1. Introduction
The production of recombinant proteins from mammalian cells for therapeutic and research purposes has always been associated with the challenge of improving production. As a result, volumetric productivity has been increased significantly in the last two decades by media optimization, careful process control [
1,
2,
3], genetic modification [
4,
5,
6], and cell line selection [
1]. Chinese hamster ovary (CHO) cells are currently the main industrial producers of recombinant therapeutic proteins [
7,
8] because of their efficient growth and high expression characteristics. These and other non-human producers, such as baby hamster kidney (BHK21) cells and murine myeloma cells (NS0 and Sp2/0) [
9], are limited by their ability to correctly perform post-translational modifications, which are important for proper activity of the produced protein [
10]. Therefore, efforts are ongoing to improve growth and expression of recombinant protein from human cell lines, such as human embryonic kidney 293 (HEK293) and fibrosarcoma HT-1080 [
11]. Recent research conducted by Su et al. looked at the possibility of improving protein expression from HEK293 cells by utilizing non-coding RNA, such as microRNA (miRNA) and small interfering RNA (siRNA) [
12,
13,
14]. The study was conducted in two directions; first, the focus was on finding microRNA that have a direct effect on protein expression; and the second was focused on finding specific genes whose inactivation by siRNA improved expression. This work was performed by implementing two independent high throughput screenings of the non-coding RNAs. By screening the effect of 23,000 different siRNAs, ten genes whose knock-down triggered higher expression of recombinant luciferase were selected for further studies. Among the ten identified genes, ornithine decarboxylase antizyme (
OAZ1) attracted special attention, because its inhibition improved the expression of luciferase with minimal effects on cell growth and viability. Additionally, the role of
OAZ1 in polyamine metabolism has been well studied [
15,
16], making it a promising target to explore the connection between
OAZ1 inhibition and the level of different polyamines. The
OAZ1 protein negatively regulates polyamine biosynthesis by degradation of ornithine decarboxylase. This enzyme catalyzes the decarboxylation of ornithine to form putrescine, a committed step in polyamine biosynthesis. This connection was investigated [
17], and preliminary information showed that transient inhibition of
OAZ1 in HEK293 cells expressing luciferase by siRNA was associated with increased luciferase expression and a higher intracellular concentration of putrescine. Creating a permanent cell line lacking
OAZ1 should, therefore, be the next step in evaluating the potential of the
OAZ1 knockout cell line as an efficient producer of recombinant proteins from HEK293, and perhaps other mammalian cell lines.
Several gene editing tools are currently available, such as transcription activator-like effector nuclease (TALENS), zinc finger nucleases (ZFNs), and the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system, which consists of a Cas endonuclease directed to cleave a target sequence by a guide RNA. The CRISPR system, with its unprecedented level of simplicity, efficiency, and ability to carry out multiplexed mutations [
18], was chosen to create an
OAZ1 deleted cell line. In this report, we evaluate the growth and production capabilities of the created cell line for production of recombinant proteins in both stable and transient transfection systems.
2. Materials and Methods
Cell line: HEK293 stably transfected with the luciferase gene of Photinus pyralis under a cytomegalovirus (CMV) promoter (CMV-Luc2-Hygro HEK293, Promega ID# CAS140901, Madison, WI, USA). This cell line will be referred to as the parental cell line in the text.
Cell culture: Cells were grown in adherent cultures in Dulbecco’s Modified Eagle Medium (DMEM Gibco cat# 11995–040, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals cat# S11150H, Flowery Branch, GA, USA), 100 units/mL of penicillin, and 100 µg/mL of streptomycin (Gibco cat# 15140–122, Grand Island, NY, USA). The cultures were incubated in a humidity-controlled incubator at 37 °C and 5% CO2.
2.1. Construction of CRISPR/Cas9 Lentiviral Particles with Single Guide RNAs Targeting the OAZ1 Gene
Three all-in-one CRISPR/Cas9 lentiviral particles with single guide RNAs (sgRNAs) targeting the second exonic region of the OAZ1 gene were designed and purified by Sigma Aldrich. All-in-one particles encode the sgRNA, Cas9 endonuclease, puromycin resistance, and green fluorescence protein (GFP), driven by a U6 promoter. The sequence of the target regions in the three guide RNAs are as follows:
taacccgggtccggggcctcgg-Sigma Aldrich cat# HS0000288774 (774)
gatcggctgaatgtaacagagg-Sigma Aldrich cat# HS0000288775 (775)
agacgccaaacgcattaactgg-Sigma Aldrich cat# HS0000288778 (778)
2.2. Transduction of Human Embryonic Kidney 293 Luciferase Expressing Cells with Lentiviral Particles and Isolation of Single Colonies
Ninety-six well plates (Corning cat #356717, Kennebunk, ME, USA) were coated with 1% Matrigel (Corning cat# 354248) and seeded with 75,000 CMV-Luc2-Hygro HEK293 cells per well in 30 µL of culture media. Plates were incubated overnight in a humidified incubator at 37 °C, 5% CO
2. Lentiviral particles, three
OAZ1 targeting lentiviral constructs, and CRISPR-Lenti non-targeting control transduction particles (Sigma-Aldrich # CRISPR12V, St Louis, MO, USA) were added the following day at a previously optimized multiplicity of infection (MOI) of 5, in 50 µL media and incubated overnight as above. The media were replaced for an additional overnight incubation. The media were replaced with selection media, containing 1 µg/mL puromycin, on day three post-transduction. The selection media were replaced every other day until confluence was achieved. Limiting dilutions were carried out from each well, as previously described [
19,
20]
, in 96-well plates. The wells were scored for the presence of GFP expressing single colonies over a period of two weeks. The wells containing single colonies were propagated and sub-cultured into larger vessels, until enough cells were available for assays (confluent T-25 flask).
