
Comprehensive Analysis of Differentially Expressed CircRNAs in the Ovaries of Low- and High-Fertility Sheep
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Laboratory Animal Samples and Collection
2.3. Total RNA Library Construction and Sequencing
2.4. Raw Data Quality Control and Transcript Assembly
2.5. CircRNA Identification
2.6. Differential Expression Analysis
2.7. Functional Enrichment Analysis of CircRNAs
2.8. Construction and TF Analysis for the Construction of Protein–Protein Interaction (PPI) Networks
2.9. Construction of DEcircRNA-DEmiRNA-DEmRNA Networks
2.10. Ovarian Granulosa Cell Isolation and Culture
2.11. RNA Pull-Down Assays
2.12. RNA Library Preparation and Sequencing
2.13. RNA Pull-Down Data Analysis
3. Results
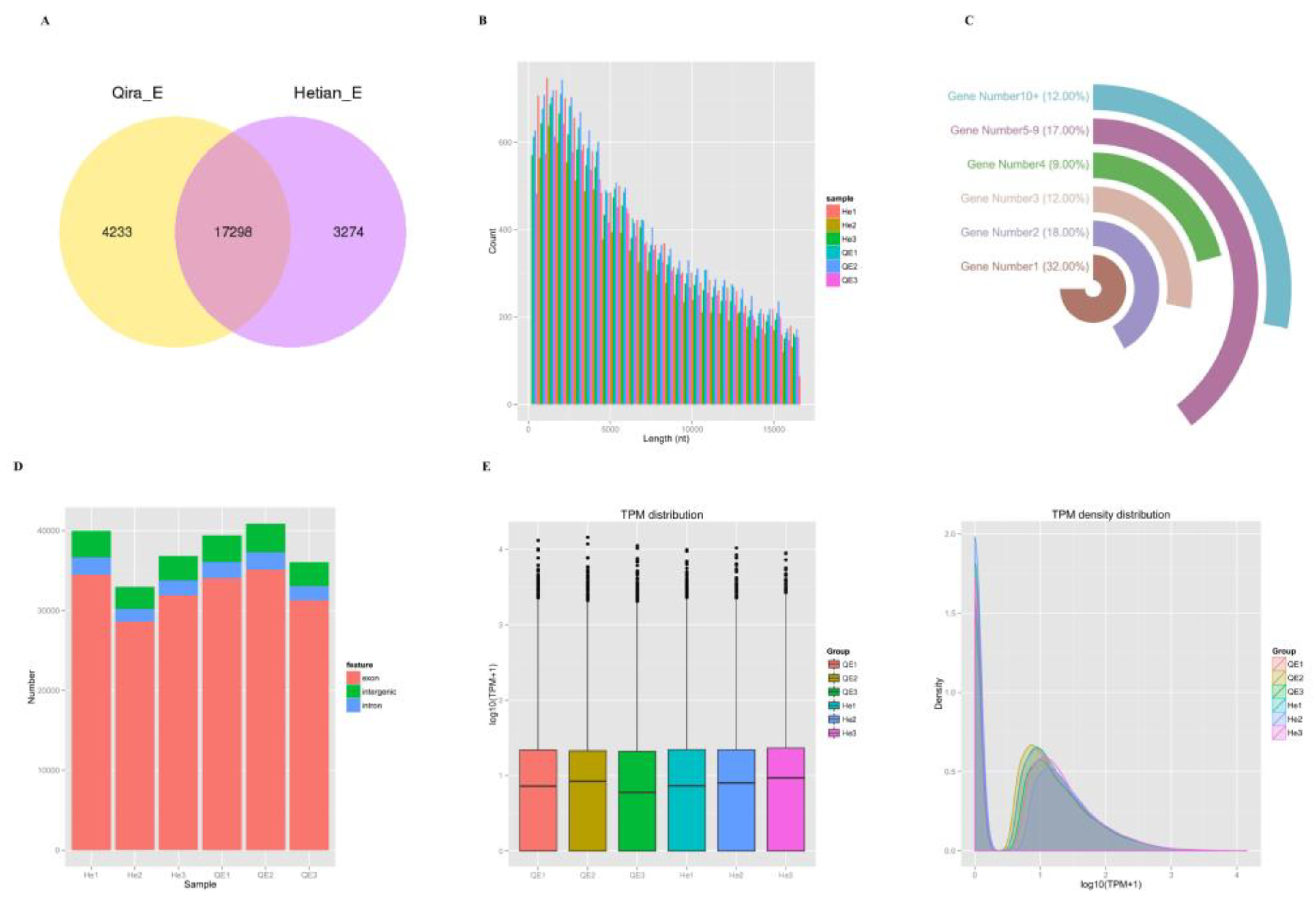
3.1. Genomic Characteristics of CircRNA in Sheep Ovaries
3.2. Analysis of DEcircRNAs
3.3. DEcircRNA Functional Enrichment Analysis
3.4. PPI Network Analysis of Host Genes
3.5. TFs Prediction and Analysis
3.6. Construction of a DEcircRNA-DEmiRNA-DEmRNA Regulatory Network
3.7. Novel-circ-0040512 May Spongify oar-miR-125b in Sheep Ovarian Granulosa Cells
3.7.1. RNA Pull-Down Calling oar-miR-125b-Binding CircRNA
3.7.2. Functional Analysis of oar-miR-125b Potential Binding DEcircRNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davis, G.H.; Montgomery, G.W.; Allison, A.J.; Kelly, R.W.; Bray, A.R. Segregation of a major gene influencing fecundity in progeny of Booroola sheep. N. Z. J. Agric. Res. 1982, 25, 525–529. [Google Scholar] [CrossRef]
- Shimasaki, S.; Zachow, R.J.; Li, D.M.; Kim, H.; Iemura, S.; Ueno, N.; Sampath, K.; Chang, R.J.; Erickson, G.F. A functional bone morphogenetic protein system in the ovary. Proc. Natl. Acad. Sci. USA 1999, 96, 7282–7287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Montiel, W.; Colli-Dula, R.C.; Ramon-Ugalde, J.P.; Martinez-Nunez, M.A.; Zamora-Bustillos, R. RNA-seq Transcriptome Analysis in Ovarian Tissue of Pelibuey Breed to Explore the Regulation of Prolificacy. Genes 2019, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Jia, R.; Ying, S.; Wang, Z.; Wang, F. Analysis of genes that influence sheep follicular development by different nutrition levels during the luteal phase using expression profiling. Anim. Genet. 2016, 47, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, K.; Peippo, J.; Honkatukia, M.; Seppala, A.; Rautiainen, J.; Ghanem, N.; Hamama, T.M.; Crowe, M.A.; Andersson, M.; Li, M.H.; et al. Integrated ovarian mRNA and miRNA transcriptome profiling characterizes the genetic basis of prolificacy traits in sheep (Ovis aries). BMC Genom. 2018, 19, 17. [Google Scholar] [CrossRef] [Green Version]
