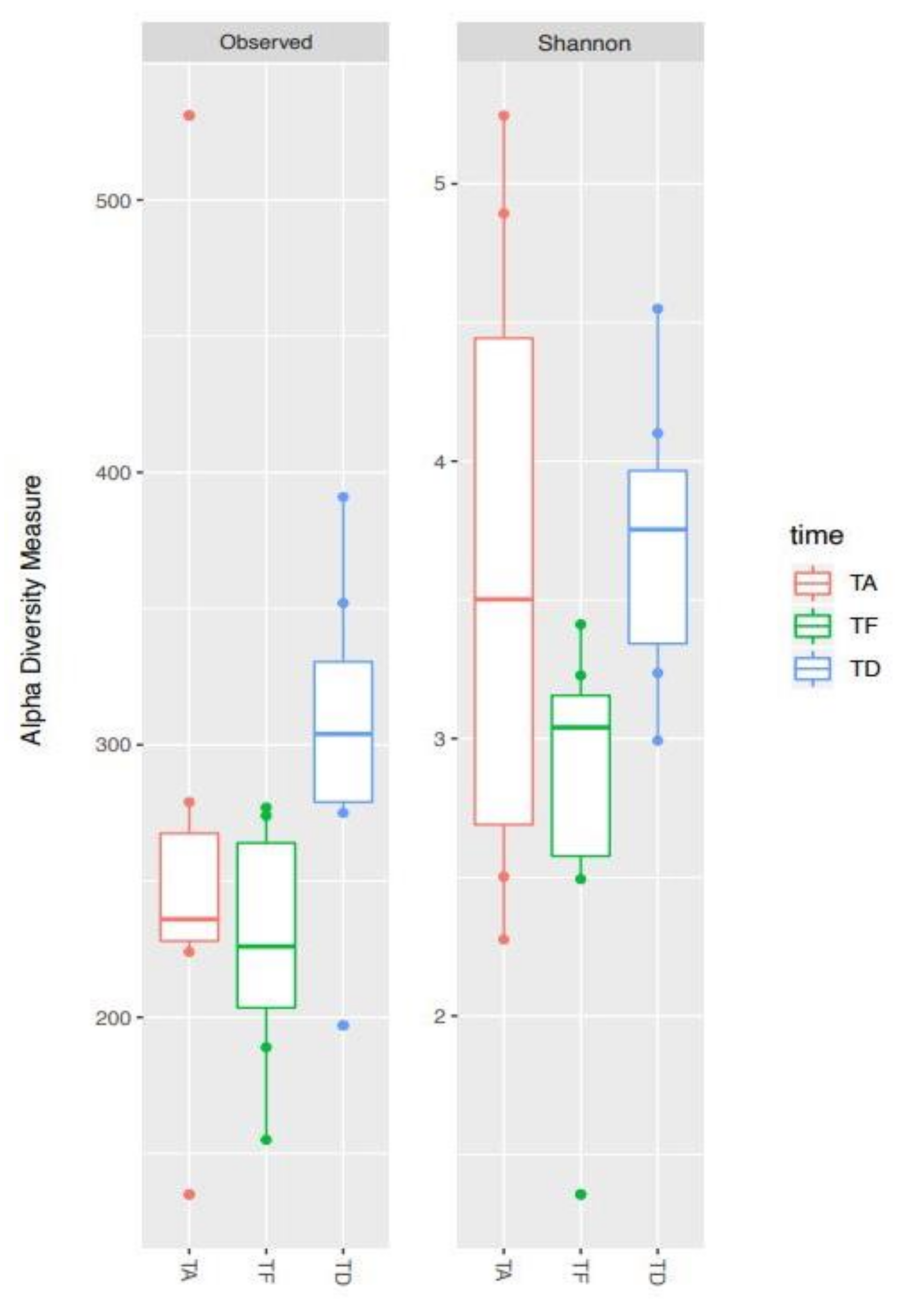
3.2. Alpha Diversity Analysis of the Bacterial Ecosystem in Relation to Antimicrobial Treatment
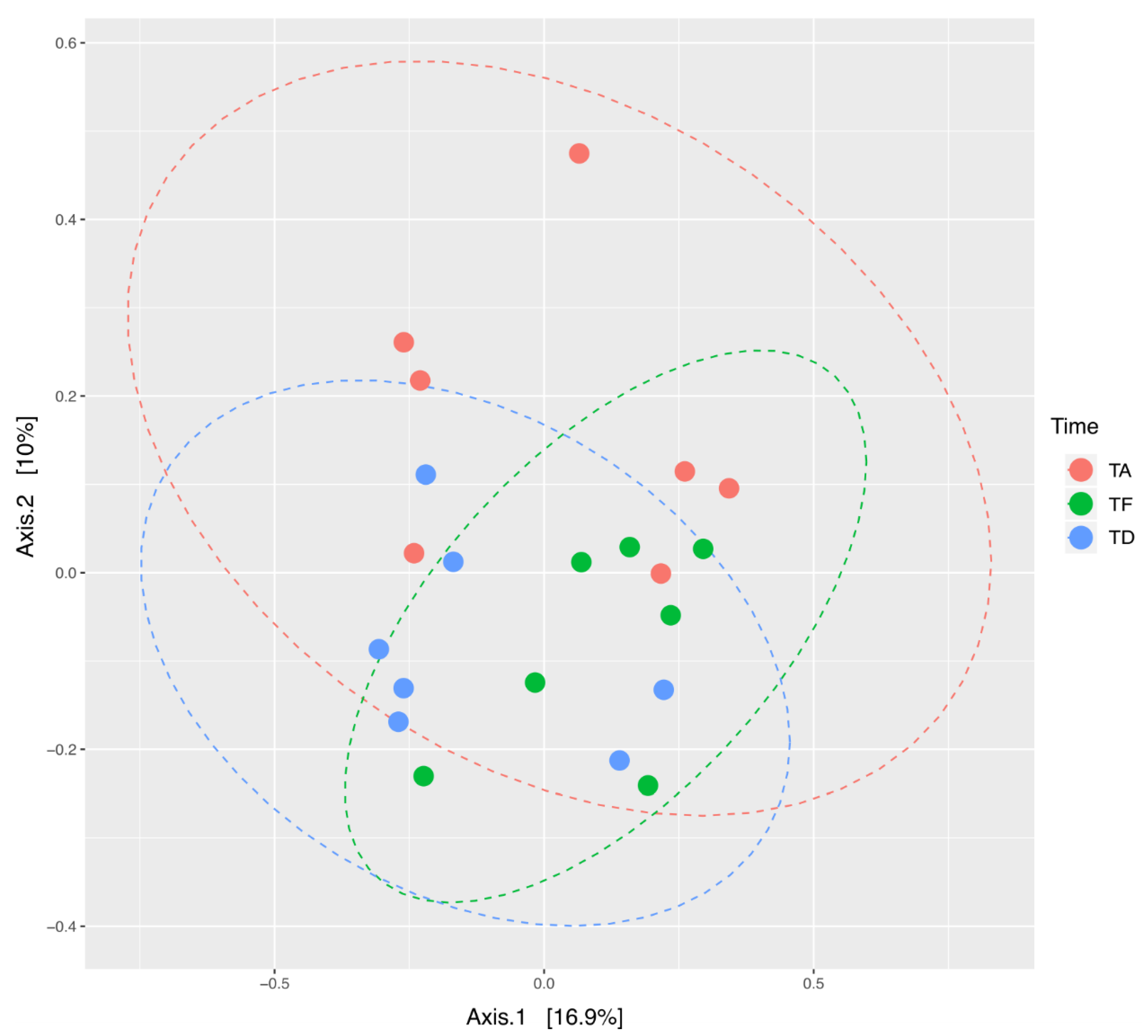
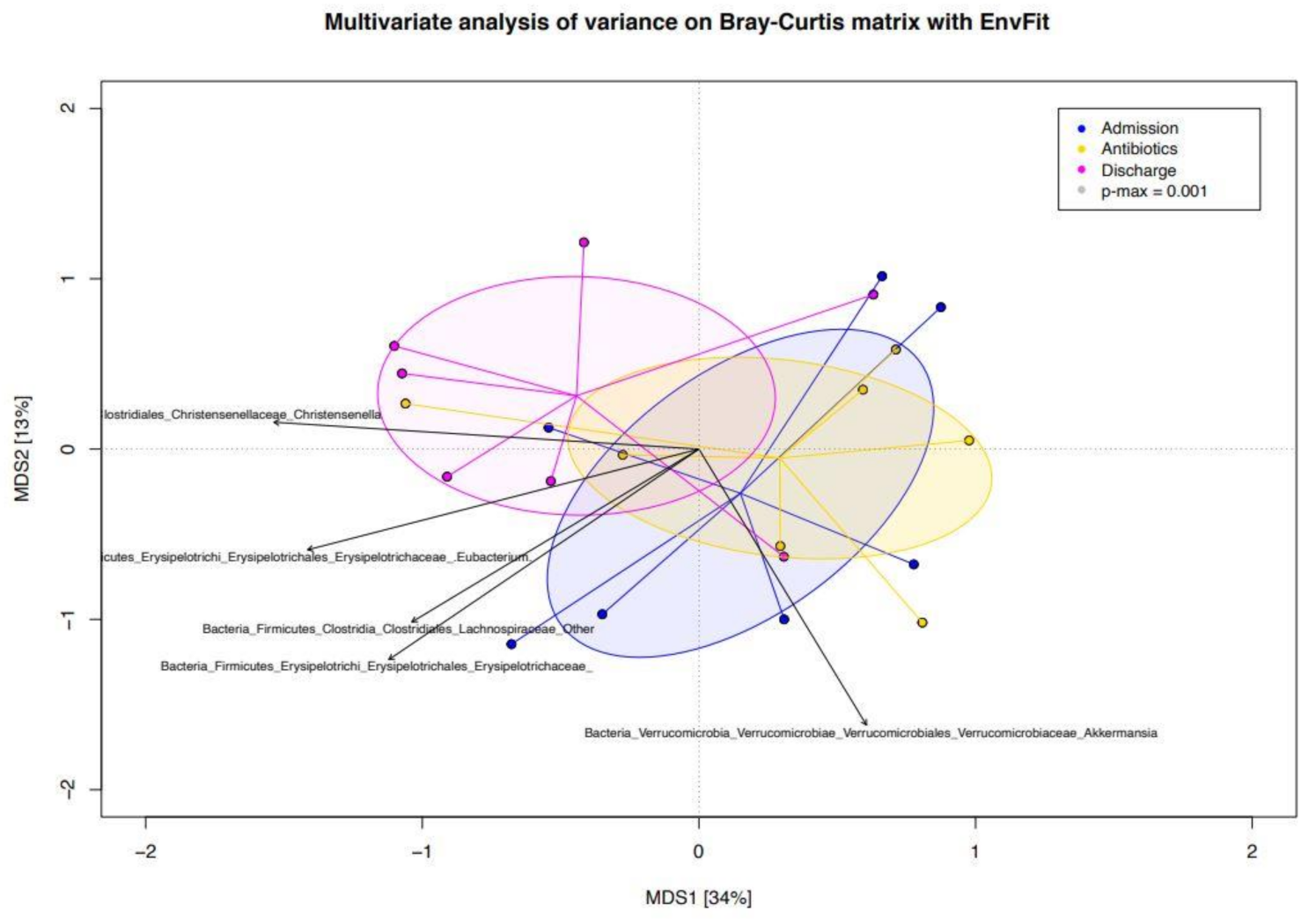
3.3. Beta Diversity Analysis of the Bacterial Ecosystem in Relation to Antimicrobial Treatment
4. Discussion
4.1. Fecal Microbiota in Neonatal Foals after IV Broad-Spectrum Antimicrobials
4.2. Fecal Microbiota in Neonatal Foals at Hospital Discharge
4.3. Limitations and Potential Confounding Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Theelen, M.J.; Wilson, W.D.; Byrne, B.A.; Edman, J.M.; Kass, P.H.; Magdesian, K.G. Initial antimicrobial treatment of foals with sepsis: Do our choices make a difference? Vet. J. 2019, 243, 74–76. [Google Scholar] [CrossRef]
- Floyd, E.F.; Easton-Jones, C.A.; Theelen, M.J.P. Systemic antimicrobial therapy in foals. Equine Vet. Educ. 2021, X, 1–8. [Google Scholar]
- Magdesian, K.G. Antimicrobial pharmacology for the neonatal foal. Vet. Clin. Equine Pract. 2017, 33, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Magdesian, K.G. Neonatal Foal Diarrhea. Vet. Clin. Equine Pract. 2005, 21, 295–312. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Collado, M.C. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050. [Google Scholar] [CrossRef] [PubMed]
- Nogacka, A.M.; Salazar, N.; Arboleya, S.; Suárez, M.; Fernández, N.; Solís, G.; de los Reyes-Gavilán, C.G.; Gueimonde, M. Early microbiota, antibiotics and health. Cell. Mol. Life Sci. 2018, 75, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Eck, A.; Rutten, N.B.; Singendonk, M.M.; Rijkers, G.T.; Savelkoul, P.H.; Meijssen, C.B.; Crijns, C.E.; Oudshoorn, J.H.; Budding, A.E.; Vlieger, A.M. Neonatal microbiota development and the effect of early life antibiotics are determined by two distinct settler types. PLoS ONE 2020, 15, e0228133. [Google Scholar] [CrossRef] [Green Version]
- Rosa, C.P.; Brancaglion, G.A.; Miyauchi-Tavares, T.M.; Corsetti, P.P.; de Almeida, L.A. Antibiotic-induced dysbiosis effects on the murine gastrointestinal tract and their systemic re-percussions. Life Sci. 2018, 207, 480–491. [Google Scholar] [CrossRef]
- Ferrer, M.; Méndez-García, C.; Rojo, D.; Barbas, C.; Moya, A. Antibiotic use and microbiome function. Biochem. Pharmacol. 2017, 134, 114–126. [Google Scholar] [CrossRef]
