The Role of the Gut Microbiome in Nonalcoholic Fatty Liver Disease


Abstract
:1. Introduction
2. NAFLD/NASH Epidemiology
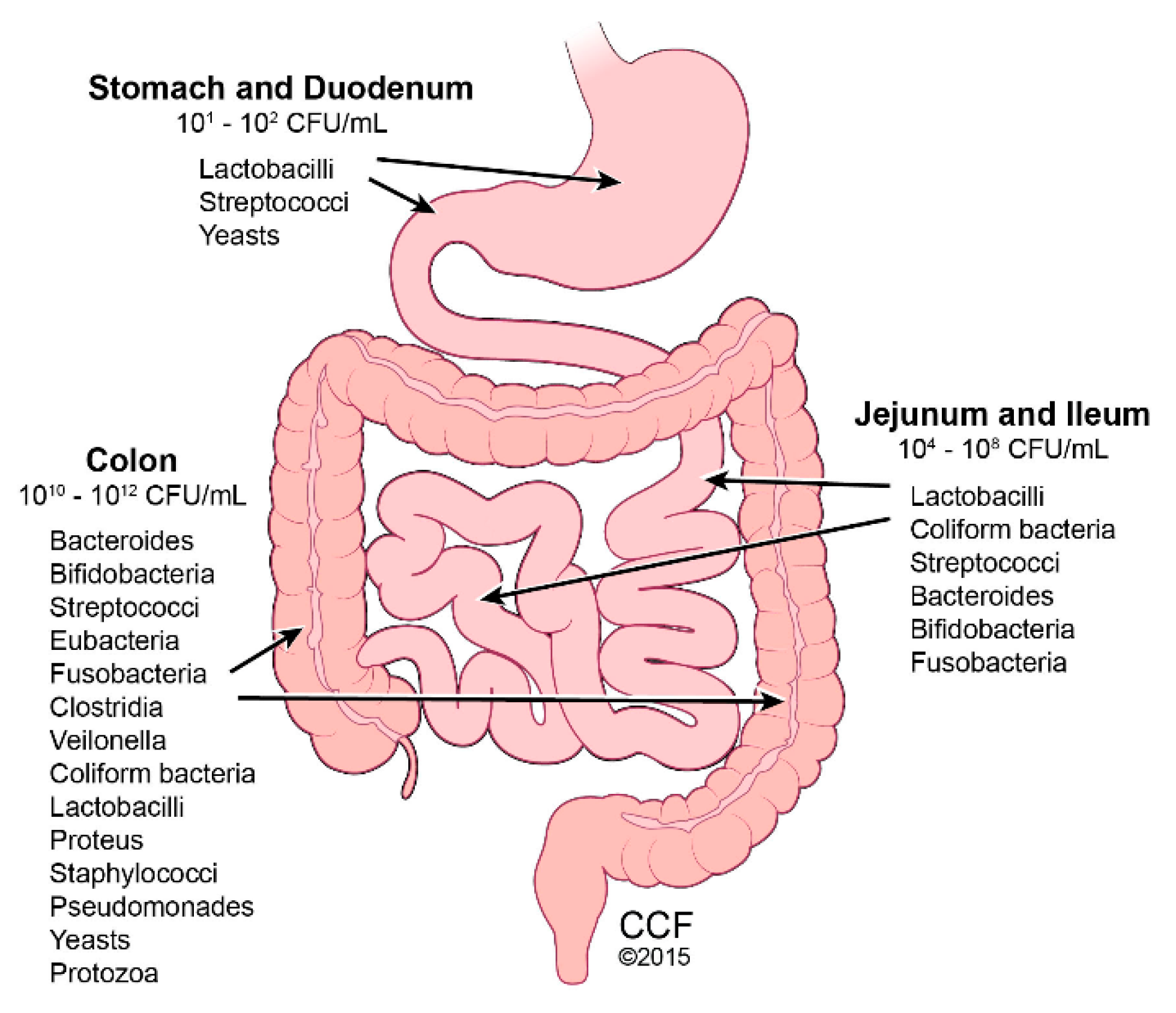
3. Gut Microbiome
4. Gut Dysbiosis and Bile Acids
5. NAFLD Pathophysiology
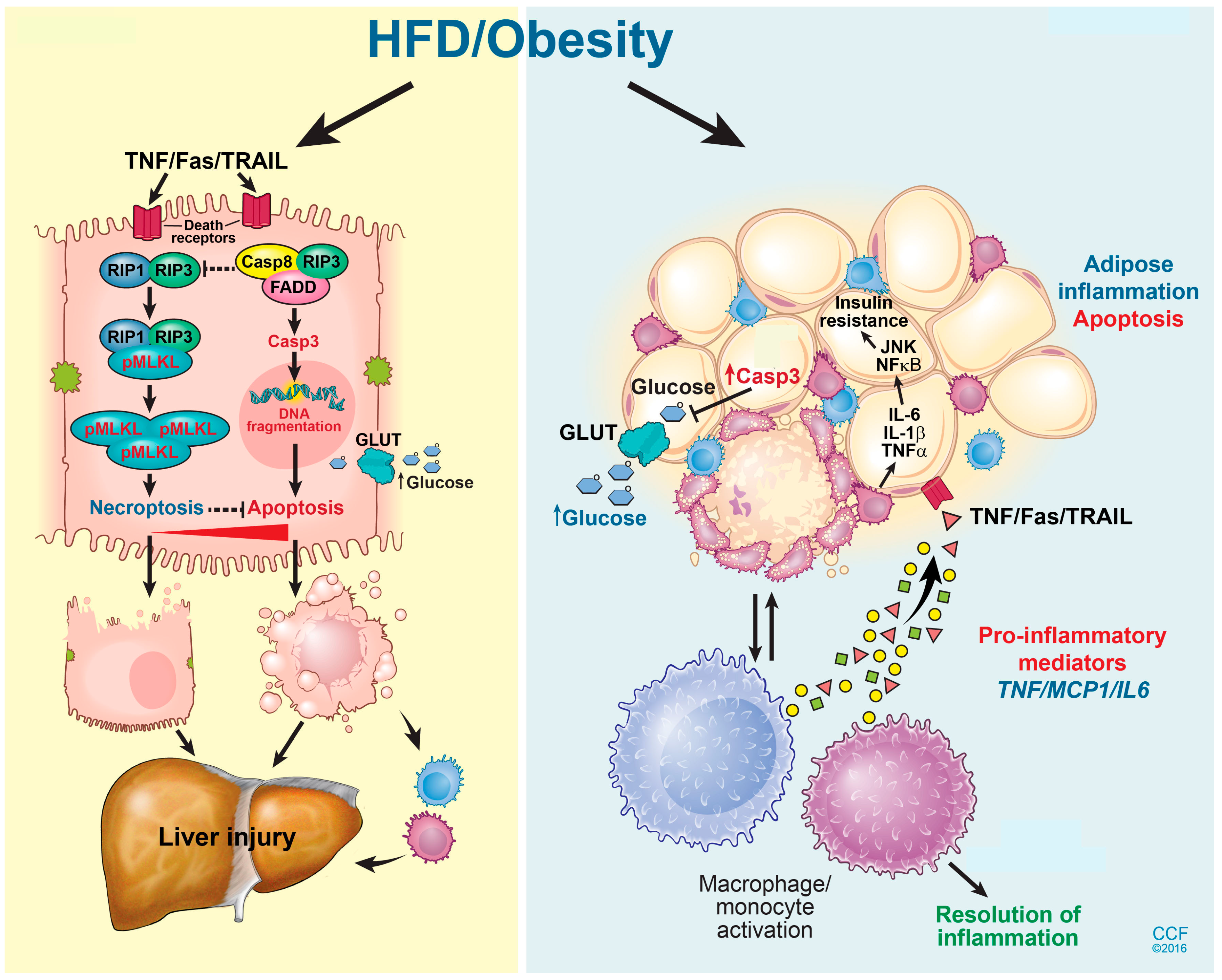
6. Adipose Tissue Dysfunction and NAFLD
7. CYP2E1 and NAFLD
8. Modes of Hepatic Cell Death in NAFLD
9. NAFLD Treatment: The Role of Gut Microbiome Manipulation
10. Summary
Funding
Conflicts of Interest
References
- Petäjä, E.M.; Yki-Järvinen, H. Definitions of Normal Liver Fat and the Association of Insulin Sensitivity with Acquired and Genetic NAFLD-A Systematic Review. Int. J. Mol. Sci. 2016, 17, 633. [Google Scholar] [CrossRef]
- Martin, F.P.; Sprenger, N.; Montoliu, I.; Rezzi, S.; Kochhar, S.; Nicholson, J.K. Metabonomics research articles. J. Proteome Res. 2010, 9, 5284–5295. [Google Scholar] [CrossRef] [PubMed]
- Than, N.N.; Newsome, P.N. A concise review of non-alcoholic fatty liver disease. Atherosclerosis 2015, 239, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. 2015, 47, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Makhlouf, H.R. Histology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in Adults and Children. Clin. Liver Dis. 2016, 20, 293–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.T.; Kleiner, D.E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism 2016, 65, 1080–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, J.P.; Pitts, A.; Younossi, Z.M. Increased overall mortality and liver-related mortality in non-alcoholic fatty liver disease. J. Hepatol. 2008, 49, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Adams, L.A.; Canbay, A.; Syn, W.K. Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology 2014, 59, 1174–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanWagner, L.B.; Rinella, M.E. Extrahepatic Manifestations of Nonalcoholic Fatty Liver Disease. Curr. Hepatol. Rep. 2016, 15, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Zaza, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Bonora, E.; Targher, G. Nonalcoholic fatty liver disease increases risk of incident chronic kidney disease: A systematic review and meta-analysis. Metabolism. Clin. Exp. 2018, 79, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, P.K.C.; Kabbany, M.N.; Nobili, V.; Alkhouri, N. Nonalcoholic Fatty Liver Disease in Children. Pediatr. Clin. N. Am. 2017, 64, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Sanmiguel, C.; Gupta, A.; Mayer, E.A. Gut Microbiome and Obesity: A Plausible Explanation for Obesity. Curr. Obes. Rep. 2015, 4, 250–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasselman, L.J.; Vernice, N.A.; DeLeon, J.; Reiss, A.B. The gut microbiome and elevated cardiovascular risk in obesity and autoimmunity. Atherosclerosis 2018, 271, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.J.; Willcox, D.C. Caloric restriction, caloric restriction mimetics, and healthy aging in Okinawa: Controversies and clinical implications. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Okeke, F.; Roland, B.C.; Mullin, G.E. The Role of the gut Microbiome in the Pathogenesis and Treatment of Obesity. Glob. Adv. Heal. Med. 2014, 3, 44–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastelli, M.; Knauf, C.; Cani, P.D. Gut Microbes and Health: A Focus on the Mechanisms Linking Microbes, Obesity, and Related Disorders. Obesity 2018, 26, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar] [PubMed]
- Moran, J.R.; Ghishan, F.K.; Halter, S.A.; Greene, H.L. Steatohepatitis in obese children: A cause of chronic liver dysfunction. Am. J. Gastroenterol. 1983, 78, 374–377. [Google Scholar] [PubMed]
- Schwimmer, J.B.; Deutsch, R.; Kahen, T.; Lavine, J.E.; Stanley, C.; Behling, C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006, 118, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Lazo, M.; Hernaez, R.; Eberhardt, M.S.; Bonekamp, S.; Kamel, I.; Guallar, E.; Koteish, A.; Brancati, F.L.; Clark, J.M. Prevalence of Nonalcoholic Fatty Liver Disease in the United States: The Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Epidemiol. 2013, 178, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Wong, V.W.-S.; Chitturi, S. NAFLD in Asia—As common and important as in the West. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Satapathy, S.; Sanyal, A. Epidemiology and Natural History of Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2015, 35, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, K.; Das, K.; Mukherjee, P.S.; Ghosh, A.; Ghosh, S.; Mridha, A.R.; Dhibar, T.; Bhattacharya, B.; Bhattacharya, D.; Manna, B.; et al. Nonobese population in a developing country has a high prevalence of nonalcoholic fatty liver and significant liver disease. Hepatology 2010, 51, 1593–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarapurkar, D.; Kamani, P.; Patel, N.; Gupte, P.; Kumar, P.; Agal, S.; Baijal, R.; Lala, S.; Chaudhary, D.; Deshpande, A. Prevalence of non-alcoholic fatty liver disease: Population based study. Ann. Hepatol. 2007, 6, 161–163. [Google Scholar] [PubMed]
- Zhu, J.-Z.; Dai, Y.-N.; Wang, Y.-M.; Zhou, Q.-Y.; Yu, C.-H.; Li, Y.-M. Prevalence of Nonalcoholic Fatty Liver Disease and Economy. Dig. Dis. Sci. 2015, 60, 3194–3202. [Google Scholar] [CrossRef] [PubMed]
