
Weighted Single-Step Genome-Wide Association Study Uncovers Known and Novel Candidate Genomic Regions for Milk Production Traits and Somatic Cell Score in Valle del Belice Dairy Sheep

 , , , , , ,
, , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotypes and Pedigree
2.2. Genotyping and Quality Control
2.3. Weighted Single-Step Genome-Wide Association Study
2.4. Detection of top SNP Windows and Functional Annotations of Candidate Genes
3. Results and Discussion
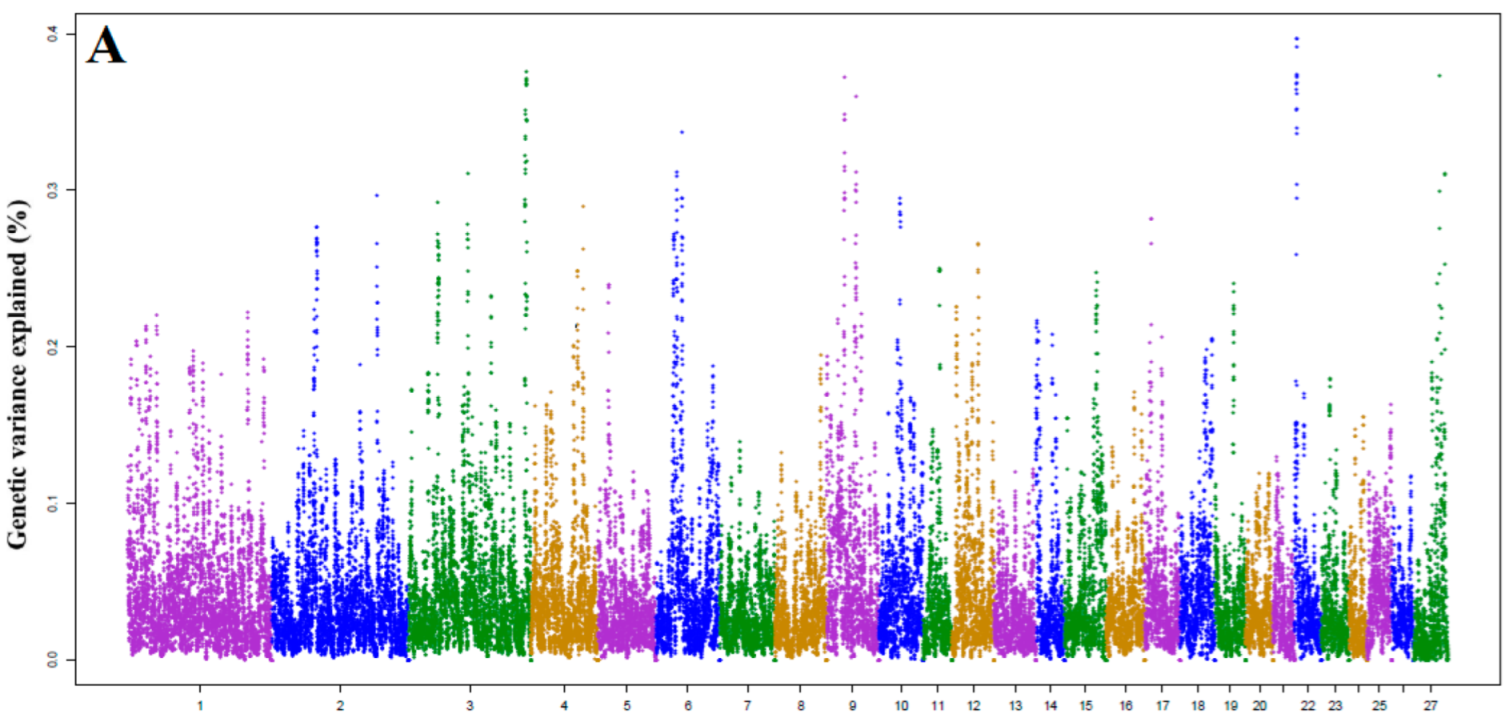
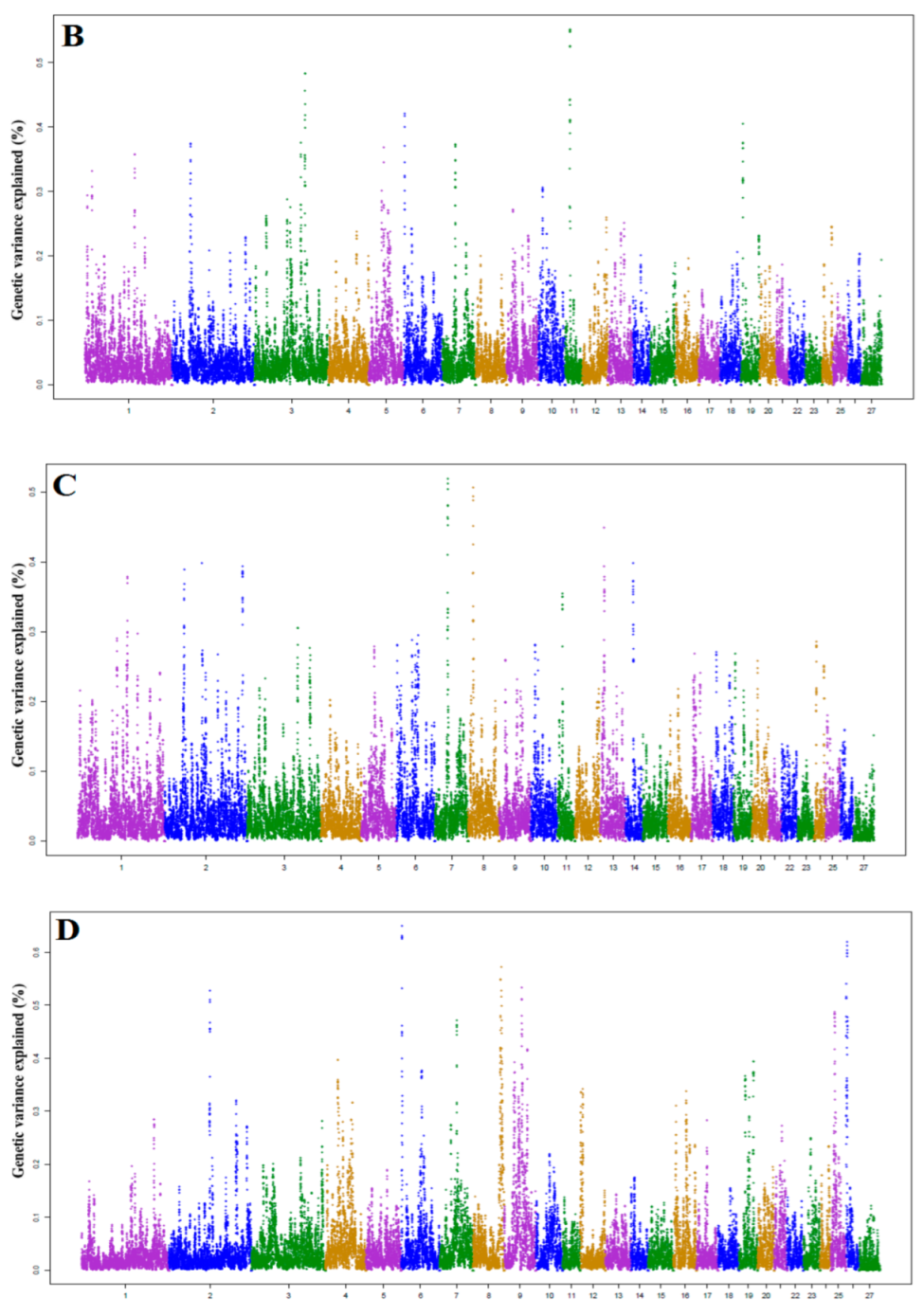
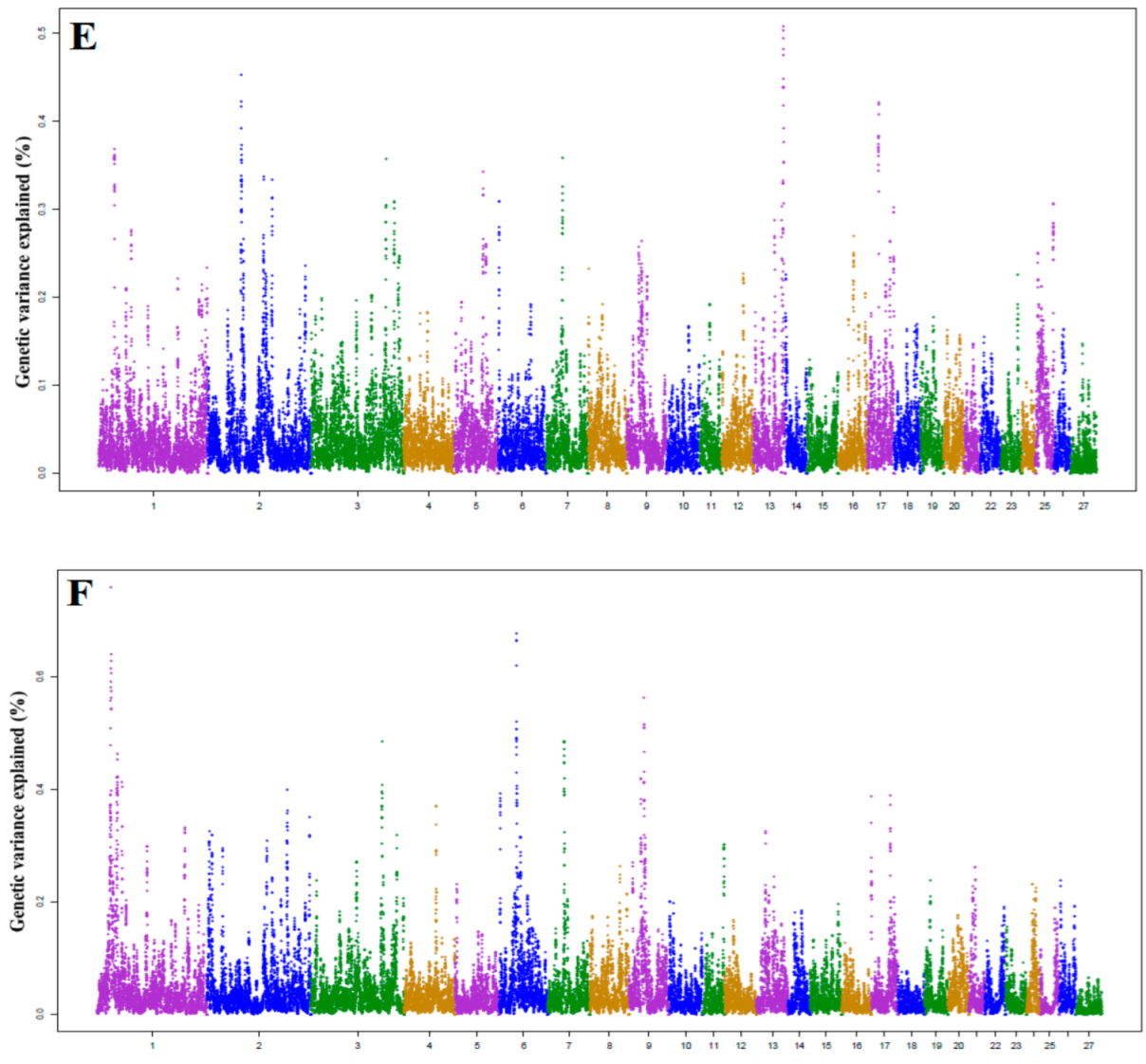
3.1. Identification of Genomic Region and Candidate Genes
3.2. Candidate Genes for Milk Yield (MY)
3.3. Candidate Genes for Milk Fat Yield (FY) and Fat Percentage (FAT%)
3.4. Candidate Genes for Milk Protein Yield and Protein Percentage
3.5. Candidate Genes for SCS
3.6. Functional Annotation of Enrichment Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buaban, S.; Lengnudum, K.; Boonkum, W.; Phakdeedindan, P. Genome-wide association