- Ling, Z.; Liu, X.; Jia, X.; Cheng, Y.; Luo, Y.; Yuan, L.; Wang, Y.; Xiang, C. Impacts of infection with different toxigenic Clostridium difficile strains on faecal microbiota in children. Sci. Rep. 2014, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Kobayashi, T.; Songjinda, P.; Tateyama, A.; Tsubouchi, M.; Kiyohara, C.; Shi-rakawa, T.; Sonomoto, K.; Nakayama, J. Influence of antibiotic exposure in the early post-natal period on the development of intestinal microbiota. FEMS Immunol. Med. Microbiol. 2009, 56, 80–87. [Google Scholar] [CrossRef] [Green Version]
- De La Cochetière, M.F.; Durand, T.; Lalande, V.; Petit, J.C.; Potel, G.; Beaugerie, L. Effect of antibiotic therapy on human fecal microbiota and the relation to the development of Clostridium difficile. Microb. Ecol. 2008, 56, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, T.M.; Levy, S.B. The impact of antibiotic use on resistance development and persistence. Drug Resist. Updat. 2000, 3, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, C.J.; Young, V.B. Antibiotic administration alters the community structure of the gastrointestinal microbiota. Gut Microbes 2010, 1, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianiro, G.; Tilg, H.; Gasbarrini, A. Antibiotics as deep modulators of gut microbiota: Between good and evil. Gut 2016, 65, 1906–1915. [Google Scholar] [CrossRef]
- Iizumi, T.; Battaglia, T.; Ruiz, V.; Perez, G.I.P. Gut microbiome and antibiotics. Arch. Med. Res. 2017, 48, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Kauter, A.; Epping, L.; Semmler, T.; Antao, E.M.; Kannapin, D.; Stoeckle, S.D.; Gehlen, H.; Lübke-Becker, A.; Günther, S.; Wiele, L.H.; et al. The gut microbiome of horses: Current research on equine enteral microbiota and future perspectives. Anim. Microbiome 2019, 1, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, A.; Hastie, P.; Murray, J.A. Factors influencing equine gut microbiota: Current knowledge. J. Equine Vet. Sci. 2020, 88, 102943. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.C.; Stämpfli, H.R.; Arroyo, L.G.; Allen-Vercoe, E.; Gomes, R.G.; Weese, J.S. Changes in the equine fecal microbiota associated with the use of systemic antimicrobial drugs. BMC Vet. Res. 2015, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- De La Torre, U.; Henderson, J.D.; Furtado, K.L.; Pedroja, M.; Elenamarie, O.M.; Mora, A.; Pechanec, M.Y.; Maga, E.A.; Mienaltowski, M.J. Utilizing the fecal microbiota to understand foal gut transitions from birth to weaning. PLoS ONE 2019, 14, e0216211. [Google Scholar]
