
EST-Microsatellite Types and Structural Scenarios in European Hake Fisheries



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Development of EST-Microsatellites
2.2. Sampling, DNA Extraction, and Microsatellite Genotyping
2.3. Selection Signature and Power Testing
2.4. Gene Diversity and Subpopulation Structure
3. Results
3.1. Development of EST-Microsatellites
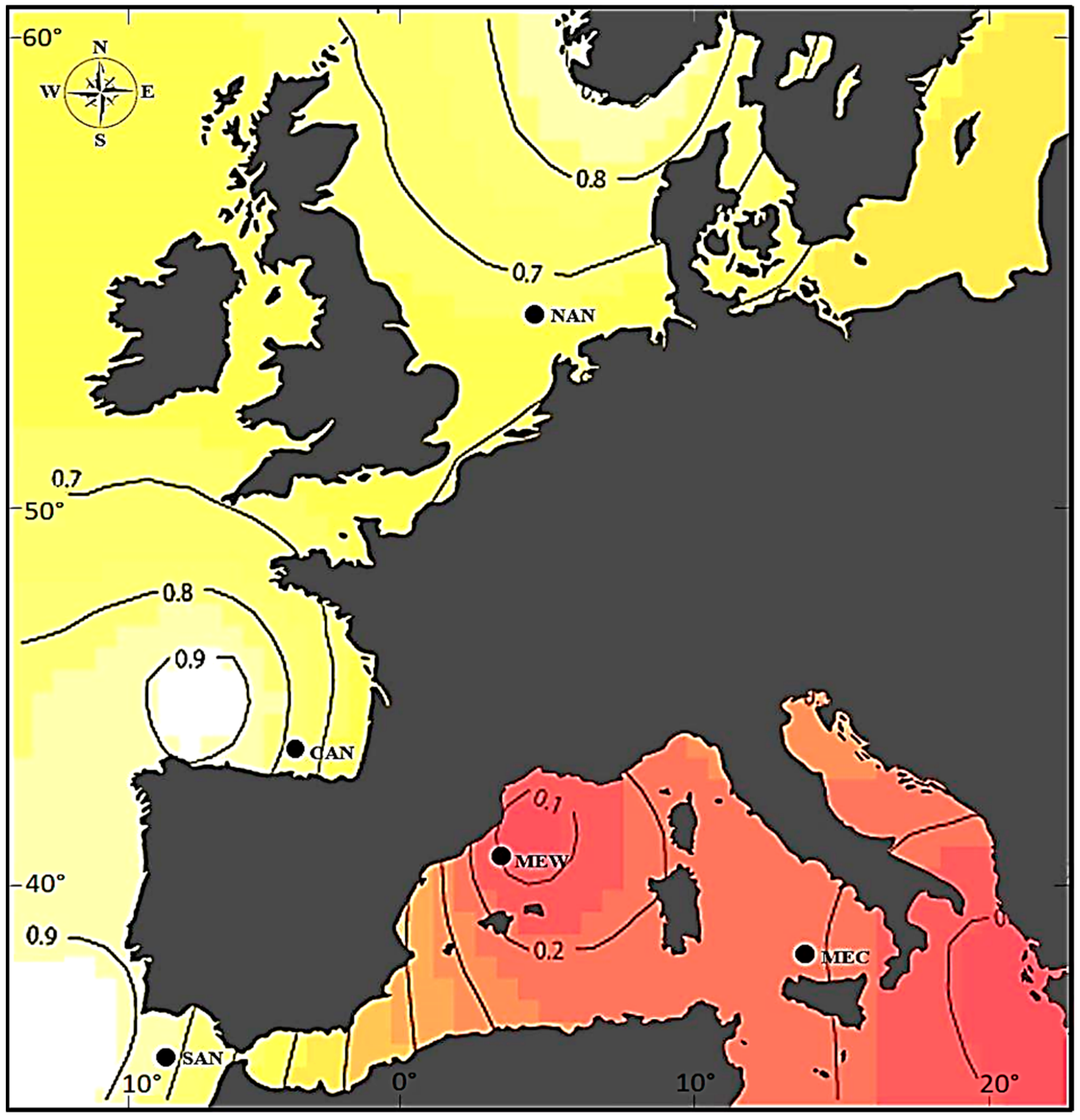
3.2. Genetic Diversity in Natural Samples
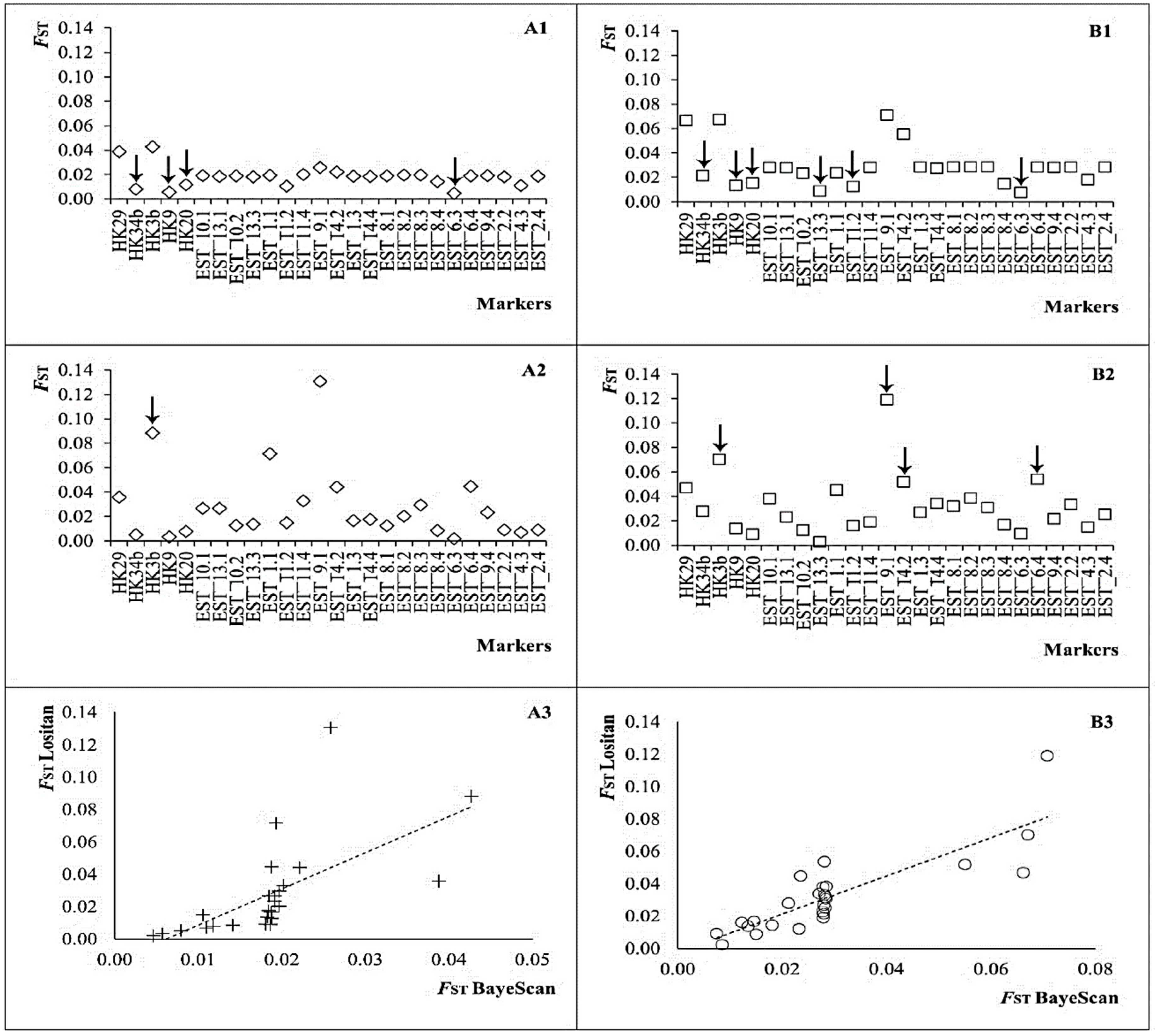
3.3. Functional Annotation, Selection Signature, and Power Testing
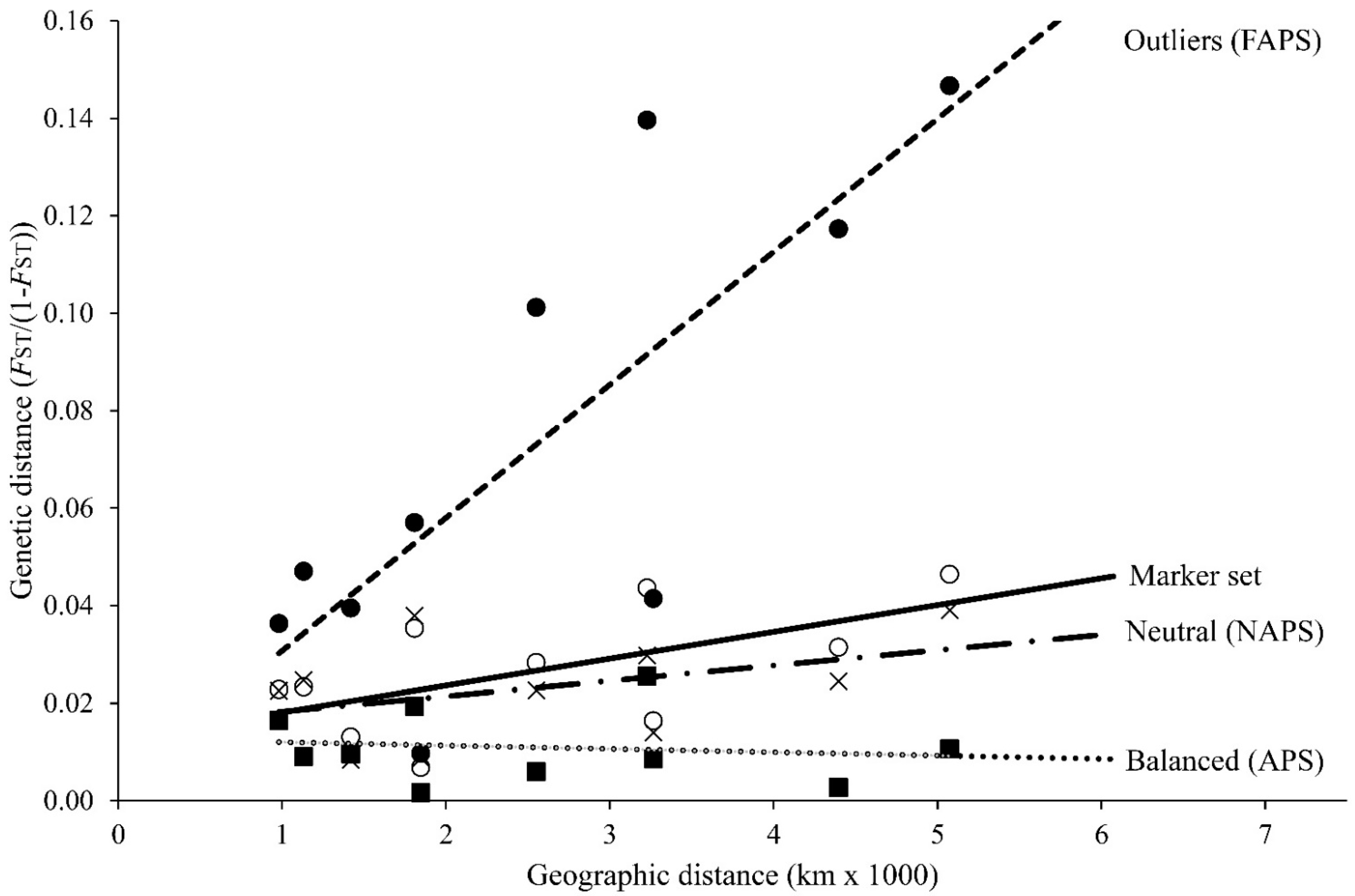
3.4. Structural Population Genetics
4. Discussion
4.1. The Polymorphic Properties of the Marker Set
4.2. Marker-Type-Dependent Structural Scenarios between Basins
4.3. Within-Basin Substructuring and Management Implications
4.4. Management Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cadrin, S.X.; Kerr, L.A.; Mariani, S. Chapter one-Stock identification methods: An overview. In Stock Identification Methods, 2nd ed.; Cadrin, S.X., Kerr, L.A., Mariani, S., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 1–5. [Google Scholar] [CrossRef]
- Enberg, K.; Jørgensen, C.; Dunlop, E.S.; Heino, M.; Dieckmann, U. Implications of fisheries-induced evolution for stock rebuilding and recovery. Evol. Appl. 2009, 2, 394–414. [Google Scholar] [CrossRef] [Green Version]
- Presa, P.; Blanco, G.; Vazquez, E.; Sanchez, J.A. Life-history transitions among Atlantic salmon (Salmo salar) morphotypes. Can. J. Anim. 1996, 76, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Presa, P.; Krieg, F.; Estoup, A.; Guyomard, R. Diversité et gestion génétique de la truite commune: Apport de l’étude du polymorphisme des locus protéiques et microsatellites. Genet. Sel. Evol. 1994, 26 (Suppl. 1), 183s–202s. [Google Scholar] [CrossRef]
- Hawkins, S.J.; Bohn, K.; Sims, D.W.; Ribeiro, P.; Faria, J.; Presa, P.; Pita, A.; Martins, G.M.; Neto, A.I.; Burrows, M.T.; et al. Fisheries stocks from an ecological perspective: Disentangling ecological connectivity from genetic interchange. Fish. Res. 2016, 179, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Lowe, W.H.; Allendorf, F.W. What can genetics tell us about population connectivity? Mol. Ecol. 2010, 19, 3038–3051. [Google Scholar] [CrossRef]
- Murawski, S.A. Rebuilding depleted fish stocks: The good, the bad, and, mostly, the ugly. ICES J. Mar. Sci. 2010, 67, 1830–1840. [Google Scholar] [CrossRef] [Green Version]