study on milk production and somatic cell score for Thai dairy cattle using weighted single-step approach with random regression test-day model. J. Dairy Sci. 2022, 105, 468–494. [Google Scholar] [CrossRef] [PubMed]
- Öner, Y.; Serrano, M.; Sarto, P.; Iguácel, L.P.; Piquer-Sabanza, M.; Estrada, O.; Juan, T.; Calvo, J.H. Genome-Wide Association Studies of Somatic Cell Count in the Assaf Breed. Animals 2021, 11, 1531. [Google Scholar] [CrossRef] [PubMed]
- Pösö, J.; Mäntysaari, E.A. Relationships between clinical mastitis, somatic cell score, and production for the first three lactations of Finnish Ayrshire. J. Dairy Sci. 1996, 79, 1284–1291. [Google Scholar] [CrossRef]
- Ilie, D.E.; Mizeranschi, A.E.; Mihali, C.V.; Neamț, R.I.; Goilean, G.V.; Georgescu, O.I.; Zaharie, D.; Carabaș, M.; Huțu, I. Genome-Wide Association Studies for Milk Somatic Cell Score in Romanian Dairy Cattle. Genes 2021, 12, 1495. [Google Scholar] [CrossRef]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet. Res. 2012, 94, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Fernando, R.L.; Vitezica, Z.; Okimoto, R.; Wing, T.; Hawken, R.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes in a single-step (ssGWAS) for 6-week body weight in broiler chickens. Front. Genet. 2014, 5, 134. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, I.; Misztal, I.; Johnson, D.L.; Legarra, A.; Tsuruta, S.; Lawlor, T.J. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef]
- Marques, D.B.D.; Bastiaansen, J.W.M.; Broekhuijse, M.L.W.J.; Lopes, M.S.; Knol, E.F.; Harlizius, B.; Guimarães, S.E.F.; Silva, F.F.; Lopes, P.S. Weighted single-step GWAS and gene network analysis reveal new candidate genes for semen traits in pigs. Genet. Sel. Evol. 2018, 50, 40. [Google Scholar] [CrossRef] [Green Version]
- Misztal, I.; Tsuruta, S.; Lourenco, D.A.L.; Masuda, Y.; Aguilar, I.; Legarra, A.; Vitezica, Z. Manual for BLUPF90 Family of Programs. University of Georgia, 2018. Available online: http://nce.ads.uga.edu/wiki/lib/exe/fetch.php?media=blupf90all7.pdf (accessed on 22 May 2014).