- Lindenberg, F.; Krych, L.; Kot, W.; Fielden, J.; Frøkiær, H.; van Galen, G.; Nielsen, D.S.; Hansen, A.K. Development of the equine gut microbiota. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Quercia, S.; Freccero, F.; Castagnetti, C.; Soverini, M.; Turroni, S.; Biagi, E.; Rampelli, S.; Lanci, A.; Mariella, J.; Chinellato, E.; et al. Early colonisation and tem-poral dynamics of the gut microbial ecosystem in Standardbred foals. Equine Vet. J. 2019, 51, 231–237. [Google Scholar] [CrossRef]
- Husso, A.; Jalanka, J.; Alipour, M.J.; Huhti, P.; Kareskoski, M.; Pessa-Morikawa, T.; Iivan-ainen, A.; Niku, M. The composition of the perinatal intestinal microbiota in horse. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, M.C.; Stämpfli, H.R.; Allen-Vercoe, E.; Weese, J.S. Development of the faecal micro-biota in foals. Equine Vet. J. 2016, 48, 681–688. [Google Scholar] [CrossRef]
- Collinet, A.; Grimm, P.; Julliand, S.; Julliand, V. Oral administration of antibiotics alters fecal ecosystem of adult horses in the long-term. J. Equine Vet. Sci. 2019, 76, 94. [Google Scholar] [CrossRef]
- Arnold, C.E.; Isaiah, A.; Pilla, R.; Lidbury, J.; Coverdale, J.S.; Callaway, T.R.; Lawhon, S.D.; Steiner, J.; Suchodolski, J.S. The cecal and fecal microbiomes and metabolomes of horses before and after metronidazole administration. PLoS ONE 2020, 15, e0232905. [Google Scholar] [CrossRef]
- Collinet, A.; Grimm, P.; Julliand, S.; Julliand, V. Multidimensional approach for investigating the effects of an antibiotic–probiotic combination on the equine hindgut ecosystem and microbial fibrolysis. Front. Microbiol. 2021, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- Knych, H.K.; Magdesian, K.G. Equine antimicrobial therapy: Current and past issues facing practitioners. J. Vet. Pharmacol. Ther. 2021, 44, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bailey, K.E.; Dyall-Smith, M.; Marenda, M.S.; Hardefeldt, L.Y.; Browning, G.F.; Gilkerson, J.R.; Billman-Jacobe, H. Faecal microbiota and antimicrobial resistance gene profiles of healthy foals. Equine Vet. J. 2021, 53, 806–816. [Google Scholar] [CrossRef]