- Matić-Skoko, S.; Šegvić-Bubić, T.; Mandić, I.; Izquierdo-Gomez, D.; Arneri, E.; Carbonara, P.; Grati, F.; Ikica, Z.; Kolitari, J.; Milone, N.; et al. Evidence of subtle genetic structure in the sympatric species Mullus barbatus and Mullus surmuletus (Linnaeus, 1758) in the Mediterranean Sea. Sci. Rep. 2018, 8, 676. [Google Scholar] [CrossRef]
- Silva, A.; Garrido, S.; Ibaibarriaga, L.; Pawlowski, L.; Riveiro, I.; Marques, V.; Ramoset, F.; Duhamel, E.; Iglesias, M.; Bryère, P.; et al. Adult-mediated connectivity and spatial population structure of sardine in the Bay of Biscay and Iberian coast. Deep. Sea Res. Part II Top. Stud. Oceanogr. 2019, 159, 62–74. [Google Scholar] [CrossRef]
- De Pontual, H.; Groison, A.L.; Piñeiro, C.; Bertignac, M. Evidence of underestimation of European hake growth in the Bay of Biscay, and its relationship with bias in the agreed method of age estimation. ICES J. Mar. Sci. 2006, 63, 1674–1681. [Google Scholar] [CrossRef]
- Tanner, S.E.; Vasconcelos, R.P.; Cabral, H.N.; Thorrold, S.R. Testing an otolith geochemistry approach to determine population structure and movements of European hake in the northeast Atlantic Ocean and Mediterranean Sea. Fish. Res. 2012, 125, 198–205. [Google Scholar] [CrossRef]
- Reis-Santos, P.; Tanner, S.E.; Aboim, M.A.; Vasconcelos, R.P.; Laroche, J.; Charrier, G.; Pérez, M.; Presa, P.; Gillanders, B.M.; Cabral, H.N. Reconciling differences in natural tags to infer demographic and genetic connectivity in marine fish populations. Sci. Rep. 2018, 8, 10343. [Google Scholar] [CrossRef]
- Cuéllar-Pinzón, J.; Presa, P.; Hawkins, S.J.; Pita, A. Genetic markers in marine fisheries: Types, tasks and trends. Fish. Res. 2016, 173, 194–205. [Google Scholar] [CrossRef]
- Pérez, M.; Cruz, F.; Presa, P. Distribution properties of polymononucleotide repeats in molluscan genomes. J. Hered. 2005, 96, 40–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haasl, R.J.; Payseur, B.A. Multi-locus inference of population structure: A comparison between single nucleotide polymorphisms and microsatellites. Heredity 2011, 106, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Chiarri, M.; Guo, X.; Tanguy, A.; He, Y.; Proestou, D. The use of-omic tools in the study of disease processes in marine bivalve mollusks. J. Invertebr. Pathol. 