- Atashi, H.; Salavati, M.; De Koster, J.; Ehrlich, J.; Crowe, M.; Opsomer, G.; GplusE Consortium; Hostens, M. Genome-wide association for milk production and lactation curve parameters in Holstein dairy cows. J. Anim. Breed Genet. 2020, 137, 292–304. [Google Scholar] [CrossRef]
- Brunes, L.C.; Baldi, F.; Lopes, F.B.; Lôbo, R.B.; Espigolan, R.; Costa, M.F.O.; Stafuzza, N.B.; Magnabosco, C.U. Weighted single-step genome-wide association study and pathway analyses for feed efficiency traits in Nellore cattle. J. Anim. Breed Genet. 2021, 138, 23–44. [Google Scholar] [CrossRef]
- Raschia, M.A.; Nani, J.P.; Carignano, H.A.; Amadio, A.F.; Maizon, D.O.; Poli, M.A.; Nacional, I.; Agropecuaria, D.T.; De Genética, I.; Favret, E.A.; et al. Weighted single-step genome-wide association analyses for milk traits in Holstein and Holstein x Jersey crossbred dairy cattle. Livest. Sci. 2020, 242, 104294. [Google Scholar] [CrossRef]
- Zhou, C.; Li, C.; Cai, W.; Liu, S.; Yin, H.; Shi, S.; Zhang, Q.; Zhang, S. Genome-Wide Association Study for Milk Protein Composition Traits in a Chinese Holstein Population Using a Single-Step Approach. Front. Genet. 2019, 10, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teissier, M.; Larroque, H.; Robert-Granie, C. Accuracy of genomic evaluation with weighted single-step genomic best linear unbiased prediction for milk production traits, udder type traits, and somatic cell scores in French dairy goats. J. Dairy Sci. 2019, 102, 3142–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Luo, H.; Huang, X.; Wei, C.; Di, J.; Tian, Y.; Fu, X.; Li, B.; Liu, G.E.; Fang, L.; et al. Integration of a single-step genome-wide association study with a multi-tissue transcriptome analysis provides novel insights into the genetic basis of wool and weight traits in sheep. Genet. Sel. Evol. 2021, 53, 56. [Google Scholar] [CrossRef] [PubMed]
- Sutera, A.M.; Moscarelli, A.; Mastrangelo, S.; Sardina, M.T.; Di Gerlando, R.; Portolano, B.; Tolone, M. Genome-Wide Association Study Identifies New Candidate Markers for Somatic Cells Score in a Local Dairy Sheep. Front. Genet. 2021, 12, 643531. [Google Scholar] [CrossRef] [PubMed]
- ICAR. International Agreement of Recording Practices. International Committee for Animal Recording. 2014. Available online: https://www.icar.org/index.php/icar-recording-guidelines/ (accessed on 1 January 2020).
- Ali, A.; Shook, G.E. An optimum transformation for somatic cell count in milk. J. Dairy Sci. 1980, 63, 487–490. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- VanRaden, P.M.; Van Tassell, C.P.; Wiggans, G.R.; Sonstegard, T.S.; Schnabel, R.D.; Taylor, J.F.; Schenkel, F.S. Invited review: Reliability of genomic predictions for North American Holstein bulls. J. Dairy Sci. 2009, 92, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Sweett, H.; Fonseca, P.A.S.; Suárez-Vega, A.; Livernois, A.; Miglior, F.; Cánovas, A. Genome-wide association study to identify genomic regions and positional candidate genes associated with male fertility in beef cattle. Sci. Rep. 2020, 10, 20102. [Google Scholar] [CrossRef]
- Otto, P.I.; Guimarães, S.E.F.; Calus, M.P.L.; Vandenplas, J.; Machado, M.A.; Panetto, J.C.C.; da Silva, M.V.G.B. Single-step genome-wide association studies (GWAS) and post-GWAS analyses to identify genomic regions and candidate genes for milk yield in Brazilian Girolando cattle. J. Dairy Sci. 2020, 103, 10347–10360. [Google Scholar] [CrossRef]
