1. Introduction
In 1912, Prof. G. Ciamician envisioned a future in which chemical processes would be mostly run by a sustainable, abundant and never-ending energy source—photons coming from our Sun [
1,
2]. More than 100 years later, we are still far from reaching that goal, but significant progress has been achieved in organic chemistry thanks to the advent of visible light photoredox catalysis. With this approach, chemical transformations (redox reactions) can be promoted in a catalytic way using energetic photons obtained from light. In contrast to non-catalyzed photochemistry, which employs powerful UV lamps, the chemical transformations of photoredox catalysis are usually achieved by photons of the visible light spectrum. For this to work efficiently, an absorbing sensitizer (photoredox catalyst) needs to be present.
Photoredox catalysis possesses the intrinsic advantage of allowing chemical reactions to proceed that are otherwise difficult to realize. One of the first examples related to photoredox catalysis was published in 1978 from Kellogg and co-workers, where a light-induced and dye-accelerated reduction of phenacyl sulfonium salts was performed in the presence of a Ru(II)(bipy)
3Cl
2 catalyst and substituted 1,4-dihydropyridine as a reducing agent [
3]. A few years later, it was found that a similar ruthenium complex was able to catalyze the photo-oxidation of carbinols to aldehydes when operating in the presence of visible light [
4]. Starting from these seminal studies, many developments have been made in the field, and today, processes involving metal complexes, organic dyes and solid materials (semi-conductors or conjugated organic polymers) as molecular photoredox catalysts are known [
5,
6,
7,
8,
9].
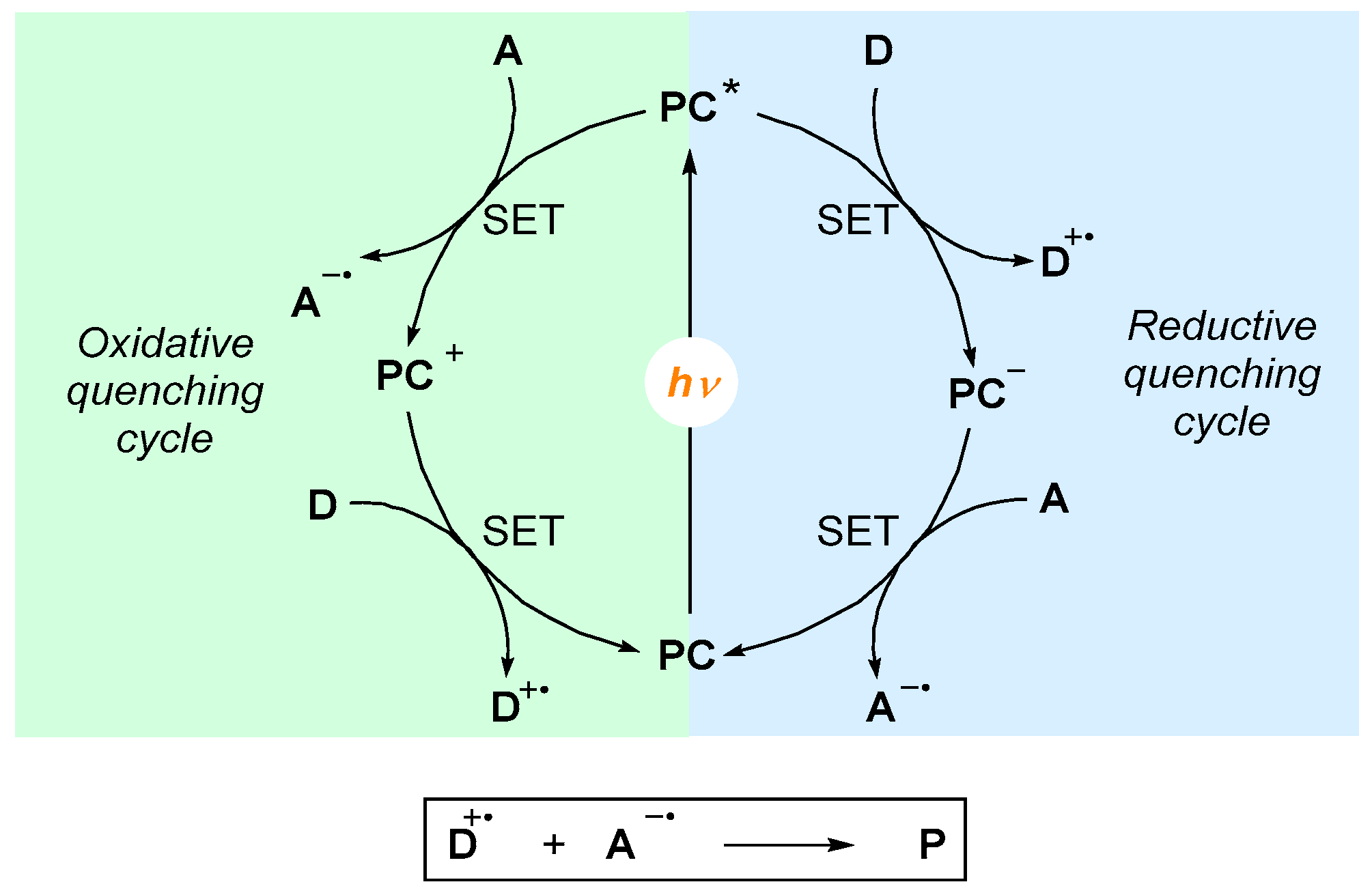
A typical photoredox catalytic cycle is shown in
Scheme 1. Usually, the photoredox catalyst