2015, 131, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Touma, J.; García, K.K.; Bravo, S.; Leiva, F.; Moya, J.; Vargas-Chacoff, L.; Reyes, A.; Vidal, R. De novo assembly and characterization of patagonian toothfish transcriptome and develop of EST-SSR markers for population genetics. Front. Mar. Sci. 2019, 6, 720. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Hofer, T.; Foll, M. Detecting loci under selection in a hierarchically structured population. Heredity 2009, 103, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, S.; Bekkevold, D. Chapter fourteen-The nuclear genome: Neutral and adaptive markers in fisheries science. In Stock Identification Methods, 2nd ed.; Cadrin, S.X., Kerr, L.A., Mariani, S., Eds.; Academic Press: San Diego, CA, USA:, 2014; pp. 297–327. [Google Scholar] [CrossRef]
- Waples, R.S. Separating the wheat from the chaff: Patterns of genetic differentiation in high gene flow species. J. Hered. 1998, 89, 438–450. [Google Scholar] [CrossRef]
- Westgaard, J.I.; Staby, A.; Aanestad Godiksen, J.; Geffen, A.J.; Svensson, A.; Charrier, G.; Svedäng, H.; André, C. Large and fine scale population structure in European hake (Merluccius merluccius) in the Northeast Atlantic. ICES J. Mar. Sci. 2017, 74, 1300–1310. [Google Scholar] [CrossRef]
- Pita, A.; Pérez, M.; Cerviño, S.; Presa, P. What can gene flow and recruitment dynamics tell us about connectivity between European hake stocks in the Eastern North Atlantic? Cont. Shelf Res. 2011, 31, 376–387. [Google Scholar] [CrossRef]
- Leone, A.; Álvarez, P.; García, D.; Saborido-Rey, F.; Rodriguez-Ezpeleta, N. Genome-wide SNP based population structure in European hake reveals the need for harmonizing biological and management units. ICES J. Mar. Sci. 2019, 76, 2260–2266. [Google Scholar] [CrossRef]
- Milano, I.; Babbucci, M.; Cariani, A.; Atanassova, M.; Bekkevold, D.; Carvalho, G.R.; Espiñeira, M.; Fiorentino, F.; Garofalo, G.; Geffen, A.J.; et al. Outlier SNP markers reveal fine-scale genetic structuring across European hake populations (Merluccius merluccius). Mol. Ecol. 2014, 23, 118–135. [Google Scholar] [CrossRef] [PubMed]
- Lo Brutto, S.; Arculeo, M.; Mauro, A.; Scalisi, M.; Cammarata, M.; Parrinello, N. Allozymic variation in Mediterranean hake Merluccius merluccius (Gadidae). Ital. J. Zool. 1998, 65, 49–52. [Google Scholar] [CrossRef]
- Roldán, M.I.; García-Marín, J.L.; Utter, F.M.; Pla, C. Population genetic structure of European hake, Merluccius merluccius. Heredity 1998, 81, 327–334. [Google Scholar] [CrossRef]