2.3. Luciferase Assay for Selection of Highly Expressing Clones
Cell viability and luciferase activity were determined using the CellTiter-Glo luminescent cell viability assay and the One-Glo luciferase assay system (Promega cat# G7570 and cat# E6110, Madison, WI, USA, respectively), following manufacturer’s protocol. Briefly, cells at a confluence of 80–90% in a 96-well plate were re-suspended in 100 µL media and transferred to a white opaque 96-well plate (Greiner Bio-one, cat# 655088, Frickenhausen, Germany) in quadruplets. Then, 100 µL of CellTiter-Glo reagent was added to two out of the four quads, mixed for 2 min on a shaker, and allowed to sit at room temperature for 10 min. At approximately 6 min into the 10-min incubation, 100 µL of One-Glo reagent was added to the remaining two wells, mixed briefly, and kept at room temperature until the end of the 10-min incubation period. The luminescence was read using the SpectraMax® microplate reader (Molecular devices, Wals, Austria) at an integration time of 250 ms.
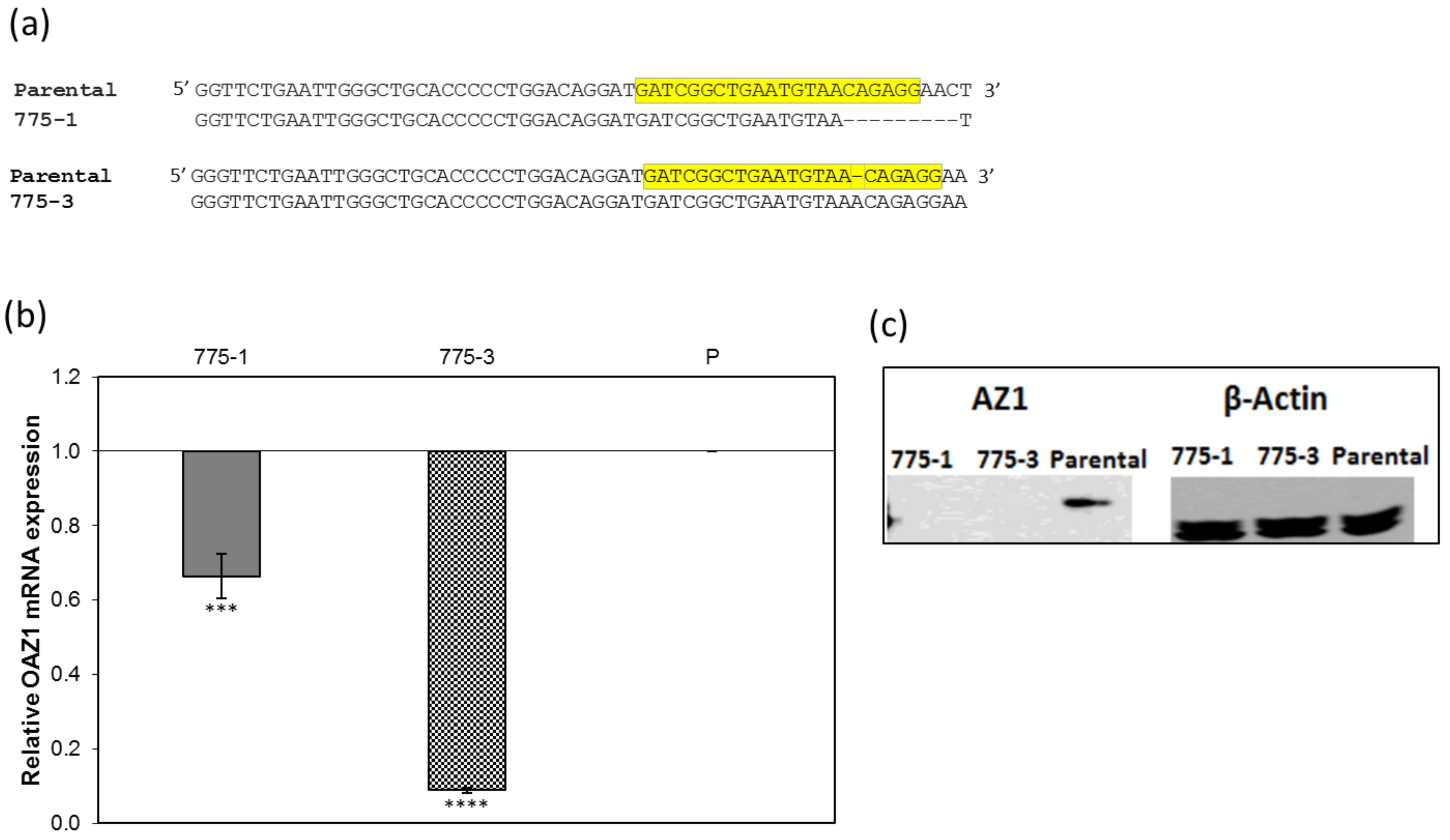
2.4. Sequencing of the Ornithine Decarboxylase Antizyme 1 Gene in Parental and Mutant Strains