- Yin, H.; Zhou, C.; Shi, S.; Fang, L.; Liu, J.; Sun, D.; Jiang, L.; Zhang, S. Weighted Single-Step Genome-Wide Association Study of Semen Traits in Holstein Bulls of China. Front. Genet. 2019, 10, 1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, K.; Lee, H.; Choi, T.Y.; Joo, Y.; Kim, S.J.; Kim, H.; Kim, J.Y.; Jahng, J.W.; Lee, S.; Choi, S.Y.; et al. Negr1 controls adult hippocampal neurogenesis and affective behaviors. Mol. Psychiatry 2019, 8, 1189–1205. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Jia, C.; Fu, D.; Chu, M.; Ding, X.; Wu, X.; Guo, X.; Pei, J.; Bao, P.; Liang, C.; et al. Analysis of Hematological Traits in Polled Yak by Genome-Wide Association Studies Using Individual SNPs and Haplotypes. Genes 2019, 10, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Gao, N.; Li, S.; Kuang, W.; Zhu, L.; Jiang, W.; Yu, W.; Guo, J.; Li, Z.; Yang, C.; et al. Genome-Wide Association Study of Meat Quality Traits in a Three-Way Crossbred Commercial Pig Population. Front. Genet. 2021, 12, 614087. [Google Scholar] [CrossRef]
- Fontanesi, L.; Calò, D.G.; Galimberti, G.; Negrini, R.; Marino, R.; Nardone, A.; Ajmone-Marsan, P.; Russo, V. A candidate gene association study for nine economically important traits in Italian Holstein cattle. Anim. Genet. 2014, 45, 576–580. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Tingzhu, Y.; Pasandideh, M.; Liang, A.; Hua, G.; Farmanullah, S.N.M.; Raza, S.H.A.; Salzano, A.; Campanile, G.; Gasparrini, B.; et al. Genetic Association of PPARGC1A Gene Single Nucleotide Polymorphism with Milk Production Traits in Italian Mediterranean Buffalo. Biomed. Res. Int. 2021, 21, 3653157. [Google Scholar] [CrossRef]
- Khatib, H.; Zaitoun, I.; Wiebelhaus-Finger, J.; Chang, Y.M.; Rosa, G.J. The association of bovine PPARGC1A and OPN genes with milk composition in two independent Holstein cattle populations. J. Dairy Sci. 2007, 90, 2966–2970. [Google Scholar] [CrossRef]
- Lindholm-Perry, A.K.; Kuehn, L.A.; Smith, T.P.; Ferrell, C.L.; Jenkins, T.G.; Freetly, H.C.; Snelling, W.M. A region on BTA14 that includes the positional candidate genes LYPLA1, XKR4 and TMEM68 is associated with feed intake and growth phenotypes in cattle(1). Anim. Genet. 2012, 43, 216–219. [Google Scholar] [CrossRef]
- Banos, G.; Clark, E.L.; Bush, S.J.; Dutta, P.; Bramis, G.; Arsenos, G.; Hume, D.A.; Psifidi, A. Genetic and genomic analyses underpin the feasibility of concomitant genetic improvement of milk yield and mastitis resistance in dairy sheep. PLoS ONE 2019, 14, e0214346. [Google Scholar] [CrossRef] [Green Version]
- Avondo, M.; Di Trana, A.; Valenti, B.; Criscione, A.; Bordonaro, S.; De Angelis, A.; Giorgio, D.; Di Gregorio, P. Leptin Gene Polymorphism in Goats Fed with Diet at Different Energy Level: Effects on Feed Intake, Milk Traits, Milk Fatty Acids Composition, and Metabolic State. Animals 2019, 9, 424. [Google Scholar] [CrossRef] [Green Version]
- Abousoliman, I.; Reyer, H.; Oster, M.; Muráni, E.; Mourad, M.; Rashed, M.A.; Mohamed, I.; Wimmers, K. Analysis of Candidate Genes for Growth and Milk Performance Traits in the Egyptian Barki Sheep. Animals 2020, 10, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trakovicka, A.; Moravcikova, N.; Minarovic, T.; Alica Navratilova, A. SNPs analyses of the bovine LEP and PIT-1 genes by multiplex PCR-RFLP method and their effect on milk performance traits in Slovak Simmental cattle. J. Cent. Eur. Agric. 2015, 16, 65–75. [Google Scholar] [CrossRef]
- Mahmoud, A.; Saleh, A.; Almealamah, N.; Ayadi, M.; Matar, A.; Abou-Tarboush, F.; Aljumaah, R.; Abouheif, M. Polymorphism of leptin gene and its association with milk traits in Najdi sheep. J. Appl. Microbiol. 2014, 8, 2953–2959. [Google Scholar]
- MacLeod, I.M.; Bowman, P.J.; Vander Jagt, C.J.; Haile-Mariam, M.; Kemper, K.E.; Chamberlain, A.J.; Schrooten, C.; Hayes, B.J.; Goddard, M.E. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genom. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, R.; MacLeod, I.M.; Daetwyler, H.D.; de Jong, G.; O’Connor, E.; Schrooten, C.; Chamberlain, A.J.; Goddard, M.E. Genome-wide fine-mapping identifies pleiotropic and functional variants that predict many traits across global cattle populations. Nat. Commun. 2021, 12, 860. [Google Scholar] [CrossRef] [PubMed]
- Lopdell, T.J.; Tiplady, K.; Couldrey, C.; Johnson, T.J.J.; Keehan, M.; Davis, S.R.; Harris, B.L.; Spelman, R.J.; Snell, R.G.; Littlejohn, M.D. Multiple QTL underlie milk phenotypes at the CSF2RB locus. Genet. Sel. Evol. 2019, 51, 3. [Google Scholar] [CrossRef] [Green Version]