- Álvarez–Narváez, S.; Berghaus, L.J.; Morris, E.R.A.; Willingham-Lane, J.M.; Slovis, N.M.; Giguere, S.; Cohen, N.D. A common practice of widespread antimicrobial use in horse pro-duction promotes multi-drug resistance. Sci. Rep. 2020, 10, 1–13. [Google Scholar]
- Vaala, W.E.; House, J.K.; Madigan, J.E. Initial management and physical examination of the neonate. In Large Animal Internal Medicine; Smith, B.P., Ed.; Mosby: St. Louis, MO, USA, 2002; pp. 277–293. [Google Scholar]
- Giguére, S.; Polkes, A.C. Immunologic disorders of neonatal foals. Vet. Clin. Equine Pract. 2005, 21, 241–272. [Google Scholar] [CrossRef]
- Castagnetti, C.; Pirrone, A.; Mariella, J.; Mari, G. Venous blood lactate evaluation in equine neonatal intensive care. Theriogenology 2010, 73, 343–347. [Google Scholar] [CrossRef]
- Wong, D.M.; Wilkins, P.A. Defining the Systemic Inflammatory Response Syndrome in Equine Neonates. Vet. Clin. Equine Pract. 2015, 31, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Toribio, R.E. Equine Neonatal Encephalopathy: Facts, Evidence, and Opinions. Vet. Clin. Equine Pract. 2019, 35, 363–378. [Google Scholar] [CrossRef]
- Knottenbelt, D.C.; Holdstock, N.; Madigan, J.E. Equine Neonatology Medicine and Surgery; Saunders: Philadelphia, PA, USA, 2004; pp. 155–363. [Google Scholar]
- Biagi, E.; Franceschi, C.; Rampelli, S.; Severgnini, M.; Ostan, R.; Turroni, S.; Consolandi, C.; Quercia, S.; Scurti, M.; Monti, D.; et al. Gut Microbiota and Extreme Longevity. Curr. Biol. 2016, 26, 1480–1485. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Morrison, M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 2004, 36, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Candela, M.; Biagi, E.; Soverini, M.; Consolandi, C.; Quercia, S.; Severgnini, M.; Peano, C.; Turroni, S.; Rampelli, S.; Pozzilli, P.; et al. Modulation of gut microbiota dysbioses in type 2 diabetic patients by macrobiotic Ma-Pi 2 diet. Br. J. Nutr. 2016, 116, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Biagi, E.; Quercia, S.; Aceti, A.; Berghetti, I.; Rampelli, S.; Turroni, S.; Faldella, G.; Candela, M.; Brigidi, P.; Corvaglia, L. Bacterial sharing between the ecosystems of mother’s milk and infant’s mouth and gut. Front. Miocrobiol. 2017, 8, 1214. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; Sankaran, K.; Fukuyama, J.A.; McMurdie, P.J.; Holmes, S.P. Bioconductor workflow for microbiome data analysis: From raw reads to community analyses. F1000Research 2016, 5, 1492. [Google Scholar] [CrossRef]