Total genomic DNA was isolated using the DNeasy kit (Qiagen, cat#69506, Hilden, Germany), following the manufacturers protocol. The genomic region flanking the CRISPR target site for the OAZ1 gene was polymerase chain reaction (PCR) amplified, using the Phusion® High-Fidelity PCR Master Mix with HF Buffer (NEB, cat# MO531S, Ipswitch, MA, USA), and the following primers: forward primer—cagcagcagtgagagttcca; reverse primer—gcttttggagagcaatggag. Amplicons were gel-extracted using the QIAquick® Gel Extraction Kit (QIAGEN, Ref# 28704, Hilden, Germany), and sequenced using capillary DNA sequencing. Sequences were aligned with the parental sequence using the Clustal Omega (European Bioinformatics Institute, Cambridge, UK) alignment program.
2.5. Growth Characterization Determination
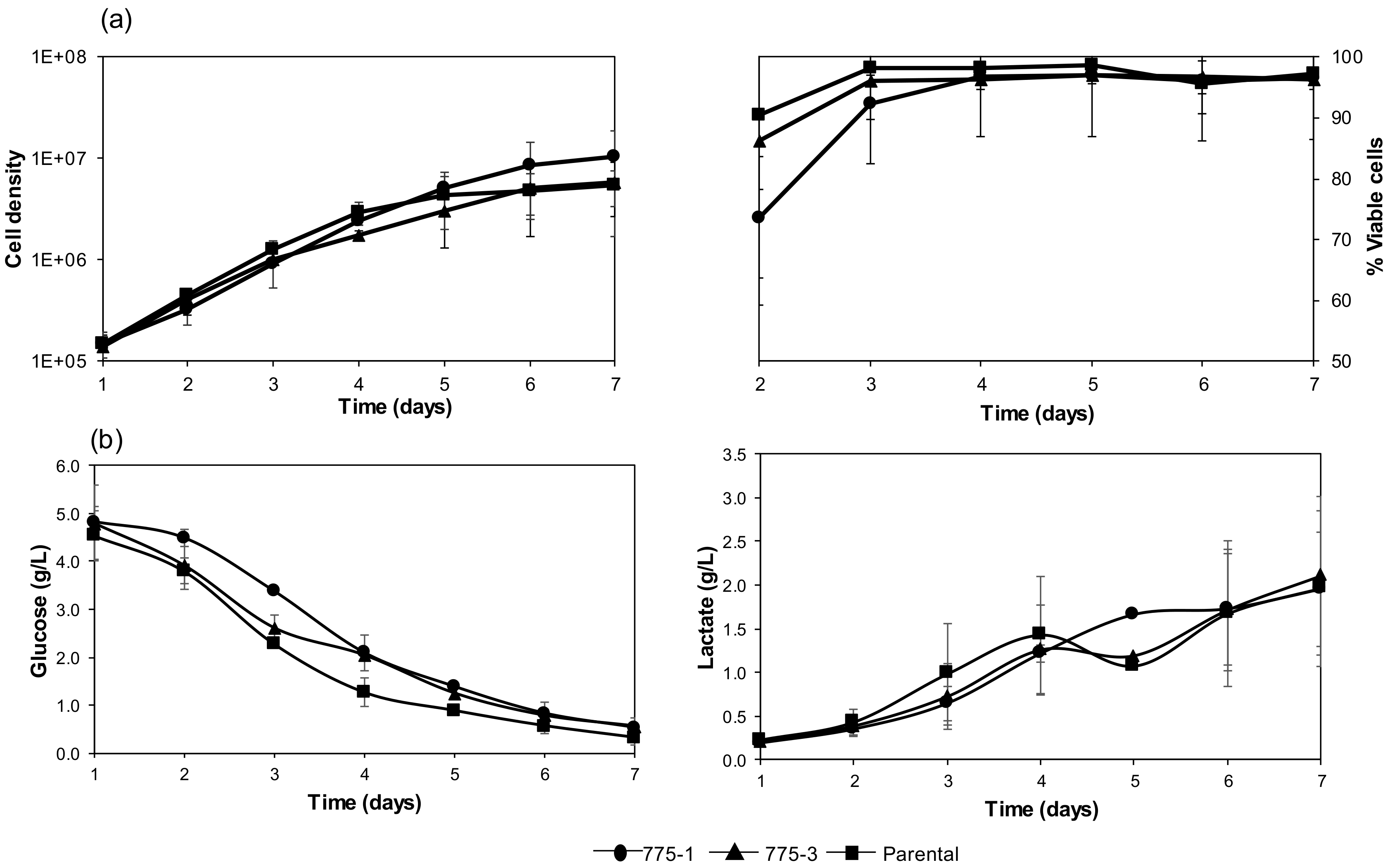
Seven T-25 flasks per cell line were each inoculated with 7 × 105 cells in 5 mL culture media and incubated, as described above. The cells were enumerated each day for seven days, using the CEDEX HiREs cell analyzer/counter (Roche # 7766, Basel, Switzerland), and the levels of glucose and lactate were quantified using the YSI 2900 bio-analyzer (Yellow Springs Instrument Co., Yellow Springs, OH, USA).
2.6. Antizyme 1 and Luciferase Western Blot
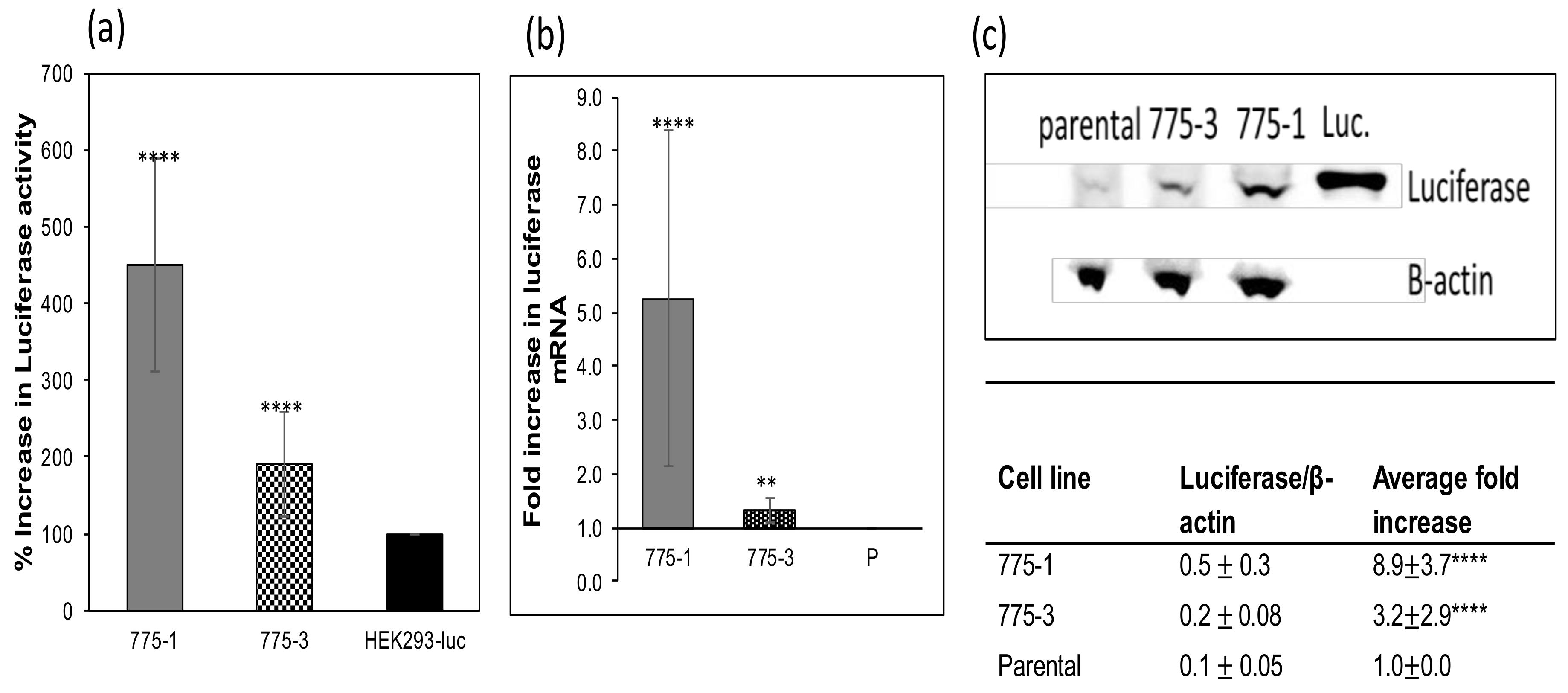
Parental and CRISPR-treated cells in confluent six-well plates were lysed using radioimmunoprecipitation assay (RIPA) cell lysis buffer (Thermo Scientific, # 89900, Rockford, IL, USA) supplemented with 1× protease and phosphatase inhibitor (Thermo Scientific #1861281, Rockford, IL, USA), and total protein was quantified at A280 using the Nanodrop Onec (Thermo Scientific, Rockford, IL, USA). Cell lysate samples containing equal amounts of protein (340 µg) and recombinant firefly luciferase protein (abcam # ab100961, Cambridge, MA, USA) were electrophoresed on a 4–12% bis-tris gel (ThermoFisher, # NP0322BOX, Rockford, IL, USA). Separated proteins were transferred onto a nitrocellulose membrane and antizyme 1 (AZ1) or luciferase was detected using rabbit anti-AZ1 polyclonal antibodies targeting the N-terminal region of the AZ1 protein (Sigma-Aldrich, cat# SAB1307119, St Louis, MO, USA )/HRP conjugated anti-rabbit secondary antibodies (ThermoFisher Scientific # 65–6120, Rockford, IL, USA); or mouse anti-luciferase polyclonal antibodies (ThermoFisher, # PA1–179, Rockford, IL, USA)/HRP conjugated goat anti-mouse antibodies (KPL # 474–1806, Gaithersburg, MD, USA). Mouse anti-β-actin monoclonal antibodies (Sigma # A2228) and HRP conjugated goat anti-mouse antibodies (KPL # 474–1806, Gaithersburg, MD, USA) were used to detect β-actin in identical samples. The signal was developed using the SuperSignal® west Pico Chemiluminescent substrate (ThermoFisher, # OD187429, Rockford, IL, USA), and visualized in a LAS-4000 Mini Luminescent Image Analyzer (GE healthcare, # 28955810, Marlborough, MA, USA). The intensity of the Western blot bands were determined using ImageJ software and the luciferase/actin ratio, and fold increase was reported.