- The Non-alcoholic Fatty Liver Disease Study Group; Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. 2015, 47, 997–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazel, Y.; Koenig, A.B.; Sayiner, M.; Goodman, Z.D.; Younossi, Z.M. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 2016, 65, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.L.C.; Lazo, M.; Selvin, E.; Clark, J.M. Racial differences in nonalcoholic fatty liver disease in the U.S. population. Obesity 2014, 22, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Milic, S.; Štimac, D. Nonalcoholic Fatty Liver Disease/Steatohepatitis: Epidemiology, Pathogenesis, Clinical Presentation and Treatment. Dig. Dis. 2012, 30, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Karpen, S.; Vos, M.B. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J. Pediatr. 2013, 162, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.; Longnecker, M.P.; Lawlor, D.A. Prevalence of elevated alanine aminotransferase among US adolescents and associated factors: NHANES 1999-2004. Gastroenterology 2007, 133, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Callaway, M.; Macdonald-Wallis, C.; Anderson, E.; Fraser, A.; Howe, L.D.; Day, C.; Sattar, N. Nonalcoholic fatty liver disease, liver fibrosis, and cardiometabolic risk factors in adolescence: A cross-sectional study of 1874 general population adolescents. J. Clin. Endocrinol. Metab. 2014, 99, E410–E417. [Google Scholar] [CrossRef] [PubMed]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Definig the Human Microbiome. NIH Manuscr. 2013, 70, 1–12. [Google Scholar] [CrossRef]
- Bibbò, S.; Ianiro, G.; Dore, M.P.; Simonelli, C.; Newton, E.E.; Cammarota, G. Gut Microbiota as a Driver of Inflammation in Nonalcoholic Fatty Liver Disease. Mediators Inflamm. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 777, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.; Turnbaugh, P.; Klein, S.; Gordon, J. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.H.; Macfarlane, G.T.; Englyst, H.N. Prebiotic digestion and fermentation. Am. J. Clin. Nutr. 2001, 73, 415S–420S. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.; Wahlström, A.; Marschall, H.U. Role of Bile Acids in Metabolic Control. Trends Endocrinol. Metab. 2018, 29, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- De Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Ganopolsky, J.G.; Labbé, A.; Prakash, S. The human microbiome and bile acid metabolism: Dysbiosis, dysmetabolism, disease and intervention. Expert Opin. Biol. Ther. 2014, 14, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, G.; Paschetta, E.; Gambino, R.; Cassader, M.; Molinaro, F. Interactions among bone, liver, and adipose tissue predisposing to diabesity and fatty liver. Trends Mol. Med. 2013, 19, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Mehal, W.Z. The Gordian Knot of dysbiosis, obesity and NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Poeta, M.; Pierri, L.; Vajro, P. Gut–Liver Axis Derangement in Non-Alcoholic Fatty Liver Disease. Children 2017, 4, 66. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Renshaw, S.A. Zebrafish screens for new colitis treatments—A bottom-up approach. FEBS J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Aboura, I.; Nani, A.; Belarbi, M.; Murtaza, B.; Fluckiger, A.; Dumont, A.; Benammar, C.; Tounsi, M.S.; Ghiringhelli, F.; Rialland, M.; et al. Protective effects of polyphenol-rich infusions from carob (Ceratonia siliqua) leaves and cladodes of Opuntia ficus-indica against inflammation associated with diet-induced obesity and DSS-induced colitis in Swiss mice. Biomed. Pharmacother. 2017. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, A.; Du Jeu, X.D.M.; Johnson, C.D.; Nektaria, A.; Feldstein, A.E. Liver Bid suppression for treatment of fibrosis associated with non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 699–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, A.; Feldstein, A.E. Adipocyte cell death, fatty liver disease and associated metabolic disorders. Dig. Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Toulis, K.A.; Goulis, D.G.; Zavos, C.; Kountouras, J. Serum total adiponectin in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Metabolism 2011, 60, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Watanabe, K.; Kubota, N.; Gunji, T.; Omata, M.; Kadowaki, T.; Ohnishi, S. Adiponectin knockout mice on high fat diet develop fibrosing steatohepatitis. J. Gastroenterol. Hepatol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Udayappan, S.D.; Kovatcheva-Datchary, P.; Bakker, G.J.; Havik, S.R.; Herrema, H.; Cani, P.D.; Bouter, K.E.; Belzer, C.; Witjes, J.J.; Vrieze, A.; et al. Intestinal Ralstonia pickettii augments glucose intolerance in obesity. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Anzenbacher, P.; Anzenbacherová, E. Cytochromes P450 and metabolism of xenobiotics. Cell. Mol. Life Sci. 2001. [Google Scholar] [CrossRef]
- Gong, Y.; Fu, Z.; Edin, M.L.; Liu, C.H.; Wang, Z.; Shao, Z.; Fredrick, T.W.; Saba, N.J.; Morss, P.C.; Burnim, S.B.; et al. Cytochrome P450 oxidase 2C inhibition adds to ω-3 long-chain polyunsaturated fatty acids protection against retinal and choroidal neovascularization. Arterioscler. Thromb. Vasc. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Armoni, M.; Harel, C.; Karnieli, E.; Pessin, J.E. Cytochrome P-450 CYP2E1 knockout mice are protected against high-fat diet-induced obesity and insulin resistance. AJP Endocrinol. Metab. 2012, 302, E532–E539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armoni, M.; Harel, C.; Ramdas, M.; Karnieli, E. CYP2E1 impairs GLUT4 gene expression and function: NRF2 as a possible mediator. Horm. Metab. Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cederbaum, A.I. Cytochrome P450s And Alcoholic Liver Disease. Curr. Pharm. Des. 2018. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: Diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.J.; Flask, C.A.; Papouchado, B.G.; Feldstein, A.E.; Nagy, L.E. Caspase-1 as a Central Regulator of High Fat Diet-Induced Non-Alcoholic Steatohepatitis. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Vucur, M.; Reisinger, F.; Cardenas, D.V.; Roderburg, C.; Koppe, C.; Kreggenwinkel, K.; Schneider, A.T.; Bartneck, M.; Neumann, U.P.; et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 2014, 6, 1062–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandelia, C.; Collyer, E.; Mansoor, S.; Lopez, R.; Lappe, S.; Nobili, V.; Alkhouri, N. Plasma cytokeratin-18 level as a novel biomarker for liver fibrosis in children with nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 2016, 63, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Thapaliya, S.; Wree, A.; Povero, D.; Inzaugarat, M.E.; Berk, M.; Dixon, L.; Papouchado, B.G.; Feldstein, A.E. Caspase 3 inactivation protects against hepatic cell death and ameliorates fibrogenesis in a diet-induced NASH model. Dig. Dis. Sci. 2014, 59, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Marin, V.; Rosso, N.; Dal Ben, M.; Raseni, A.; Boschelle, M.; Degrassi, C.; Nemeckova, I.; Nachtigal, P.; Avellini, C.; Tiribelli, C.; et al. An animal model for the juvenile non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. PLoS ONE 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Manning, G. Necroptosis and Inflammation. Annu. Rev. Biochem. 2016, 85, 743–763. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, S.; McCullough, R.L.; Sanz-Garcia, C.; Saikia, P.; Alkhouri, N.; Matloob, A.; Pollard, K.A.; McMullen, M.R.; Croniger, C.M.; Nagy, L.E. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology 2016, 64, 1518–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonso, M.B.; Rodrigues, P.M.; Carvalho, T.; Caridade, M.; Borralho, P.; Cortez-Pinto, H.; Castro, R.E.; Rodrigues, C.M.P. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci. 2015, 129, 721–739. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Vucur, M.; Schneider, A.T.; Severi, I.; Roderburg, C.; Roy, S.; Bartneck, M.; Schrammen, P.; Diaz, M.B.; Ehling, J.; et al. The necroptosis-inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Beier, J.I.; Banales, J.M. Pyroptosis: An inflammatory link between NAFLD and NASH with potential therapeutic implications. J. Hepatol. 2018, 68, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; McGeough, M.D.; Inzaugarat, M.E.; Eguchi, A.; Schuster, S.; Johnson, C.D.; Peña, C.A.; Geisler, L.J.; Papouchado, B.G.; Hoffman, H.M.; et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology 2018, 67, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Paolella, G.; Mandato, C.; Pierri, L.; Poeta, M.; Di Stasi, M.; Vajro, P. Gut-liver axis and probiotics: Their role in non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 15518–15531. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; Desimone, C.; Song, X.