- Anderson, M.J.; Walsh, D.C.I. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol. Monogr. 2013, 83, 557–574. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Oksanen, M.J. Package ‘vegan’. Community Ecology Package, Version. 2013, Volume 2, pp. 1–295. Available online: http://sortie-admin.readyhosting.com/lme/R%20Packages/vegan.pdf (accessed on 20 May 2021).
- Wickham, H. Ggplot2: Elegant graphics for data analysis. J. R. Stat. Soc. Ser. A 2011, 174, 245–246. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Cobas, A.E.; Gosalbes, M.J.; Friedrichs, A.; Knecht, H.; Artacho, A.; Eismann, K.; Otto, W.; Rojo, D.; Bargiela, R.; von Bergen, M.; et al. Gut microbiota disturbance during antibiotic therapy: A multiomic approach. Gut 2014, 62, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, C.; Morrow, A.L.; Lagomarcino, A.J.; Altaye, M.; Taft, D.H.; Yu, Z.; Newburg, D.S.; Ward, D.V.; Schibler, K.R. Early empiric antibiotic use in preterm infants is associated with lower bacterial diversity and higher relative abundance of Enterobacter. J. Pediatr. 2014, 165, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.C.; Derrien, M.; Isolauri, E.; de Vos, W.M.; Salminen, S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl. Environ. Microbiol. 2007, 73, 7767–7770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, M.; Collado, M.C.; Ben-Amor, K.; Salminen, S.; de Vos, W.M. The Mucin degrader Akkermansia muciniphila is an abundant resident of the human intestinal tract. Appl. Environ. Microbiol. 2008, 74, 1646–1648. [Google Scholar] [CrossRef] [Green Version]
- Ottman, N.; Geerlings, S.Y.; Aalvink, S.; de Vos, W.M.; Belzer, C. Action and function of Akkermansia muciniphila in microbiome ecology, health and disease. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 637–642. [Google Scholar] [CrossRef] [Green Version]
- Arboleya, S.; Sánchez, B.; Solís, G.; Fernández, N.; Suárez, M.; Hernández-Barranco, A.M.; Milani, C.; Margolles, A.; de los Reyes-Gavilán, C.G.; Ventura, M.; et al. Impact of prematurity and perinatal antibiotics on the developing intestinal microbiota: A functional inference study. Int. J. Mol. Sci. 2016, 17, 649. [Google Scholar] [CrossRef] [PubMed]