- Rovadoscki, G.A.; Pertile, S.F.N.; Alvarenga, A.B.; Cesar, A.S.M.; Pértille, F.; Petrini, J.; Franzo, V.; Soares, W.V.B.; Morota, G.; Spangler, M.L.; et al. Estimates of genomic heritability and genome-wide association study for fatty acids profile in Santa Inês sheep. BMC Genom. 2018, 1, 375. [Google Scholar] [CrossRef]
- Do, D.N.; Schenkel, F.S.; Miglior, F.; Zhao, X.; Ibeagha-Awemu, E.M. Genome wide association study identifies novel potential candidate genes for bovine milk cholesterol content. Sci. Rep. 2018, 1, 13239. [Google Scholar] [CrossRef]
- Yamamoto, N.; Kashiwagi, M.; Ishihara, M.; Kojima, T.; Maturana, A.D.; Kuroda, S.; Niimi, T. Robo2 contains a cryptic binding site for neural EGFL-like (NELL) protein 1/2. J. Biol. Chem. 2019, 294, 4693–4703. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Uemoto, Y.; Kikuchi, T.; Egawa, S.; Kohira, K.; Saito, T.; Sakuma, H.; Miyashita, S.; Arata, S.; Suzuki, K. Genome-wide association studies reveal additional related loci for fatty acid composition in a Duroc pig multigenerational population. Anim. Sci. J. 2017, 10, 1482–1490. [Google Scholar] [CrossRef]
- Goshu, H.A.; Xiaoyun, W.; Chu, M.; Pengjia, B.; Xue, D.X.; Zhi, P. Novel copy number variations of the CHRM3 gene associated with gene expression and growth traits in Chinese Datong yak (Bos grunniens). J. Appl. Anim. Res. 2020, 1, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Mei, C.; Wang, H.; Liao, Q.; Khan, R.; Raza, S.H.A.; Zhao, C.; Wang, H.; Cheng, G.; Tian, W.; Li, Y.; et al. Genome-wide analysis reveals the effects of artificial selection on production and meat quality traits in Qinchuan cattle. Genomics 2019, 6, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Ibeagha-Awemu, E.M.; Peters, S.O.; Akwanji, K.A.; Imumorin, I.G.; Zhao, X. High density genome wide genotyping-by-sequencing and association identifies common and low frequency SNPs, and novel candidate genes influencing cow milk traits. Sci. Rep. 2016, 6, 31109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, H. Impact of Genetics on Meat Quality of Pigs and Beef Cattle. Ph.D. Thesis, University of Alberta, Edmonton, AB, Canada, 2019; pp. 1–275. [Google Scholar]
- Brindle, N.P.; Saharinen, P.; Alitalo, K. Signaling and functions of angiopoietin-1 in vascular protection. Circ. Res. 2006, 8, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Liu, L.; Gao, Y.; Shi, L.; Li, Y.; Liang, W.; Sun, D. Determination of genetic associations between indels in 11 candidate genes and milk composition traits in Chinese Holstein population. BMC Genet. 2019, 1, 48. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Liu, L.; Augustino, S.M.A.; Duan, T.; Hall, T.J.; MacHugh, D.E.; Dou, J.; Zhang, Y.; Wang, Y.; Yu, Y. Identification of novel molecular markers of mastitis caused by Staphylococcus aureus using gene expression profiling in two consecutive generations of Chinese Holstein dairy cattle. J. Anim. Sci. Biotechnol. 2020, 11, 98. [Google Scholar] [CrossRef]
- Pinedo, P.J.; Buergelt, C.D.; Donovan, G.A.; Melendez, P.; Morel, L.; Wu, R.; Langaee, T.Y.; Rae, D.O. Candidate gene polymorphisms (BoIFNG, TLR4, SLC11A1) as risk factors for paratuberculosis infection in cattle. Prev. Vet. Med. 2009, 4, 189–196. [Google Scholar] [CrossRef]
- Thompson-Crispi, K.A.; Sargolzaei, M.; Ventura, R.; Abo-Ismail, M.; Miglior, F.; Schenkel, F.; Mallard, B.A. A genome-wide association study of immune response traits in Canadian Holstein cattle. BMC Genom. 2014, 1, 559. [Google Scholar] [CrossRef] [Green Version]
- Knappe, A.; Hör, S.; Wittmann, S.; Fickenscher, H. Induction of a novel cellular homolog of interleukin-10, AK155, by transformation of T lymphocytes with herpesvirus saimiri. J. Virol. 2000, 8, 3881–3887. [Google Scholar] [CrossRef] [Green Version]