- Song, P.Y.; Yue, Q.X.; Fu, Q.; Li, X.Y.; Li, X.J.; Zhou, R.Y.; Chen, X.Y.; Tao, C.Y. Integrated analysis of miRNA-mRNA interaction in ovaries of Turpan Black Sheep during follicular and luteal phases. Reprod. Domest. Anim. 2021, 56, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Chen, H.Y.; Jia, B.; Han, G.H.; Zhang, Y.S.; Zeng, X.C. Characterization and expression analysis of microRNAs in Qira black sheep and Hetian sheep ovaries using Solexa sequencing. Genet. Mol. Res. 2015, 14, 7356–7367. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Lu, M. Circular RNA: Functions, applications and prospects. ExRNA 2020, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Zong, Z.H.; Liu, Y.; Chen, S.; Wang, L.L.; Zhao, Y. circPUM1 Promotes Tumorigenesis and Progression of Ovarian Cancer by Sponging miR-615-5p and miR-6753-5p. Mol. Ther.-Nucleic Acids 2019, 18, 882–892. [Google Scholar] [CrossRef]
- Liu, X.R.; Zhang, L.; Yang, L.C.; Cui, J.Z.; Che, S.C.; Liu, Y.X.; Han, J.C.; An, X.P.; Cao, B.Y.; Song, Y.X. miR-34a/c induce caprine endometrial epithelial cell apoptosis by regulating circ-8073/CEP55 via the RAS/RAF/MEK/ERK and PI3K/AKT/mTOR pathways. J. Cell. Physiol. 2020, 235, 10051–10067. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.X.; Yuan, B.; Su, M.T.; Zheng, Y.; Zhang, J.Y.; Han, D.X.; Wang, H.Q.; Huang, Y.J.; Jiang, H.; Zhang, J.B. Identification of Circular RNAs in the Anterior Pituitary in Rats Treated with GnRH. Animals 2021, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.C.; Xu, B.; Wu, J. Circular RNA expression profiles of mouse ovaries during postnatal development and the function of circular RNA epidermal growth factor receptor in granulosa cells. Metab.-Clin. Exp. 2018, 85, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.F.; He, X.Y.; Zhang, X.S.; Zhang, J.L.; Guo, X.F.; Sun, W.; Chu, M.X. Analysis of Expression Profiles of CircRNA and MiRNA in Oviduct during the Follicular and Luteal Phases of Sheep with Two Fecundity (FecB Gene) Genotypes. Animals 2021, 11, 2826. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.J.J.; Wang, H.B.; Li, Z.; Li, F.W.; Liang, L.L.; Zou, Y.R.; Shen, H.; Li, J.; Xia, Y.; Cheng, Z.J.; et al. Circular RNA ACTN4 promotes intrahepatic cholangiocarcinoma progression by recruiting YBX1 to initiate FZD7 transcription. J. Hepatol. 2022, 76, 135–147. [Google Scholar] [CrossRef]