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Hua, J.; Li, Z. Probiotics improve high fat diet-induced hepatic steatosis and insulin resistance by increasing hepatic NKT cells. J. Hepatol. 2008, 49, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Karahan, N.; Işler, M.; Koyu, A.; Karahan, A.G.; Başyığıt Kiliç, G.; Cırış, I.M.; Sütçü, R.; Onaran, I.; Cam, H.; Keskın, M. Effects of probiotics on methionine choline deficient diet-induced steatohepatitis in rats. Turk. J. Gastroenterol. 2012, 23, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Mattace Raso, G.; Simeoli, R.; Iacono, A.; Santoro, A.; Amero, P.; Paciello, O.; Russo, R.; D’Agostino, G.; Di Costanzo, M.; Berni Canani, R.; et al. Effects of a Lactobacillus paracasei B21060 based synbiotic on steatosis, insulin signaling and toll-like receptor expression in rats fed a high-fat diet. J. Nutr. Biochem. 2014, 25, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Lirussi, F.; Mastropasqua, E.; Orando, S.; Orlando, R. Probiotics for non-alcoholic fatty liver disease and/or steatohepatitis. Cochrane Database Syst. Rev. 2007, CD005165. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-Y.; Li, L.; Yu, C.-H.; Shen, Z.; Chen, L.-H.; Li, Y.-M. Effects of probiotics on nonalcoholic fatty liver disease: A meta-analysis. World J. Gastroenterol. 2013, 19, 6911. [Google Scholar] [CrossRef] [PubMed]
- Eslamparast, T.; Poustchi, H.; Zamani, F.; Sharafkhah, M.; Malekzadeh, R.; Hekmatdoost, A. Synbiotic supplementation in nonalcoholic fatty liver disease: A randomized, double-blind, placebo-controlled pilot study. Am. J. Clin. Nutr. 2014, 99, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Vajro, P.; Mandato, C.; Licenziati, M.R.; Franzese, A.; Vitale, D.F.; Lenta, S.; Caropreso, M.; Vallone, G.; Meli, R. Effects of Lactobacillus rhamnosus Strain GG in Pediatric Obesity-related Liver Disease. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 740–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alisi, A.; Bedogni, G.; Baviera, G.; Giorgio, V.; Porro, E.; Paris, C.; Giammaria, P.; Reali, L.; Anania, F.; Nobili, V. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2014, 39, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Famouri, F.; Shariat, Z.; Hashemipour, M.; Keikha, M.; Kelishadi, R. Effects of Probiotics on Nonalcoholic Fatty Liver Disease in Obese Children and Adolescents. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Finelli, C. Systematic review on intervention with prebiotics/probiotics in patients with obesity-related nonalcoholic fatty liver disease. Future Microbiol. 2015, 10, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Prakash, S. Oral supplementation with probiotic L. reuteri NCIMB 30242 increases mean circulating 25-hydroxyvitamin D: A post hoc analysis of a randomized controlled trial. J. Clin. Endocrinol. Metab. 