PC is excited with a photon of a suitable wavelength to generate the excited state
PC*. This new species can be then subjected to a reductive or oxidative step according to the nature of the reaction partners. In the first case,
PC* could receive a single electron from reactant
D (“donor”,
reductive quenching cycle) to form a
PC− species. This species, after a second single electron transfer process, can be oxidized to
PC in the presence of reactant
A. In this way, the initial photoredox catalyst is regenerated, closing the catalytic cycle. Conversely, in an
oxidative quenching cycle, activated
PC* species can be subjected to an abstraction of a single electron in the presence of reactant
A (“acceptor”). This generates the oxidized species
PC+, which can be reduced by reactant
D to regenerate the photoredox catalyst
PC.
Independently of the quenching cycle involved, the net charge of the photoredox catalyst does not change, while formally, the radical cation and anion of reactants A and D recombine to form the product P.
Most photoredox catalytic reactions employ heavy metal complexes as sensitizers, which are toxic, harmful to the environment, expensive and not sustainable for industrial processes. Organic dyes such as Eosin Y (EY) are very preferable compared to heavy metal complexes, since they can be obtained at low cost and present good availability since they can be synthesized from the live feedstock. Furthermore, organic dyes generally possess no toxic properties and are environmentally benign. Considering the numerous reviews published in the last few years in this very wide field [
10,
11,
12], in the present contribution, only the most recent and significant applications of Eosin Y as an organo-photocatalyst in homogeneous reactions performed under fluidic conditions and in solid-supported catalyst reactions will be discussed.
The most important employment of Eosin Y is related to its use as a filler in the production of dye-sensitized solar cells [