- Li, C.Y.; Li, X.Y.; Ma, Q.M.; Zhang, X.Y.; Cao, Y.; Yao, Y.; You, S.; Wang, D.W.; Quan, R.Z.; Hou, X.X.; et al. Genome-wide analysis of circular RNAs in prenatal and postnatal pituitary glands of sheep. Sci. Rep. 2017, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.J.; Chen, X.Y.; Liu, M.H.; Zhang, L.M.; Ma, X.F.; Tian, S.J. Differential Expression and Functional Analysis of CircRNA in the Ovaries of Low and High Fecundity Hanper Sheep. Animals 2021, 11, 20. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, X.R.; Che, S.C.; Cui, J.Z.; Ma, X.N.; An, X.P.; Cao, B.Y.; Song, Y.X. Endometrial Epithelial Cell Apoptosis Is Inhibited by a ciR8073-miR181a-Neurotensis Pathway during Embryo Implantation. Mol. Ther.-Nucleic Acids 2019, 14, 262–273. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.B.; Tang, J.S.; He, X.Y.; Zhu, M.X.; Gan, S.Q.; Guo, X.F.; Zhang, X.S.; Zhang, J.L.; Hu, W.P.; Chu, M.X. Comparative Transcriptomics Identify Key Hypothalamic Circular RNAs that Participate in Sheep (Ovis aries) Reproduction. Animals 2019, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Shen, H.; Jia, B.; Zhang, Y.S.; Wang, X.H.; Zeng, X.C. Differential Gene Expression in Ovaries of Qira Black Sheep and Hetian Sheep Using RNA-Seq Technique. PLoS ONE 2015, 10, 15. [Google Scholar] [CrossRef]
- Li, X.D.; Yao, X.L.; Xie, H.Q.; Zhang, G.M.; Deng, M.T.; Deng, K.P.; Gao, X.X.; Bao, Y.J.; Li, K.; Wang, F. PPP2R2A affects embryonic implantation by regulating the proliferation and apoptosis of Hu sheep endometrial stromal cells. Theriogenology 2021, 176, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xing, Q.; Mao, J.Y.; Sun, H.K.; Teng, W.P.; Shan, Z.Y. miRNA-125b-5p Suppresses Hypothyroidism Development by Targeting Signal Transducer and Activator of Transcription 3. Med. Sci. Monitor 2018, 24, 5041–5049. [Google Scholar] [CrossRef]
- Chen, X.; Chen, H.Y.; Jiang, S.; Shen, H.; Li, C.C.; Zeng, X.C. Expression profile analysis of microRNAs during the oestrous cycle of Qira black sheep. Thai J. Vet. Med. 2021, 51, 451–459. [Google Scholar]
- Pan, X.C.; Gong, W.T.; He, Y.T.; Li, N.A.; Zhang, H.; Zhang, Z.; Li, J.Q.; Yuan, X.L. Ovary-derived circular RNAs profile analysis during the onset of puberty in gilts. BMC Genom. 2021, 22, 12. [Google Scholar] [CrossRef]