- Massacci, F.R.; Clark, A.; Ruet, A.; Lansade, L.; Costa, M.; Mach, N. Inter-breed diversity and temporal dynamics of the faecal microbiota in healthy horses. J. Anim. Breed. Genet. 2020, 137, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.; Bloomfield, F.; O’Sullivan, J. Factors Affecting Gastrointestinal Microbiome Development in Neonates. Nutrients 2018, 10, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, L.A.; Brown, R.; Poxton, I.R. A comparative study of the intestinal microbiota of healthy horses and those suffering from equine grass sickness. Vet. Microbiol. 2002, 87, 81–88. [Google Scholar] [CrossRef]
- Costa, M.C.; Arroyo, L.G.; Allen-Vercoe, E.; Stampfli, H.R.; Kim, P.T.; Sturgeon, A.; Weese, S.J. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PLoS ONE 2012, 7, e41484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, M.M.; Eades, S.C.; Reinemeyer, C.R.; Fugaro, M.N.; Onishi, J.C. Illumina sequencing of the V4 hypervariable region 16S rRNA gene reveals extensive changes in bacterial communities in the cecum following carbohydrate oral infusion and development of early-stage acute laminitis in the horse. Vet. Microbiol. 2014, 168, 436–441. [Google Scholar] [CrossRef]
- Weese, J.S.; Holcombe, S.J.; Embertson, R.M.; Kurtz, K.A.; Roessner, H.A.; Jalali, M.; Wismer, S.E. Changes in the faecal microbiota of mares precede the development of post partum colic. Equine Vet. J. 2015, 47, 641–649. [Google Scholar] [CrossRef]
- Elzinga, S.E.; Weese, J.S.; Adams, A.A. Comparison of the fecal microbiota in horses with equine metabolic syndrome and metabolically normal controls fed a similar all-forage diet. J. Equine Vet. Sci. 2016, 44, 9–16. [Google Scholar] [CrossRef]
- Schoster, A.; Staempfli, H.R.; Guardabassi, L.G.; Jalali, M.; Weese, J.S. Comparison of the fecal bacterial microbiota of healthy and diarrheic foals at two and four weeks of life. BMC Vet. Res. 2017, 13, 144. [Google Scholar] [CrossRef] [Green Version]
- Stewart, H.L.; Southwood, L.L.; Indugu, N.; Vecchiarelli, B.; Engiles, J.B.; Pitta, D. Differences in the equine faecal microbiota between horses presenting to a tertiary referral hospital for colic compared with an elective surgical procedure. Equine Vet. J. 2019, 51, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Schoster, A. Probiotic Use in Equine Gastrointestinal Disease. Vet. Clin. Equine Pract. 2018, 34, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weese, J.S.; Maureen, E.C.; Anderson, A.L.; Monteith, G.J. Preliminary investigation of the probiotic potential of Lactobacillus rhamnosus strain GG in horses: Fecal recovery following oral administration and safety. Can. Vet. J. 2003, 44, 299–302. [Google Scholar]