2.7. Quantitative PCR Analysis
Real time quantitative PCR was done using the SYBR GREEN protocol. Total RNA was extracted from OAZ1 knockout and parental cells using the RNeasy kit (QIAGEN, cat# 74101, Hilden, Germany). First strand cDNA was synthesized from total RNA using the Maxima First strand cDNA synthesis kit for real time quantitative PCR (RT-qPCR) (Thermo Scientific, # K1642, Vilnius Lithuania). Then, 20 ng of cDNA was amplified in a qPCR using the following primers, OAZ1: forward primer—GGAACCGTAGACTCGCTCAT, reverse primer—TCGGAGTGAGCGTTTATTTG; and Luc: forward primer—GTGGTGTGCA GCGAGAATAG, reverse primer—CGCTCGTTGTAGATGTCGTTAG.
Threshold and threshold cycle (Ct) values were determined automatically by the RQ ManagerTM Software (Applied Biosystems, Foster City, CA, USA) using default parameters. The comparative cycle threshold (2−ΔΔCt method) was used to analyze the expression levels of genes examined in this study. The abundance of each gene transcript was normalized by glyceraldehyde phosphate dehydrogenase (GAPDH) or β Actin (ACTB) gene expression levels and expressed in arbitrary units (AU). The relative quantization of gene expression was performed in triplicates for each sample.