- Tintoré, J.; La Violette, P.E.; Blade, I.; Cruzado, A. A study of an intense density front in the eastern Alboran Sea: The Almería–Oran front. J. Phys. Oceanogr. 1988, 18, 1384–1397. [Google Scholar] [CrossRef]
- Ouagajjou, Y.; Presa, P. The connectivity of Mytilus galloprovincialis in northern Morocco: A gene flow crossroads between continents. Estuar. Coast. Shelf Sci. 2015, 152, 1–10. [Google Scholar] [CrossRef]
- Nielsen, E.E.; Cariani, A.; Aoidh, E.M.; Maes, G.E.; Milano, I.; Ogden, R.; Taylor, M.; Hemmer-Hansen, J.; Babbucci, M.; Bargelloni, L.; et al. Gene-associated markers provide tools for tackling illegal fishing and false eco-certification. Nat. Commun. 2012, 3, 851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaux, F.; Bohn, S.; Hyde, J.R.; O’Malley, K.G. Adaptive markers distinguish North and South Pacific Albacore amid low population differentiation. Evol. Appl. 2021, 14, 1343–1364. [Google Scholar] [CrossRef] [PubMed]
- Milano, I.; Babbucci, M.; Panitz, F.; Ogden, R.; Nielsen, R.O.; Taylor, M.I.; Helyar, S.J.; Carvalho, G.R.; Espiñeira, M.; Atanassova, M.; et al. Novel tools for conservation genomics: Comparing two high-throughput approaches for SNP discovery in the transcriptome of the European hake. PLoS ONE 2011, 6, e28008. [Google Scholar] [CrossRef] [PubMed]
- Meglécz, E.; Costedoat, C.; Dubut, V.; Gilles, A.; Malausa, T.; Pech, N.; Martin, J.F. QDD: A user-friendly program to select microsatellite markers and design primers from large sequencing projects. Bioinformatics 2010, 26, 403–404. [Google Scholar] [CrossRef] [Green Version]
- Estoup, A.; Presa, P.; Krieg, F.; Vaiman, D.; Guyomard, R. (CT)n and (GT)n microsatellites: A new class of genetic markers for Salmo trutta L. (brown trout). Heredity 1993, 71, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalendar, R.; Lee, D.; Schulman, A.H. Java web tools for PCR, in silico PCR, and oligonucleotide assembly and analysis. Genomics 2011, 98, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, M.; Presa, P. FENOSALT: Un método sintético para la extracción de ADN de peces y moluscos. In Métodos y Técnicas en Investigación Marina; García-Estévez, J.M., Olabarría, C., Pérez, S., Rolán-Álvarez, E., Rosón, G., Eds.; Tecnos: Madrid, Spain, 2011; pp. 79–88. [Google Scholar]
- Moran, P.; Lundy, C.; Rico, C.; Hewitt, G.M. Isolation and characterization of microsatellite loci in European hake, Merlucius merlucius (Merlucidae, Teleostei). Mol. Ecol. 1999, 8, 1357–1358. [Google Scholar] [CrossRef]