13,
14,
15,
16] and as a photoredox catalyst in organic synthesis [
10,
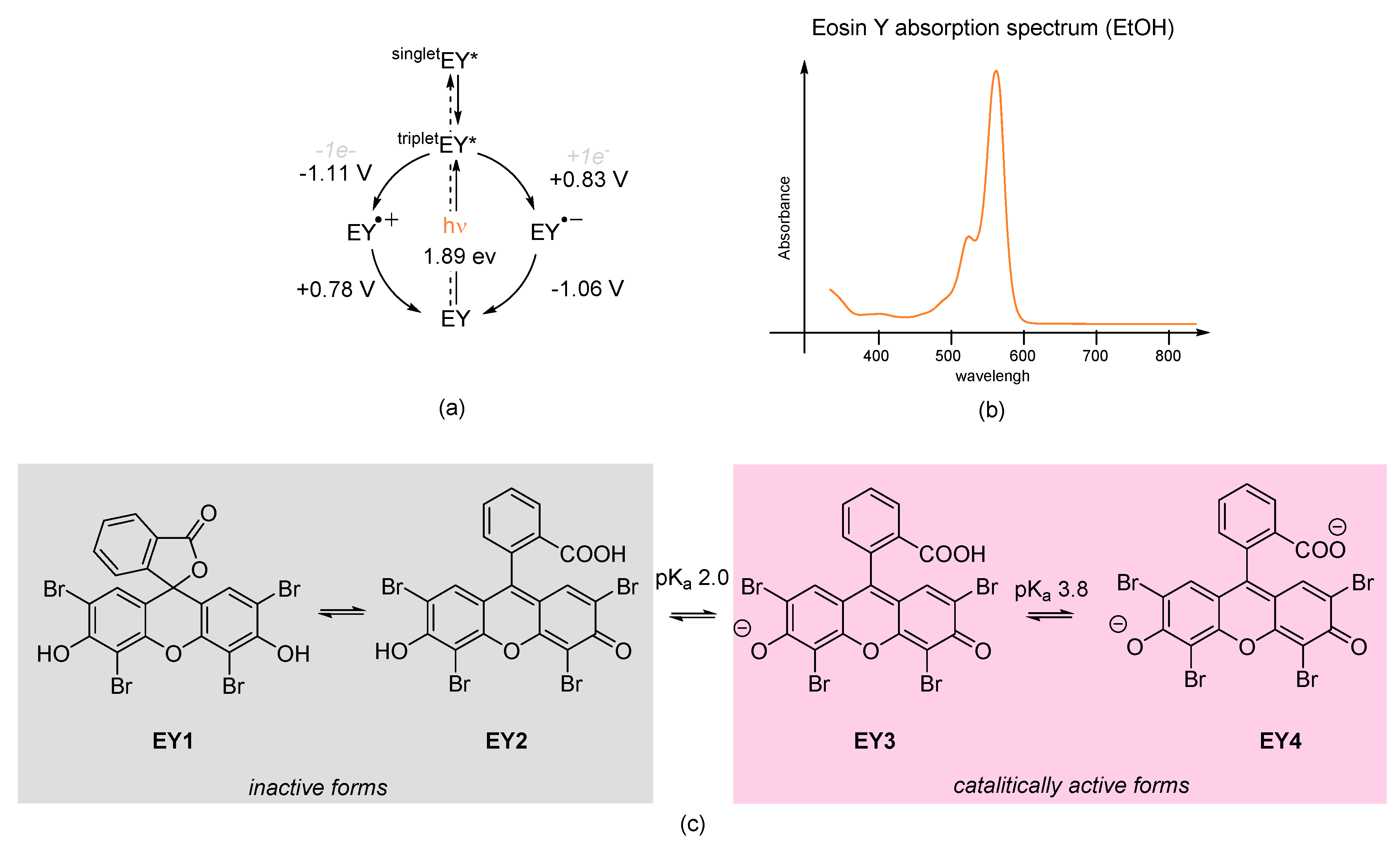
11]. In the latter case, the catalytic properties of Eosin Y rely on its redox potential: the oxidation potential range is between −1.06 V and 1.10 V, whereas the reduction potential is between +0.78 V and +0.83 V (
Scheme 2a). The maximum of absorption sits at 539 nm, with a molar extinction coefficient ε = 60 803 M
−1 cm
−1 (
Scheme 2b) [
17,
18,
19], and for this reason, green LEDs are usually employed as convenient light sources for Eosin Y activation (maximum emission around 530 nm). Upon excitation by light, Eosin Y undergoes a rapid intersystem crossing from the ground state to the lowest energy triplet state. Direct singlet–triplet transition is not allowed, but light excites Eosin Y electrons to a higher excited singlet state from where they relax quickly to the lowest excited singlet state; only at this point do excited electrons undergo intersystem crossing to the high reactive triplet state, which has a lifetime of 24 ms [
18,
19]. When in solution, Eosin Y equilibrates into four different forms due to the presence of two relatively acidic protons (pKa 2.0 and 3.8 in water,
Scheme 2c). All these forms are pH dependent: at pH < 2, the protonated spirocyclic form
EY1 is in equilibrium with the neutral form
EY2, whereas at 2 < pH > 3.8, the monoanionic form
EY3 is in equilibrium with the dianionic form
EY4, which becomes predominant at pH > 3.8. Only
EY3 and
EY4 are catalytically active, but this behavior has generated a lack of clarity in many publications regarding the nature of the dye involved in the transformation [
20]. In order to ensure the presence of the dianionic form
EY4, Eosin Y sodium salt is employed as a photocatalyst, although the experimental reaction conditions of chemical transformation should be taken into account.