2013, 98, 2944–2951. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Ganopolsky, J.G.; Sulemankhil, I.; Ghali, P.; Prakash, S. Improvement of gastrointestinal health status in subjects consuming Lactobacillus reuteri NCIMB 30242 capsules: A post-hoc analysis of a randomized controlled trial. Expert Opin. Biol. Ther. 2013, 13, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Parent, M.; Prakash, S. Cholesterol-lowering efficacy of a microencapsulated bile salt hydrolase-active Lactobacillus reuteri NCIMB 30242 yoghurt formulation in hypercholesterolaemic adults. Br. J. Nutr. 2012, 107, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Bagherniya, M.; Nobili, V.; Blesso, C.N.; Sahebkar, A. Medicinal plants and bioactive natural compounds in the treatment of non-alcoholic fatty liver disease: A clinical review. Pharmacol. Res. 2018, 130, 213–240. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Chu, Q.; Yan, F.; Yang, Y.; Han, W.; Zheng, X. Red pitaya betacyanins protects from diet-induced obesity, liver steatosis and insulin resistance in association with modulation of gut microbiota in mice. J. Gastroenterol. Hepatol. 2016, 31, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, K.; Deng, Y.; Chen, R.; Liang, S.; Xie, H.; He, Y.; Chen, Y.; Yang, Q. Effects of shenling baizhu powder herbal formula on intestinal microbiota in high-fat diet-induced NAFLD rats. Biomed. Pharmacother. 2018, 102, 1025–1036. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Feeding Models | Outcome | Phenotype | Reference |
|---|---|---|---|
| Western Diet | Protective | Protects against hepatocyte injury, steatosis and fibrosis | [75] |
| Choline deficient High fat diet (Research Diets; D05010402) | Protective | Suppress adipocyte apoptosis and inflammation | [77] |
| Methyl choline deficient diet | Deleterious | Induces hepatocyte injury | [68] |
| Choline-deficient high fat diet | Deleterious | Induces liver injury, inflammation, steatosis and fibrosis | [76] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roychowdhury, S.; Selvakumar, P.C.; Cresci, G.A.M. The Role of the Gut Microbiome in Nonalcoholic Fatty Liver Disease. Med. Sci. 2018, 6, 47. https://doi.org/10.3390/medsci6020047
Roychowdhury S, Selvakumar PC, Cresci GAM. The Role of the Gut Microbiome in Nonalcoholic Fatty Liver Disease. Medical Sciences. 2018; 6(2):47. https://doi.org/10.3390/medsci6020047
Chicago/Turabian StyleRoychowdhury, Sanjoy, Praveen Conjeevaram Selvakumar, and Gail A.M. Cresci. 2018. "The Role of the Gut Microbiome in Nonalcoholic Fatty Liver Disease" Medical Sciences 6, no. 2: 47. https://doi.org/10.3390/medsci6020047
APA StyleRoychowdhury, S., Selvakumar, P. C., & Cresci, G. A. M. (2018). The Role of the Gut Microbiome in Nonalcoholic Fatty Liver Disease. Medical Sciences, 6(2), 47. https://doi.org/10.3390/medsci6020047