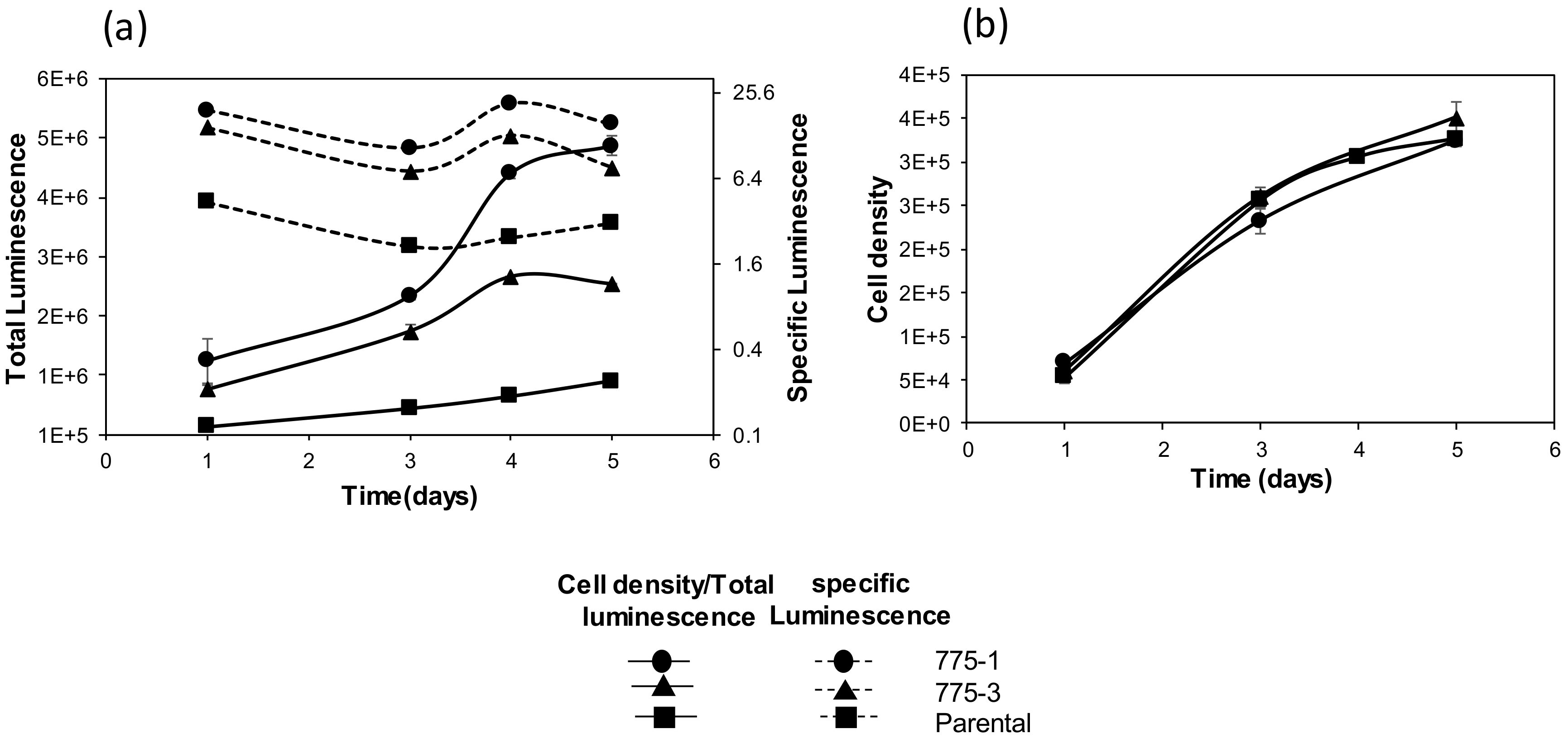
2.8. Transfection of Cells with Secreted Alkaline Phosphatase Plasmid and Quantification of SEAP Activity
Twenty-four well plates were seeded with 200,000 cells per well in culture media one day prior to transfection. The following day, cells were transfected with 500 ng/well of the pSELECT-zeo-SEAP plasmid, encoding the embryonic secreted alkaline phosphatase (SEAP) driven by a EF-1α/HTLV composite promoter (InvivoGen # psetz-seap, San Diego, CA, USA), using the Lipofectamine® 2000 transfection reagent (Invitrogen # 11668-019, Carlsbad, CA, USA), and following the manufacturer’s protocol. After two days, cell culture media were replaced with selection media (culture media supplemented with zeocin 200 µg/mL), and samples were collected for analysis on day three and day five. Alkaline phosphatase activity in the culture supernatant was quantified using the Quanti-Blue colorimetric assay (InvivoGen #rep-qb1, San Diego, CA, USA), cells were enumerated as described above, and total and specific SEAP activity was determined.
2.9. Polyamine Quantification
Five million cells per strain were washed twice in phosphate buffered saline (PBS) and pelleted at 1200 rpm for 5 min. The cells were disrupted with 2.3 mm zirconium beads in a mixture of methanol/water (50:50; v/v) acidified with 0.1% formic acid. Polyamines in cell extract were quantified using Ultra-High Performance Liquid Chromatography (UHPLC) analysis (Agilent 1290, Agilent Technologies, Santa Clara CA, USA), coupled to hybrid triple quadrupole/ion trap mass spectrometer (QTRAP 5500 from AB Sciex, Vaughan, ON, Canada), on Zorbax Eclipse Plus C18, rapid resolution high density (RRHD) column (2.1 × 50 mm 1.8 micron, Agilent Technologies). The chromatography was performed using ultrapure water containing 0.1% formic acid with 1 mM perfluoro heptanoic acid as solvent B and 0.1% formic acid plus 1 mM perfluoro heptanoic acid in 100% methanol as solvent A. The Liquid chromatograph tandem mass-spectrometry (LC-MS/MS) was run for 8.0 min with a flow rate of 300 μL/min. The gradient elution was performed at 50% solvent A for 0.00–4.50 min, for 4.50–5.25 min 100%, 5.25–6.75 min 50%, and 6.75–8.00 min 50%. The mass spectra were acquired using a Turbo Spray Ionization of 2500 V in positive ion mode, and multiple reaction monitoring (MRM). The curtain gas (nitrogen), CAD (collision activated dissociation), nebulizing, and heating gas were set to 40 psi, medium, 50 psi, and 60 psi, respectively. The temperature of the source was fixed at 500 °C. The mass spectrometer was set to have a dwell time of 50 ms. LC-MS/MS data were processed using Analyst 1.6.1 software (AB Sciex, Vaughan, ON, Canada). The results were compared to internal standard of 1,6-hexanediamine and standard curves for putrescine, spermidine, and spermine protein quantification were carried out in parallel using the modified Lowry assay.