2. Eosin Y as Organocatalyst in Photochemical Flow Reactions
According to the
Beer–Lambert–Bouguer Law [
21], a photochemical transformation performed in a traditional batch reactor will be exposed to a non-uniform radiation distribution due to the attenuation effect of photon transport. The middle of the reaction mixture is subjected to a lower intensity irradiation compared to the mixture located close to the reactor walls simply because it is further away from the irradiating source. This phenomenon causes the formation of different reactor zones characterized by different reaction rates. Due to this non-uniform situation, the use of large photochemical reactors should be discouraged in order to prevent the over-irradiation of external zones, which can facilitate the formation of undesired byproducts.
Photochemical transformations conducted in micro- or meso-reactors under fluidic conditions can overcome this problem, since the narrow channels allow the entire reaction mixture to be irradiated in a homogeneous and controlled manner. For this reason, continuous flow photochemical reactions show shorter reaction times, lower catalyst loadings, significantly less byproduct formation and increased productivity if compared to traditional batch processes [
22]. These advantages are in addition to those commonly associated with flow chemistry processes such as an increased mass and heat transfer, enhanced safety, the possibility of continuous monitoring the synthesis with integrated analysis devices and the possibility to work with gaseous reactants and hazardous intermediates in a safe manner [
23,
24]. Although micro- and meso-reactors are not suitable for the production of the large amounts of materials required by an industrial company, the scalability of continuous flow synthesis can be achieved through two essential strategies: by increasing the flow rate and the length of the reactor to allow a higher throughput or by a numbering-up approach, which involves the use of multiple reactors in a parallel manner.
In the last decade, important progress has been made in photochemistry in developing novel synthetic strategies using microflow technology [