- Wu, X.M.; Zhen, H.M.; Liu, Y.; Li, L.; Luo, Y.Z.; Liu, X.; Li, S.B.; Hao, Z.Y.; Li, M.N.; Hu, L.Y.; et al. Tissue-Specific Expression of Circ_015343 and Its Inhibitory Effect on Mammary Epithelial Cells in Sheep. Front. Vet. Sci. 2022, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS ONE 2012, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Liu, X.; Xue, Y.; Gong, W.; Ma, J.; Xi, Z.; Que, Z.; Liu, Y. TTBK2 circular RNA promotes glioma malignancy by regulating miR-217/HNF1β/Derlin-1 pathway. J. Hematol. Oncol. 2017, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.L.; Zhang, J.W.; Tian, Y.H.; Gao, Y.; Dong, X.; Chen, W.B.; Yuan, X.N.; Yin, W.N.; Xu, J.J.; Chen, K.; et al. CircRNA inhibits DNA damage repair by interacting with host gene. Mol. Cancer 2020, 19, 19. [Google Scholar] [CrossRef]
- Luo, X.Y.; Chang, H.M.; Yi, Y.Y.; Leung, P.C.K.; Sun, Y.P. Bone morphogenetic protein 2 upregulates SERPINE2 expression through noncanonical SMAD2/3 and p38 MAPK signaling pathways in human granulosa-lutein cells. Faseb J. 2021, 35, 18. [Google Scholar] [CrossRef]
- Dos Santos, E.C.; Lalonde-Larue, A.; Antoniazzi, A.Q.; Barreta, M.H.; Price, C.A.; Goncalves, P.B.D.; Portela, V.M.; Zamberlam, G. YAP signaling in preovulatory granulosa cells is critical for the functioning of the EGF network during ovulation. Mol. Cell. Endocrinol. 2022, 541, 12. [Google Scholar] [CrossRef]
- Tang, A.; Shi, P.L.; Song, A.Y.; Zou, D.Y.; Zhou, Y.; Gu, P.Y.; Huang, Z.; Wang, Q.H.; Lin, Z.Y.; Gao, X. PP2A regulates kinetochore-microtubule attachment during meiosis I in oocyte. Cell Cycle 2016, 15, 1450–1461. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.A.; He, J.C.; Fan, D.D.; Gu, Y.; Wang, J.Y.; Li, H.M.; Zhu, X.X.; Du, Y.; Tian, Y.; Liu, B.Y.; et al. Circular RNA circTmem241 drives group III innate lymphoid cell differentiation via initiation of Elk3 transcription. Nat. Commun. 2022, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.X.; Zhou, T.P.; Shao, L.; Zhang, B.; Liu, K.L.; Gao, C.; Gao, L.; Liu, J.Y.; Cui, Y.G.; Chian, R.C. Gene expression profiles in mouse cumulus cells derived from in vitro matured oocytes with and without blastocyst formation. Gene Expr. Patterns 2017, 25–26, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Ning, H.J.; Deng, J.Y.; Chen, F.; Liu, Y.F.; Kong, D.L.; Shan, L.N.; Zhang, Z.; Hu, T.H. Beta-arrestin1 inhibits hypoxic injury-induced autophagy in human pulmonary artery endothelial cells via the Akt/mTOR signaling pathway. Int. J. Biochem. Cell Biol. 2020, 125, 12. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.L.; Zhang, T.; Kong, Z.Z.; Mai, X.; Lan, C.X.; Chen, D.; Liu, Y.; Zeng, Z.W.; Cai, C.; Deng, T.; et al. Beta-arrestin1 promotes epithelial-mesenchymal transition via modulating GSK-3 beta/beta-catenin pathway in prostate cancer cells. Biochem. Biophys. Res. Commun. 2016, 479, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, X.Q.; Qiukai, E.; Shang, Y.X.; Zhang, X.Q.; Liu, S.T.; Zhang, X.S. Long non-coding RNA Xist regulates oocyte loss via suppressing miR-23b-3p/miR-29a-3p maturation and upregulating STX17 in perinatal mouse ovaries. Cell Death & Disease 2021, 12, 14. [Google Scholar] [CrossRef]