- Tanner, S.E.; Pérez, M.; Presa, P.; Thorrold, S.R.; Cabral, H.N. Integrating microsatellite DNA markers and otolith geochemistry to assess population structure of European hake (Merluccius merluccius). Estuar. Coast. Shelf Sci. 2014, 142, 68–75. [Google Scholar] [CrossRef]
- Antao, T.; Lopes, A.; Lopes, R.J.; Beja-Pereira, A.; Luikart, G.A. Workbench to detect molecular adaptation based on a Fst -outlier method. BMC Bioinform. 2008, 9, 323. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, M.A.; Nichols, R.A. Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society of London. Ser. B Biol. Sci. 1996, 263, 1619–1626. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Ryman, N.; Palm, S. POWSIM: A computer program for assessing statistical power when testing for genetic differentiation. Mol. Ecol. Notes 2006, 6, 600–602. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT (Version 1.2): A computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Rousset, F. Genepop’007: A complete re-implementation of the Genepop software for Windows and Linux. Mol. Ecol. Res. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Guillot, G. Inference of structure in subdivided populations at low levels of genetic differentiation—the correlated allele frequencies model revisited. Bioinformatics 2008, 24, 2222–2228. [Google Scholar] [CrossRef] [Green Version]
- Guillot, G.; Estoup, A.; Mortier, F.; Cosson, J.F. A spatial statistical model for landscape genetics. Genetics 2005, 170, 1261–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillot, G.; Mortier, F.; Estoup, A. GENELAND: A computer package for landscape genetics. Mol. Ecol. Notes 2005, 5, 712–715. [Google Scholar] [CrossRef]
- Corander, J.; Sirén, J.; Arjas, E. Bayesian spatial modeling of genetic population structure. Comput. Stat. 2008, 23, 111–129. [Google Scholar] [CrossRef]
- Corander, J.; Marttinen, P.; Sirén, J.; Tang, J. Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinform. 2008, 9, 539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corander, J.; Waldmann, P.; Marttinen, P.; Sillanpää, M.J. BAPS 2: Enhanced possibilities for the analysis of genetic population structure. Bioinformatics 2004, 20, 2363–2369. [Google Scholar] [CrossRef] [Green Version]
- Foll, M. BayeScan v2.1 User Manual. Available online: http://cmpg.unibe.ch/software/BayeScan/files/BayeScan2.1_manual.pdf (accessed on 22 May 2020).