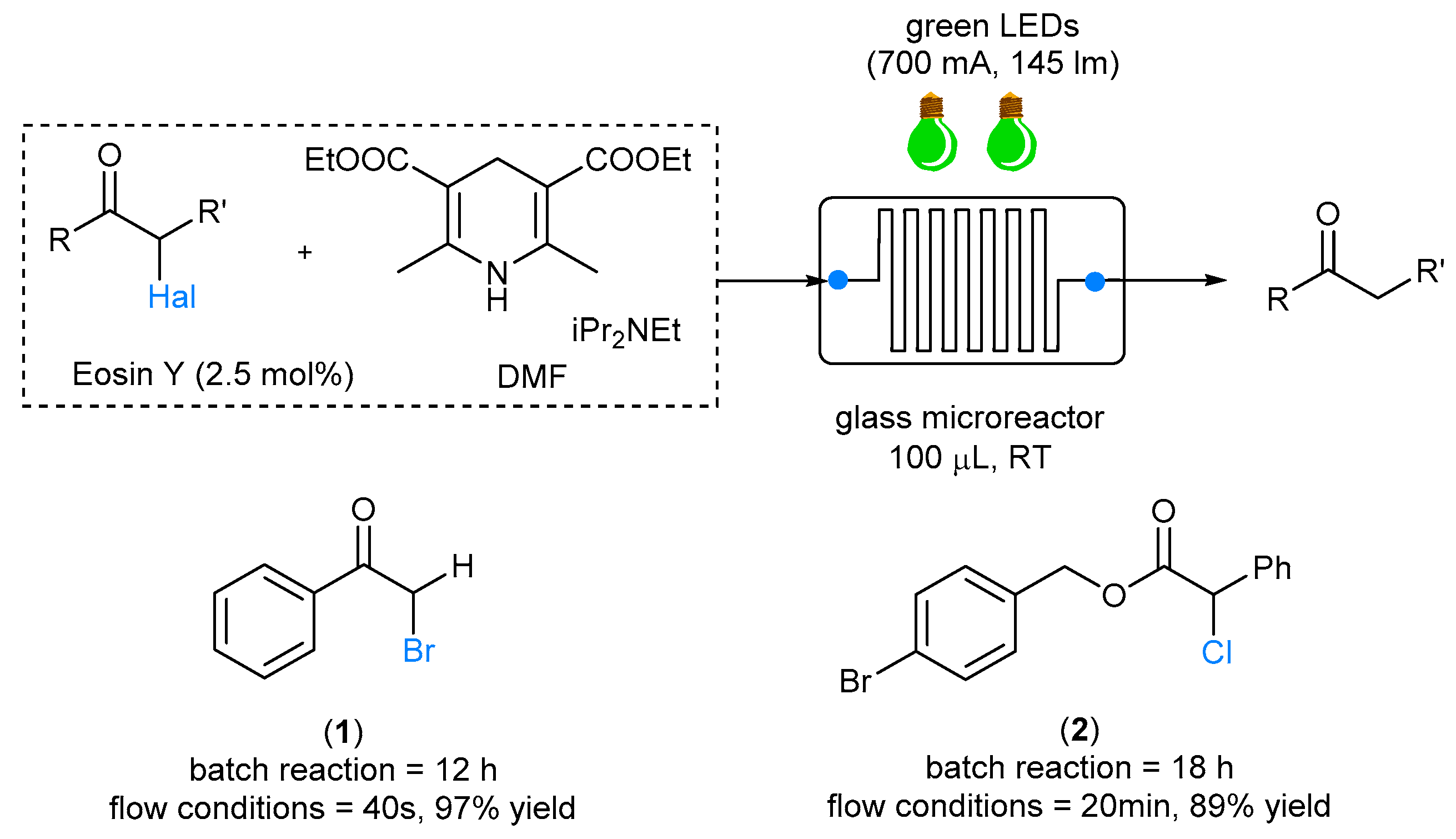
22]. Furthermore, organic dyes such as Eosin Y have been widely studied and proved to be economically and ecologically superior to metal-based catalysts in photocatalyzed organic synthesis. One of the first examples of photochemical transformation performed under fluidic conditions was reported in 2012 by Zeitler, who reported a photocatalytic reductive α-dehalogenation of carbonyl compounds catalyzed by Eosin Y and promoted by a Hantzsch ester [
25]. Eosin Y was successfully employed in the dehalogenation of activated halogenides using a microreactor setup under fluidic conditions. A premixed
N,
N-dimethylformamide (DMF) solution of halogenated compound (1) or (2), Eosin Y and iPr
2Net was fed into a commercially available glass microreactor (100 μL reaction volume, rectangular channel shape, 500 × 600 μm) equipped with two green LEDs (operating at 530 nm). Under these conditions, a tremendous acceleration of the dehalogenation process was observed compared to that in the traditional batch approach, and in the case of bromoacetophenone (1), the desired halogenated ketone was obtained in 40 s of residence time only (
Scheme 3). Interestingly, when compound (2) was used, no aromatic bromide dehalogenation was observed.
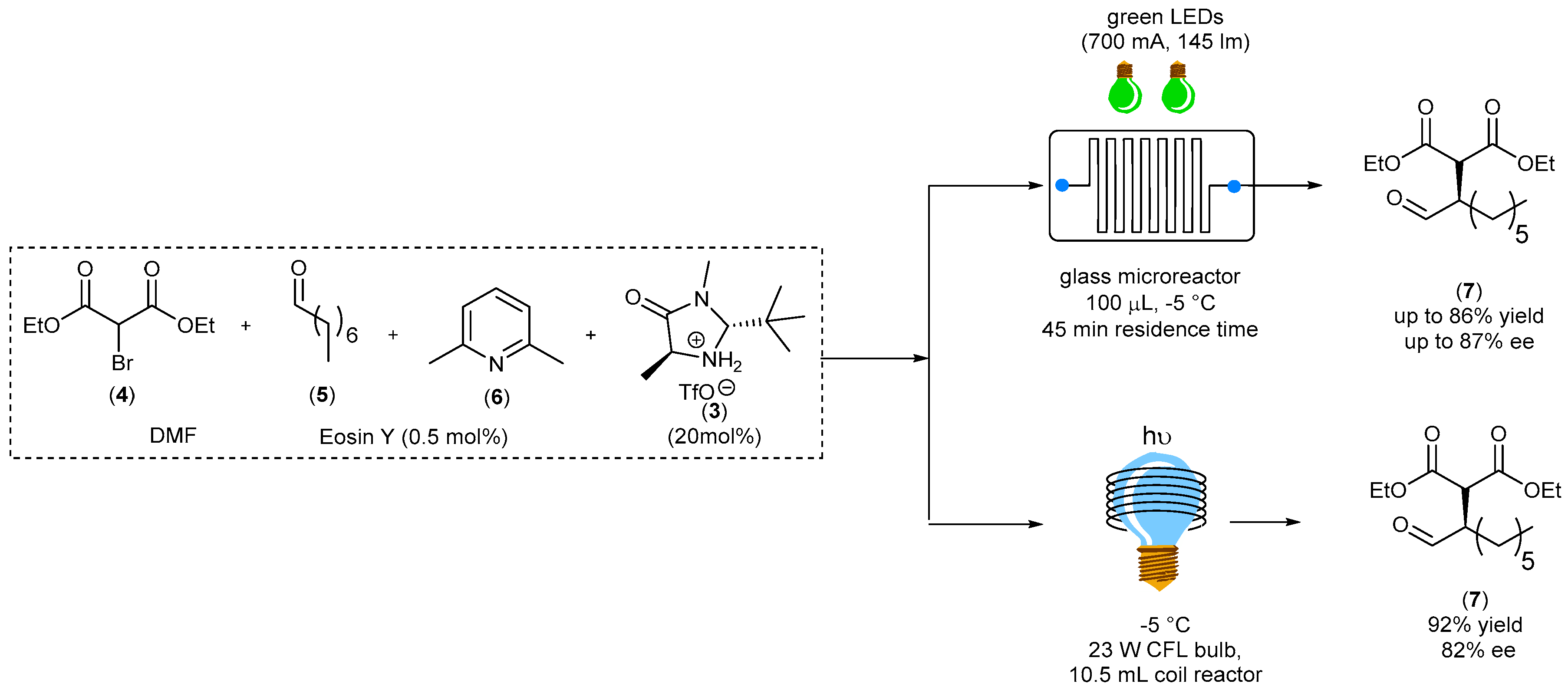
In the same paper, Zeitler reported also the α-alkylation of aliphatic aldehydes promoted by Eosin Y (0.5 mol%) in the presence of a catalytic amount of the second-generation MacMillan imidazolidinone triflate salt chiral catalyst (3). At the moment, this is the only example known related to the use of an organo-dye catalyst in a stereoselective transformation performed under fluidic conditions. Bromo-malonate (4) reacts with octanal (5) in the presence of catalyst (3), Eosin Y and 2,6-lutidine (6) to afford the corresponding α-alkylated aldehyde (7) under fluidic conditions (
Scheme 4).