One batch of three samples (5 × 105 cells) of each cell line, prepared from the same passage, was analyzed. The polyamine content and protein concentration was determined for each sample.
2.10. Statistical Analysis
Mean values and standard deviation or standard error were calculated using standard methods. p-values were determined using the Chi-square test.
4. Discussion
Mammalian cell lines are the producers of choice for many recombinant therapeutic proteins. Among the currently utilized producing cell lines, human cell lines are becoming more relevant because of their innate ability to perform the correct post-translation modifications needed for stable and functional proteins [
21,
22]. However, compared with their non-human counterparts, their productivity is relatively low, necessitating the development of efficient cell lines using genetic engineering, and the improvement of growth and expression strategies.
In an effort to identify genes potentially affecting recombinant protein expression, a high throughput screen evaluating the effect of siRNA gene knock down on luciferase expression from HEK293 cells was conducted [
17]. From a total of approximately 20,000 evaluated genes,
OAZ1 was identified as a promising candidate, whose deletion could improve protein expression, and was confirmed by transient transfection of siRNA against
OAZ1.
The work presented here describes the creation of the HEK293 cell line with
OAZ1 deletion using CRISPR genome editing. Two single clone-derived HEK293 luciferase expressing cell lines deficient in the
OAZ1 gene product, antizyme 1, were created by targeting the second exon of
OAZ1 for disruption. The premise of targeting exon 2 (
OAZ1 has six exons) was to cause disruptions early in the sequence, which would likely result in a truncated or functionally inactive protein, because the ornithine decarboxylase (ODC) binding site is in the internal (122–144) and C-terminal (211–218) portions of the protein [
23]. As a result, two mutants with the predicated truncated antizyme 1 sequences of 103 and 128 amino acids, structurally incapable of binding ODC, were selected.
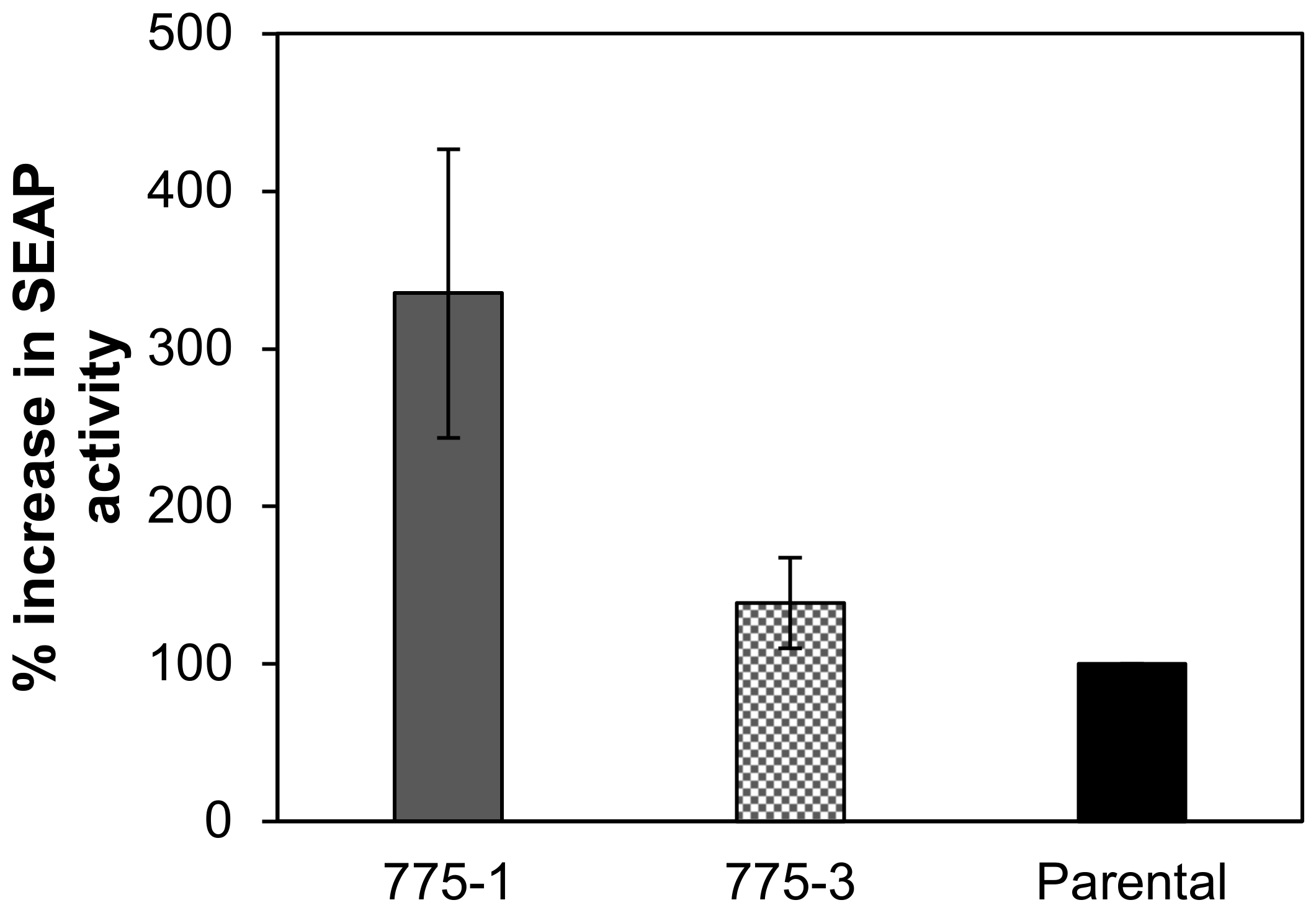
Compared with the parental cell line, both engineered cell lines demonstrated higher expression of luciferase, a stably transfected cytoplasmic protein, and alkaline phosphatase, a transiently transfected secreted protein. The growth kinetics and the metabolic activity of the mutant cell lines were comparable to the parental cell line and, as expected, their intracellular polyamine content was higher.
The improved expression of recombinant proteins exhibited by these
OAZ1 knockout cell lines can be attributed to the observed increase in the level of polyamines, which is likely the result of a missing functional
OAZ1 gene. Antizyme 1, the protein product of
OAZ1, is known to regulate polyamine production by inhibiting ODC [
15,
16]. Inhibiting AZ1 has been shown to cause an increase in intracellular polyamines levels, particularly putrescine [
24]. The disproportionate effect of
OAZ1 knockout on the different polyamines can be attributed to the