- Guo, Y.; Wu, Y.B.; Shi, J.H.; Zhuang, H.; Ci, L.; Huang, Q.; Wan, Z.P.; Yang, H.; Zhang, M.J.; Tan, Y.T.; et al. miR-29a/b(1) Regulates the Luteinizing Hormone Secretion and Affects Mouse Ovulation. Front. Endocrinol. 2021, 12, 18. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.D.; Zhou, X.Y.; Chen, S.L.; Chen, X.; Zhe, J.; Zhang, J.; Zhang, Q.Y.; Chen, Y.X. MiR-29a regulates the proliferation, aromatase expression, and estradiol biosynthesis of human granulosa cells in polycystic ovary syndrome. Mol. Cell. Endocrinol. 2019, 498, 10. [Google Scholar] [CrossRef]
- Xu, M.Q.; Jiang, H.; Zhang, L.Q.; Sun, X.L.; Luo, D.; Fu, Y.; Gao, Y.; Yuan, B.; Zhang, J.B. MiR-29b affects the secretion of PROG and promotes the proliferation of bovine corpus luteum cells. PLoS ONE 2018, 13, 14. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.B.; Wei, X.J.; Wu, B.B.; Yuan, C.H.; Li, C.; Dai, Y.D.; Chen, J.M.; Zhou, F.; Lin, X.; Zhang, S.Y. Molecular Signatures Correlated With Poor IVF Outcomes: Insights From the mRNA and lncRNA Expression of Endometriotic Granulosa Cells. Front. Endocrinol. 2022, 13, 13. [Google Scholar] [CrossRef]
- Mihalas, B.P.; Western, P.S.; Loveland, K.L.; McLaughlin, E.A.; Holt, J.E. Changing expression and subcellular distribution of karyopherins during murine oogenesis. Reproduction 2015, 150, 485–496. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Wang, J.J.; Zhang, P.P.; Hao, H.S.; Pang, Y.W.; Wang, H.Y.; Du, W.H.; Zhao, S.J.; Ruan, W.M.; Zou, H.Y.; et al. Oocyte IVM or vitrification significantly impairs DNA methylation patterns in blastocysts as analysed by single-cell whole-genome methylation sequencing. Reprod. Fertil. Dev. 2020, 32, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Song, Y.; Yan, S.; Cao, M.R.; Huang, J.; Jia, D.X.; Liu, Y.C.; Zhang, S.; Fan, W.N.; Cai, L.; et al. CUEDC1 inhibits epithelial-mesenchymal transition via the T beta RI/Smad signaling pathway and suppresses tumor progression in non-small cell lung cancer. Aging-US 2020, 12, 20047–20068. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.Y.; Zhu, L.P.; Shen, H.; Lu, J.F.; Zou, Q.Y.; Huang, C.; Li, H.; Huang, B.X. Exosomal miRNA-17-5p derived from human umbilical cord mesenchymal stem cells improves ovarian function in premature ovarian insufficiency by regulating SIRT7. Stem Cells 2020, 38, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.N.; Wang, L.; Wang, L.; Chen, Y.R.; Li, F.G. miR-17-5p affects porcine granulosa cell growth and oestradiol synthesis by targeting E2F1 gene. Reprod. Domest. Anim. 2019, 54, 1459–1469. [Google Scholar] [CrossRef]