- Diz, A.P.; Presa, P. Regional patterns of microsatellite variation in Mytilus galloprovincialis from the Iberian Peninsula. Mar. Biol. 2008, 154, 277–286. [Google Scholar] [CrossRef]
- Rodríguez, J.; García, A.; Rodriguez, V. Zooplanktonic communities of the divergence zone in the Northwestern Alboran Sea. Mar. Ecol. 1982, 3, 133–142. [Google Scholar] [CrossRef]
- Pita, A.; Pérez, M.; Balado, M.; Presa, P. Out of the Celtic cradle: The genetic signature of European hake connectivity in South-western Europe. J. Sea Res. 2014, 93, 90–100. [Google Scholar] [CrossRef]
- García-De León, F.J.; Galvan-Tirado, C.; Sanchez Velasco, L.; Silva-Segundo, C.A.; Hernandez-Guzman, R.; Barriga-Sosa, I.D.L.A.; Jaimes, P.D.; Canino, M.; Cruz-Hernández, P. Role of oceanography in shaping the genetic structure in the North Pacific hake Merluccius productus. PLoS ONE 2018, 13, e0194646. [Google Scholar] [CrossRef] [Green Version]
- Riccioni, G.; Cariani, A.; Ferrara, G.; Cannas, R.; Melis, R.; Stagioni, M.; Addis, P.; Tinti, F. Evolutionary constraints limiting the variation of Expressed Sequence Tag-linked microsatellite loci, prevent the detection of local adaptation in Mediterranean Bluefin tuna. Fish. Res. 2017, 190, 157–163. [Google Scholar] [CrossRef]
- Ahrens, C.W.; Rymer, P.D.; Stow, A.; Bragg, J.; Dillon, S.; Umbers, K.D.; Dudaniec, R.Y. The search for loci under selection: Trends, biases and progress. Mol. Ecol. 2018, 27, 1342–1356. [Google Scholar] [CrossRef] [Green Version]
- Lotterhos, K.E.; Whitlock, M.C. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Mol. Ecol. 2014, 23, 2178–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaumont, M.A.; Balding, D.J. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 2004, 13, 969–980. [Google Scholar] [CrossRef]
- Wahlund, S. Zusammensetzung von populationen und korrelationserscheinungen vom standpunkt der vererbungslehre aus betrachtet. Hereditas 1928, 11, 65–106. [Google Scholar] [CrossRef]
- Murua, H.; L. Motos, L. Reproductive strategy and spawning activity of the European hake Merluccius merluccius L. in the Bay of Biscay. J. Fish Biol. 2006, 69, 1288–1303. [Google Scholar] [CrossRef]
- Sánchez, F.; Gil, J. Hydrographic mesoscale structures and Poleward Current as a determinant of hake (Merluccius merluccius) recruitment in southern Bay of Biscay. ICES J. Mar. Sci. 2000, 57, 152–170. [Google Scholar] [CrossRef] [Green Version]
- Barrett, R.D.; Hoekstra, H.E. Molecular spandrels: Tests of adaptation at the genetic level. Nat. Rev. Genet. 2011, 12, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Ray, N. Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol. Evol. 2008, 23, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Cushman, S.A.; Landguth, E.L. Spurious correlations and inference in landscape genetics. Mol. Ecol. 2010, 19, 3592–3602. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.; Bradshaw, W.E.; Holzapfel, C.M. Testing for causality in covarying traits: Genes and latitude in a molecular world. Mol. Ecol. 2011, 20, 2471–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundy, C.J.; Moran, P.; Rico, C.; Milner, R.S.; Hewitt, G.M. Macrogeographical population differentiation in oceanic environments: A case study of European hake (Merluccius merluccius), a commercially important fish. Mol. Ecol. 1999, 8, 1889–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schunter, C.; Carreras-Carbonell, J.; Macpherson, E.; Tintoré, J.; Vidal-Vijande, E.; Pascual, A.; Guidetti, P.; Pascual, M. Matching genetics with oceanography: Directional gene flow in a Mediterranean fish species. Mol. Ecol. 2011, 20, 5167–5181. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, T.E.; Hahn, M.W. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 2014, 23, 3133–3157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waples, R.S.; Gaggiotti, O. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol. Ecol. 2006, 15, 1419–1439. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.G.; Alvarez, P.; Garcia-Vazquez, E. Population structure of Merluccius merluccius along the Iberian Peninsula coast. ICES J. Mar. Sci. 2005, 62, 1699–1704. [Google Scholar] [CrossRef] [Green Version]
- Castillo, A.G.; Martinez, J.L.; Garcia-Vazquez, E. Fine spatial structure of Atlantic hake (Merluccius merluccius) stocks revealed by variation at microsatellite loci. Mar. Biotechnol. 2004, 6, 299–306. [Google Scholar] [CrossRef]
- Lo Brutto, S.; Arculeo, M.; Parrinello, N. Congruence in genetic markers used to describe Mediterranean and Atlantic populations of European hake (Merluccius merluccius L. 1758). J. Appl. Ichthyol. 2004, 20, 81–86. [Google Scholar] [CrossRef]
- Buehler, D.; Holderegger, R.; Brodbeck, S.; Schnyder, E.; Gugerli, F. Validation of outlier loci through replication in independent data sets: A test on Arabis alpina. Ecol. Evol. 2014, 4, 4296–4306. [Google Scholar] [CrossRef]
- Hedgecock, D. Does variance in reproductive success limit effective population sizes of marine organisms? In Genetics and Evolution of Aquatic Organisms; Beaumont, A., Ed.; Springer: Amsterdam, The Netherlands, 1994; pp. 122–134. [Google Scholar]
- Landguth, E.L.; Cushman, S.A.; Murphy, M.A.; Luikart, G. Relationships between migration rates and landscape resistance assessed using individual-based simulations. Mol. Ecol. Res. 2010, 10, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Jiggins, C.D. Supergenes and their role in evolution. Heredity 2014, 113, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobson, C.M.; She, R.; Jarosz, D.F. Pervasive function and evidence for selection across standing genetic variation in S. cerevisiae. Nat. Commun. 2019, 10, 1222. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, P.; Canino, M.; Bailey, K.; Bentzen, P. Inverse relationship between FST and microsatellite polymorphism in the marine fish, walleye pollock (Theragra chalcogramma): Implications for resolving weak population structure. Mol. Ecol. 2004, 13, 1799–1814. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| BayeScan (Balancing Selection) | Lositan (Directional Selection) | |||||||
|---|---|---|---|---|---|---|---|---|
| Atlantic vs. Mediterranean | Five Regions | Atlantic vs. Mediterranean | Five Regions | |||||
| Molecular Marker | FST | q-Value | FST | q-Value | FST | p-Value | FST | p-Value |
| Anonymous microsatellites | ||||||||
| Mmer-hk29 | 0.0388 | 0.3783 | 0.0657 | 0.2341 | 0.0358 | 0.9652 | 0.0468 | 0.9905 |
| Mmer-hk34b | 0.0080 | 0.0019 | 0.0212 | 0.0001 | 0.0052 | 0.5267 | 0.0278 | 0.9332 |
| Mmer-hk3b | 0.0427 | 0.2404 | 0.0666 | 0.3780 | 0.0883 | 0.9992 | 0.0702 | 0.9990 |
| Mmer-hk9b | 0.0057 | 0.0000 | 0.0134 | 0.0000 | 0.0035 | 0.5174 | 0.0135 | 0.6675 |
| Mmer-hk20 | 0.0118 | 0.0232 | 0.0154 | 0.0000 | 0.0079 | 0.6143 | 0.0088 | 0.1896 |
| EST-microsatellites | ||||||||
| Mmer-EST_1.1 | 0.0193 | 0.7856 | 0.0236 | 0.2094 | 0.0716 | 0.9976 | 0.0448 | 0.9805 |
| Mmer-EST_11.2 | 0.0106 | 0.1563 | 0.0123 | 0.0154 | 0.0149 | 0.8418 | 0.0159 | 0.6808 |