- Hasenauer, F.C.; Rossi, U.A.; Caffaro, M.E.; Raschia, M.A.; Maurizio, E.; Poli, M.A.; Rossetti, C.A. Association of TNF rs668920841 and INRA111 polymorphisms with caprine brucellosis: A case-control study of candidate genes involved in innate immunity. Genomics 2020, 6, 3925–3932. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chou, Y.H.; Hsieh, N.T.; Chen, H.H.; Lee, M.F. MED28 regulates MEK1-dependent cellular migration in human breast cancer cells. J. Cell Physiol. 2012, 12, 3820–3827. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Ju, Z.; Wang, J.; Li, Q.; Huang, J.; Zhang, A.; Zhong, J.; Wang, C. Single nucleotide polymorphisms, haplotypes and combined genotypes of LAP3 gene in bovine and their association with milk production traits. Mol. Biol. Rep. 2011, 6, 4053–4061. [Google Scholar] [CrossRef] [PubMed]
- Kloetzel, P.M.; Ossendorp, F. Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. Curr. Opin. Immunol. 2004, 1, 76–81. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Traits | Number | Mean ± SD | CV(%) | Min-Max | h2 |
|---|---|---|---|---|---|
| MY (g) | 15,008 | 1318 ± 552 | 41.91 | 62–4140 | 0.10 |
| FY (g) | 15,008 | 91.06 ± 34.91 | 38.34 | 3.91–393.53 | 0.06 |
| FAT (%) | 15,008 | 7.08 ± 1.09 | 15.41 | 2.53–10.80 | 0.11 |
| PY (g) | 15,008 | 75.43 ± 29.81 | 39.52 | 2.93–238.98 | 0.09 |
| PROT (%) | 15,008 | 5.80 ± 0.65 | 11.16 | 2.14–8.10 | 0.15 |
| SCS | 15,008 | 2.67 ± 0.72 | 0.27 | 1–5.31 | 0.04 |
| Trait | OAR | Start (bp) | Stop (bp) | % VE | Genes |
|---|---|---|---|---|---|
| MY | 22 | 5,250,187 | 6,225,066 | 0.396 | PCDH15, U6 |
| 3 | 214,344,054 | 215,239,751 | 0.375 | EPS8, PTPRO, RERG, PDE6H, ARHGDIB, ERP27, MGP | |
| 27 | 106,515,599 | 107,465,396 | 0.373 | - | |
| 9 | 31,897,584 | 32,871,881 | 0.372 | C9H8orf76, TBC1D31, DERL1, ZHX2 | |
| 9 | 54,661,246 | 55,632,383 | 0.359 | TMEM70, LY96, JPH1, GDAP1, MIR2052 | |
| 3 | 212,632,779 | 213,605,435 | 0.351 | LMO3, MGST1 | |
| 6 | 47,499,788 | 48,496,456 | 0.337 | PPARGC1A | |
| 6 | 40,445,167 | 41,445,859 | 0.311 | LYPLA1, FAM13A, NAP1L5 | |
| 3 | 107,748,212 | 108,707,604 | 0.310 | LYG2, MRPL30, MITD1, C2orf15, MYH9, TSGA10, MGAT4A | |
| 4 | 92,470,191 | 93,383,584 | 0.291 | KCND2, LEP, TSPAN12 |
| Trait | Chr | Start (bp) | Stop (bp) | % VE | Genes |
|---|---|---|---|---|---|
| FY | 11 | 17,769,576 | 18,742,505 | 0.551 | GOSR2, RPRML, LYZL6, RDM1, PLEKHM1, ARHGAP27, MAP3K14, FMNL1, HEXIM2, HEXIM1, ACBD4, PLCD3, NMT1 |
| 3 | 151,003,018 | 151,972,475 | 0.483 | ANO6, DBX2, NELL2 | |
| 6 | 3,644,480 | 4,610,354 | 0.420 | BBS7, CCNA2, EXOSC9, SMIM43 | |
| 19 | 5,602,339 | 6,570,519 | 0.404 | GADL1, OSBPL10, STT3B | |
| 3 | 229,350,131 | 230,350,131 | 0.376 | ATP6V1E1, TUBA8, CDC42EP1, LGALS2, GGA1, CACNA1C, U6, PDXP | |
| 2 | 56,504,189 | 57,464,796 | 0.373 | - | |
| 7 | 37,844,731 | 38,839,931 | 0.373 | SNAP23, LRRC57, HAUS2, CDAN1, TTBK2, UBR1, TMEM62, CCNDBP1, EPB42 | |
| 5 | 47,771,920 | 48,762,985 | 0.368 | TCF7, CDKL3, UBE2B, JADE2, SAR1B, SEC24A, CAMLG, DDX46, C5orf24 | |
| 1 | 156,102,645 | 157,082,096 | 0.357 | ROBO2, U2 | |
| 13 | 81,534,944 | 82,519,944 | 0.301 | UBE2V1, CEBPB, PTPN1, PARD6B, BCAS4, DPM1, KCNG1, MOCS3 | |
| FAT% | 6 | 3,563,877 | 4,559,472 | 0.649 | NAF1, BBS7, CCNA2, EXOSC9, SMIM43 |
| 26 | 3,879,792 | 4,875,104 | 0.619 | - | |
| 8 | 82,898,823 | 83,853,076 | 0.571 | ESR1, SYNE1, MYCT1, VIP | |
| 8 | 79,925,767 | 80,905,873 | 0.550 | UST, TAB2, ZC3H12D, PPIL4, GINM1, KATNA1, LATS1, NUP43, PCMT1, LRP11 | |
| 26 | 700,916 | 1,677,676 | 0.540 | DLGAP2, CLN8, CETN2, KBTBD11, MYOM2 | |
| 9 | 53,991,923 | 54,979,070 | 0.533 | RDH10, ELOC, TMEM70, LY96 | |
| 2 | 124,285,685 | 125,280,477 | 0.527 | FAM168B, PLEKHB2 | |
| 25 | 12,322,443 | 13,278,443 | 0.483 | ZNF248, BMS1, CHRM3, ZNF33B | |
| 7 | 50,459,248 | 51,446,200 | 0.470 | RORA, ICE2, ANXA2 | |
| 25 | 45,303,771 | 46,293,771 | 0.419 | DRGX, ERCC6, SLC18A3, C25H10orf53, OGDHL, PARG, TIMM23B, SNORA74, MARCHF8, ZFAND4 |
| Trait | Chr | Start (bp) | Stop (bp) | % VE | Genes |