- Efimenko, E.; Padua, M.B.; Manuylov, N.L.; Fox, S.C.; Morse, D.A.; Tevosian, S.G. The transcription factor GATA4 is required for follicular development and normal ovarian function. Dev. Biol. 2013, 381, 144–158. [Google Scholar] [CrossRef] [Green Version]
- Manuylov, N.L.; Smagulova, F.O.; Leach, L.; Tevosian, S.G. Ovarian development in mice requires the GATA4-FOG2 transcription complex. Development 2008, 135, 3731–3743. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.; Wu, Y.G.; Gossen, J.; Zhou, P.; Stocco, C. Loss of GATA-6 and GATA-4 in Granulosa Cells Blocks Folliculogenesis, Ovulation, and Follicle Stimulating Hormone Receptor Expression Leading to Female Infertility. Endocrinology 2012, 153, 2474–2485. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Reads | Clean Reads | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|
| QE1 | 78,346,928 | 76,379,904 | 0.03 | 94.93 | 88.52 | 61.88 |
| QE2 | 69,340,482 | 67,725,550 | 0.03 | 95.06 | 88.78 | 61.16 |
| QE3 | 74,602,968 | 73,065,816 | 0.04 | 94.67 | 88.03 | 62.52 |
| HE1 | 79,269,182 | 77,666,732 | 0.03 | 94.94 | 88.51 | 62.62 |
| HE2 | 75,472,380 | 73,136,040 | 0.03 | 95.6 | 89.99 | 62.7 |
| HE3 | 72,610,104 | 70,164,462 | 0.03 | 95.81 | 90.38 | 61.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Chen, H.; Zhang, Y.; Jiang, S.; Zeng, X.; Shen, H. Comprehensive Analysis of Differentially Expressed CircRNAs in the Ovaries of Low- and High-Fertility Sheep. Animals 2023, 13, 236. https://doi.org/10.3390/ani13020236
Wang J, Chen H, Zhang Y, Jiang S, Zeng X, Shen H. Comprehensive Analysis of Differentially Expressed CircRNAs in the Ovaries of Low- and High-Fertility Sheep. Animals. 2023; 13(2):236. https://doi.org/10.3390/ani13020236
Chicago/Turabian StyleWang, Jinglei, Hanying Chen, Yongsheng Zhang, Song Jiang, Xiancun Zeng, and Hong Shen. 2023. "Comprehensive Analysis of Differentially Expressed CircRNAs in the Ovaries of Low- and High-Fertility Sheep" Animals 13, no. 2: 236. https://doi.org/10.3390/ani13020236
APA StyleWang, J., Chen, H., Zhang, Y., Jiang, S., Zeng, X., & Shen, H. (2023). Comprehensive Analysis of Differentially Expressed CircRNAs in the Ovaries of Low- and High-Fertility Sheep. Animals, 13(2), 236. https://doi.org/10.3390/ani13020236