| Mmer-EST_11.4 | 0.0202 | 0.6186 | 0.0278 | 0.5875 | 0.0330 | 0.9322 | 0.0189 | 0.5675 |
| Mmer-EST_10.1 | 0.0191 | 0.7434 | 0.0277 | 0.4736 | 0.0267 | 0.7689 | 0.0379 | 0.8114 |
| Mmer-EST_13.1 | 0.0185 | 0.6490 | 0.0276 | 0.5076 | 0.0267 | 0.8155 | 0.0227 | 0.6193 |
| Mmer-EST_10.2 | 0.0188 | 0.7767 | 0.0233 | 0.2656 | 0.0128 | 0.6488 | 0.0122 | 0.3886 |
| Mmer-EST_13.3 | 0.0183 | 0.5818 | 0.0085 | 0.0228 | 0.0136 | 0.6440 | 0.0023 | 0.0793 |
| Mmer-EST_9.1 | 0.0258 | 0.4060 | 0.0748 | 0.1050 | 0.1307 | 0.9972 | 0.1190 | 0.9998 |
| Mmer-EST_14.2 | 0.0221 | 0.4754 | 0.0551 | 0.0834 | 0.0442 | 0.9832 | 0.0519 | 0.9987 |
| Mmer-EST_1.3 | 0.0187 | 0.7667 | 0.0279 | 0.5642 | 0.0166 | 0.7701 | 0.0268 | 0.8609 |
| Mmer-EST_14.4 | 0.0184 | 0.6743 | 0.0271 | 0.3316 | 0.0175 | 0.7887 | 0.0339 | 0.9178 |
| Mmer-EST_8.1 | 0.0188 | 0.7293 | 0.0282 | 0.3874 | 0.0126 | 0.6657 | 0.0318 | 0.8890 |
| Mmer-EST_8.2 | 0.0197 | 0.6955 | 0.0282 | 0.4341 | 0.0203 | 0.7983 | 0.0382 | 0.9459 |
| Mmer-EST_8.3 | 0.0197 | 0.7136 | 0.0282 | 0.6083 | 0.0295 | 0.9056 | 0.0306 | 0.9053 |
| Mmer-EST_8.4 | 0.0142 | 0.3072 | 0.0153 | 0.0542 | 0.0085 | 0.5983 | 0.0167 | 0.6904 |
| Mmer-EST_6.3 † | 0.0046 | 0.0033 | 0.0075 | 0.0001 | 0.0020 | 0.3831 | 0.0092 | 0.2386 |
| Mmer-EST_6.4 | 0.0187 | 0.8017 | 0.0279 | 0.6601 | 0.0447 | 0.9853 | 0.0537 | 0.9997 |
| Mmer-EST_9.4 | 0.0192 | 0.7939 | 0.0277 | 0.5378 | 0.0233 | 0.9137 | 0.0214 | 0.8320 |
| Mmer-EST_2.2 | 0.0181 | 0.5358 | 0.0280 | 0.6445 | 0.0092 | 0.6133 | 0.0330 | 0.9786 |
| Mmer-EST_4.3 | 0.0110 | 0.2150 | 0.0188 | 0.1373 | 0.0070 | 0.5505 | 0.0144 | 0.5493 |
| Mmer-EST_2.4 | 0.0186 | 0.7557 | 0.0280 | 0.6273 | 0.0090 | 0.5873 | 0.0251 | 0.7479 |
| Directional Loci (LOSITAN) | Neutral Loci (LOSITAN) | Balanced Loci (BAYESCAN) | Neutral Loci (BAYESCAN) | Neutral Loci (Both Algorithms) | |
|---|---|---|---|---|---|
| Between basins | |||||
| No. loci = 26 (100%) | 1 (4) | 25 (96) | 4 (16) | 22 (84) | 21 (80) |
| FCT = 0.026 * | 0.091 * | 0.024 * | 0.005 ns | 0.031 * | 0.028 * |
| FCT change (%) Causal loci | +250 1 directional | −8 4 balanced | −81 4 balanced | +19% 1 directional | + 8% neutral |
| BAPs pools (k) † | 2 | 1 | 1 | 1 | 1 |
| Geneland pools (k) † | 2 | 2 | 1 | 2 | 2 |
| Among regions ‡ | |||||
| No. loci = 26 (100%) | 4 (15%) | 22 (85%) | 6 (23%) | 20 (77%) | 16 (62%) |
| FSC = 0.020 * | 0.037 * | 0.018 * | 0.012 * | 0.023 * | 0.020 * |
| FCT change (%) Causal loci | + 83% 4 directional | −10 6 balanced | −40% 6 balanced | +15 4 directional | 0 neutral |
| BAPs pools (k) § | 2 ¶ | 1 | 1 | 1 | 1 |
| Geneland pools (k) § | 3 ¥ | 1 | 2 » | 2 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pita, A.; Fernández-Míguez, M.; Presa, P. EST-Microsatellite Types and Structural Scenarios in European Hake Fisheries. Animals 2022, 12, 1462. https://doi.org/10.3390/ani12111462
Pita A, Fernández-Míguez M, Presa P. EST-Microsatellite Types and Structural Scenarios in European Hake Fisheries. Animals. 2022; 12(11):1462. https://doi.org/10.3390/ani12111462
Chicago/Turabian StylePita, Alfonso, María Fernández-Míguez, and Pablo Presa. 2022. "EST-Microsatellite Types and Structural Scenarios in European Hake Fisheries" Animals 12, no. 11: 1462. https://doi.org/10.3390/ani12111462
APA StylePita, A., Fernández-Míguez, M., & Presa, P. (2022). EST-Microsatellite Types and Structural Scenarios in European Hake Fisheries. Animals, 12(11), 1462. https://doi.org/10.3390/ani12111462