|---|---|---|---|---|---|
| PY | 7 | 37,844,731 | 38,839,931 | 0.518 | SNAP23, LRRC57, HAUS2, CDAN1, TTBK2, UBR1, TMEM62, CCNDBP1 |
| 8 | 14,481,631 | 15,471,954 | 0.505 | HDDC2, TPD52L1, RNF217, NKAIN2 | |
| 13 | 12,488,726 | 13,487,350 | 0.449 | SFMBT2, ITIH5, ITIH2, ATP5F1C, TAF3, GATA3 | |
| 2 | 111,987,606 | 112,986,964 | 0.398 | KIF13B, MSRA, PRSS51, PRSS55, PINX1 | |
| 14 | 25,500,650 | 26,497,592 | 0.397 | MT1A, MT1C, MT2, OGFOD1, OGFOD1, NUDT21, AMFR, GNAO1, CES5A | |
| 2 | 63,313,028 | 64,303,381 | 0.393 | VPS13A, FOXB2, GCNT1, RFK, PCSK5 | |
| 2 | 59,313,028 | 60,303,381 | 0.388 | - | |
| 1 | 156,123,514 | 157,114,945 | 0.379 | ROBO2, U2 | |
| 9 | 77,706,323 | 78,625,358 | 0.354 | ANGPT1, ABRA, OXR1 | |
| 3 | 151,003,018 | 151,972,475 | 0.305 | ANO6, DBX2, NELL2 | |
| PROT% | 13 | 77,917,677 | 78,910,066 | 0.507 | DNTTIP1, TNNC2, SNX21, ACOT8, CTSA, PLTP, PCIF1, ZNF335, MMP9 |
| 2 | 80,632,041 | 81,604,796 | 0.452 | KDM4C, DMAC1 | |
| 17 | 29,494,464 | 30,441,072 | 0.420 | - | |
| 1 | 38,670,059 | 39,649,763 | 0.368 | TM2D1, PATJ, KANK4 | |
| 7 | 37,916,711 | 38,872,804 | 0.357 | CDAN1, TTBK2, UBR1, TMEM62, EPB42 | |
| 3 | 179,367,669 | 180,265,170 | 0.357 | - | |
| 5 | 72,168,942 | 73,159,451 | 0.342 | SOX30, THG1L, LSM11, CLINT1 | |
| 2 | 134,606,095 | 135,598,790 | 0.337 | TTC21B, GALNT3 | |
| 2 | 154,466,090 | 155,460,561 | 0.332 | ZNF804A | |
| 13 | 76,376,357 | 77,356,228 | 0.332 | YWHAB, PABPC1L, STK4, KCNS1, MATN4 |
| Trait | Chr | Start (bp) | Stop (bp) | % VE | Genes |
|---|---|---|---|---|---|
| SCS | 1 | 33,955,437 | 34,931,948 | 0.758 | U6, OMA1, TACSTD2 |
| 6 | 41,728,563 | 42,727,105 | 0.677 | LAP3, MED28, FAM184B, DCAF16, NCAPG, LCORL | |
| 9 | 34,510,191 | 35,480,917 | 0.562 | PRKDC, MCM4, EFCAB1, SNAI2, PPDPFL | |
| 1 | 32,365,838 | 33,350,564 | 0.557 | PLPP3, PRKAA2, C8A, DAB1 | |
| 7 | 38,236,462 | 39,205,426 | 0.485 | UBR1, TMEM62, CCNDBP1, EPB42 | |
| 3 | 213,347,420 | 214,351,547 | 0.484 | LMO3, MGST1, SLC15A5, PEX26, STRAP | |
| 1 | 49,775,153 | 50,755,499 | 0.463 | NEGR1 | |
| 3 | 161,701,025 | 162,705,420 | 0.421 | CPM, SLC35E3, NUP107, RAP1B, MDM1, IL22, IL26, IFNG | |
| 9 | 28,511,250 | 29,492,867 | 0.420 | LRATD2 | |
| 1 | 61,734,830 | 62,648,480 | 0.413 | ADGRL2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammadi, H.; Farahani, A.H.K.; Moradi, M.H.; Mastrangelo, S.; Di Gerlando, R.; Sardina, M.T.; Scatassa, M.L.; Portolano, B.; Tolone, M. Weighted Single-Step Genome-Wide Association Study Uncovers Known and Novel Candidate Genomic Regions for Milk Production Traits and Somatic Cell Score in Valle del Belice Dairy Sheep. Animals 2022, 12, 1155. https://doi.org/10.3390/ani12091155
Mohammadi H, Farahani AHK, Moradi MH, Mastrangelo S, Di Gerlando R, Sardina MT, Scatassa ML, Portolano B, Tolone M. Weighted Single-Step Genome-Wide Association Study Uncovers Known and Novel Candidate Genomic Regions for Milk Production Traits and Somatic Cell Score in Valle del Belice Dairy Sheep. Animals. 2022; 12(9):1155. https://doi.org/10.3390/ani12091155
Chicago/Turabian StyleMohammadi, Hossein, Amir Hossein Khaltabadi Farahani, Mohammad Hossein Moradi, Salvatore Mastrangelo, Rosalia Di Gerlando, Maria Teresa Sardina, Maria Luisa Scatassa, Baldassare Portolano, and Marco Tolone. 2022. "Weighted Single-Step Genome-Wide Association Study Uncovers Known and Novel Candidate Genomic Regions for Milk Production Traits and Somatic Cell Score in Valle del Belice Dairy Sheep" Animals 12, no. 9: 1155. https://doi.org/10.3390/ani12091155
APA StyleMohammadi, H., Farahani, A. H. K., Moradi, M. H., Mastrangelo, S., Di Gerlando, R., Sardina, M. T., Scatassa, M. L., Portolano, B., & Tolone, M. (2022). Weighted Single-Step Genome-Wide Association Study Uncovers Known and Novel Candidate Genomic Regions for Milk Production Traits and Somatic Cell Score in Valle del Belice Dairy Sheep. Animals, 12(9), 1155. https://doi.org/10.3390/ani12091155