- Schoster, A.; Guardabassi, L.; Staempfli, H.R.; Abrahams, M.; Jalali, M.; Weese, J.S. The Longitudinal Effect of a Multi-Strain Probiotic on the Intestinal Bacterial Microbiota of Neonatal Foals. Equine Vet. J. 2016, 48, 689–696. [Google Scholar] [CrossRef]
- Schoster, A.; Staempfli, H.R.; Abrahams, M.; Jalali, M.; Weese, J.S.; Guardabassi, L. Effect of a probiotic on prevention of diarrhea and Clostridium difficile and Clostridium perfringens shedding in foals. JVIM 2015, 29, 925–931. [Google Scholar]
- Furr, M.; Cohen, N.D.; Axon, J.E.; Sanchez, L.C.; Pantaleon, L.; Haggett, E.; Campbell, R.; Tennent-Brown, B. Treatment with histamine-type 2 receptor antagonists and omeprazole increase the risk of diarrhoea in neonatal foals treated in intensive care units. Equine Vet. J. 2012, 44, s80–s86. [Google Scholar] [CrossRef]
- Cerri, S.; Taminiau, B.; de Lusancay, A.H.C.; Lecoq, L.; Amory, H.; Daube, G.; Cesarini, C. Effect of oral administration of omeprazole on the microbiota of the gastric glandular mucosa and feces of healthy horses. JVIM 2020, 34, 2727. [Google Scholar]
- Tyma, J.F.; Epstein, K.L.; Whitfield-Cargile, C.M.; Cohen, N.D.; Giguère, S. Investigation of effects of omeprazole on the fecal and gastric microbiota of healthy adult horses. Am. J. Vet. Res. 2019, 80, 79–86. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.A.; Bedenice, D.; Pacheco, A.P.; Oliveira, B.C.; Paradis, M.R.; Mazan, M.; Widmer, G. Assessment of clinical and microbiota responses to fecal microbial transplantation in adult horses with diarrhea. PLoS ONE 2021, 16, e0244381. [Google Scholar] [CrossRef] [PubMed]
- Mullen, K.R.; Yasuda, K.; Divers, T.J.; Weese, J.S. Equine faecal microbiota transplant: Current knowledge, proposed guidelines and future directions. Equine Vet. Educ. 2018, 30, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Palacio, S.D.; Montes, S.A.; Mancabelli, L.; et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. MMBR 2017, 81, e00036-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Foal | CM | GRA | FDT | GR | PIL | RH | TR |