OAZ1-independent catabolism of spermine and spermidine, but not of putrescine [
25]. Adding exogenous polyamines to cell culture of HEK293 has also been shown to enhance recombinant protein expression in a concentration dependent manner [
17].
Improved expression of recombinant proteins from the
OAZ1 deficient cells was observed both in cells stably expressing luciferase, and from the same cells transiently transfected with secreted alkaline phosphatase, although to a lesser extent. This suggests that the presented approach for improved expression can be applied to different recombinant proteins, and perhaps different cell lines, although likely with different levels of enhancement depending on the properties of the expressed proteins. The increase in luciferase activity of the knockout cell lines is accompanied by an increase in both mRNA transcription and protein expression. By comparing the increase of mRNA and protein, it was concluded that the increase in expression was engendered at the transcriptional level, with little to no post-transcriptional effects. This observation is different from the initial results obtained from the siRNA silencing of the
OAZ1 gene, in which there was no increase in luciferase mRNA [
17].
Improved protein expression is often accompanied by undesirable side effects, such as growth and metabolic disadvantages [
26,
27], caused by increased metabolic load on the cells. This was not observed in the
OAZ1 deficient or knockout cell lines, where no significant differences in growth and nutrient utilization were observed. These observations can be explained by the known role of
OAZ1 as a negative regulator of cell growth [
28], meaning its absence might offset the possible negative effects on cell growth engendered by improved protein expression. It is also possible that the effect of metabolic load is cell line dependent [
29]. A peculiar phenomenon is that although both knockout cell lines have higher intracellular polyamine concentrations than the parental cell line, the observed relationship between polyamine concentration and recombinant protein expression is not proportional. The lower producing cell line, 775–3, has at least a two-fold higher polyamine concentration than the higher producing cell line, 775–1. This discrepancy can be explained by possible toxic effects of elevated intracellular polyamine levels, which were not high enough to cause significant cytotoxicity, on the rate of protein synthesis [
30]. The previous report by Xiao et al. also showed that improved expression by addition of external polyamines is concentration-dependent up to an optimal concentration, above which higher concentrations have negative effects on the expression [
17].
The work presented here was done in anchorage dependent cells growing in serum supplemented media, which are, therefore, limited in their capacity to scale up. The next step would be to investigate the effect of the OAZ1 deletion in cells growing in suspension, which are more relevant to large-scale recombinant protein production.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}