|---|---|---|---|---|---|---|---|
| Breed | Arab | WB | WB | Stb | Stb | Stb | Stb |
| Place of birth | Farm | Farm | Farm | VTH | VTH | VTH | VTH |
| Ta (age in hours) | 30 | 42 | 10 | 0 | 18 | 18 | 18 |
| IgG (mg/dL) | 135 | 426 | 1535 | 827 | 337 | 677 | 135 |
| Diagnosis | Sepsis, Dysmaturiy, FPT | PAS, FPT | PAS | PAS | FPT | FPT | FPT |
| T/P antimicrobials | T | P | P | P | P | P | P |
| Antimicrobials association | ampicillin/ gentamicin | ampicillin/ amikacin | ampicillin/ gentamicin | ampicillin/ amikacin | ampicillin/ amikacin | ampicillin/ amikacin | ampicillin/ amikacin |
| Antimicrobials treatm duration (days) | 12 | 8 | 5 | 6 | 5 | 6 | 8 |
| Te–Td (days) | 3 | 2 | 9 | 3 | 3 | 5 | 5 |
| Diarrhoea | Yes | Yes | Yes | No | No | No | Yes |
| FMT n°; timeframe | 3; Ta–Te | No | 2; Te–Td | No | No | No | 1; Ta–Te (F) |
| Other treatments | ome/sucr | ome/sucr | ome/sucr | sucr | / | / | ome/sucr |
| Source of milk | M/R | M | M/R | M | M | M | M/R/F |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freccero, F.; Lanci, A.; Mariella, J.; Viciani, E.; Quercia, S.; Castagnetti, A.; Castagnetti, C. Changes in the Fecal Microbiota Associated with a Broad-Spectrum Antimicrobial Administration in Hospitalized Neonatal Foals with Probiotics Supplementation. Animals 2021, 11, 2283. https://doi.org/10.3390/ani11082283
Freccero F, Lanci A, Mariella J, Viciani E, Quercia S, Castagnetti A, Castagnetti C. Changes in the Fecal Microbiota Associated with a Broad-Spectrum Antimicrobial Administration in Hospitalized Neonatal Foals with Probiotics Supplementation. Animals. 2021; 11(8):2283. https://doi.org/10.3390/ani11082283
Chicago/Turabian StyleFreccero, Francesca, Aliai Lanci, Jole Mariella, Elisa Viciani, Sara Quercia, Andrea Castagnetti, and Carolina Castagnetti. 2021. "Changes in the Fecal Microbiota Associated with a Broad-Spectrum Antimicrobial Administration in Hospitalized Neonatal Foals with Probiotics Supplementation" Animals 11, no. 8: 2283. https://doi.org/10.3390/ani11082283
APA StyleFreccero, F., Lanci, A., Mariella, J., Viciani, E., Quercia, S., Castagnetti, A., & Castagnetti, C. (2021). Changes in the Fecal Microbiota Associated with a Broad-Spectrum Antimicrobial Administration in Hospitalized Neonatal Foals with Probiotics Supplementation. Animals, 11(8), 2283. https://doi.org/10.3390/ani11082283