The reaction was performed at −5 °C using two different fluidic devices: in one case, the reaction took place in a 100 μL glass microreactor equipped with two green LEDs, whereas in a second approach, a simple refrigerated HPLC tubing wrapped in coils around a compact fluorescent lamp (CFL) was used (inner diameter, 0.8 mm). In both cases, the desired product (7) was obtained with a high yield (88% using the microreactor vs. 92% using the coil system) and high level of enantioselectivity (88% and 82% ee, respectively) in short reaction time (45 min). It must be noticed that the same level of yield was obtained under traditional batch conditions only after 18 h.
2.1. Continuous Flow Photocatalytic Perfluoroalkylation Mediated by Eosin Y
In 2014, Rincón and Kappe developed an important example of a continuous flow two-step procedure for the synthesis of α-CF
3-substituted carbonyl compounds under photoredox catalysis [
26]. In their study, Eosin Y was proved the best catalyst in terms of conversion and selectivity for the α-trifluoromethylation of ketones compared to the Ru(bpy)
3Cl
2 catalyst employed a few years earlier by MacMillan to promote the same chemical transformation [
27]. Initially, the desired ketones were converted into the corresponding silylenol ethers using trimethylsilyl triflate (TMSOTf) and diisopropylethylamine (
iPr
2NEt) under fluidic conditions. The silylenol ether so generated was then fed into a second fluidic device and subjected to visible light in order to promote the radical trifluoromethylation using Eosin Y as a photoredox catalyst and triflyl chloride (CF
3SO
2Cl) as a source of CF
3 radicals (
Scheme 5).
The formation of the desired silylenol ether was achieved in transparent fluorinated ethylene propylene (FEP) tubing (1/8″ in diameter) at room temperature (RT) in only 2 min using tetrahydrofuran (THF) as a solvent. Eosin Y was introduced together with the substrate in order to avoid early interaction with the trifluoromethylation reagent. The silylenol ether was not isolated and was directly combined with triflyl chloride into a second flow reactor coiled around a glass cylinder in which a commercial cool-white household compact fluorescent lamp was inserted. This two-step process required 20 min only for completion and led to the formation of the corresponding trifluoromethylated ketones with high yields (56–87%), using low amounts of Eosin Y (0.5 mol%).
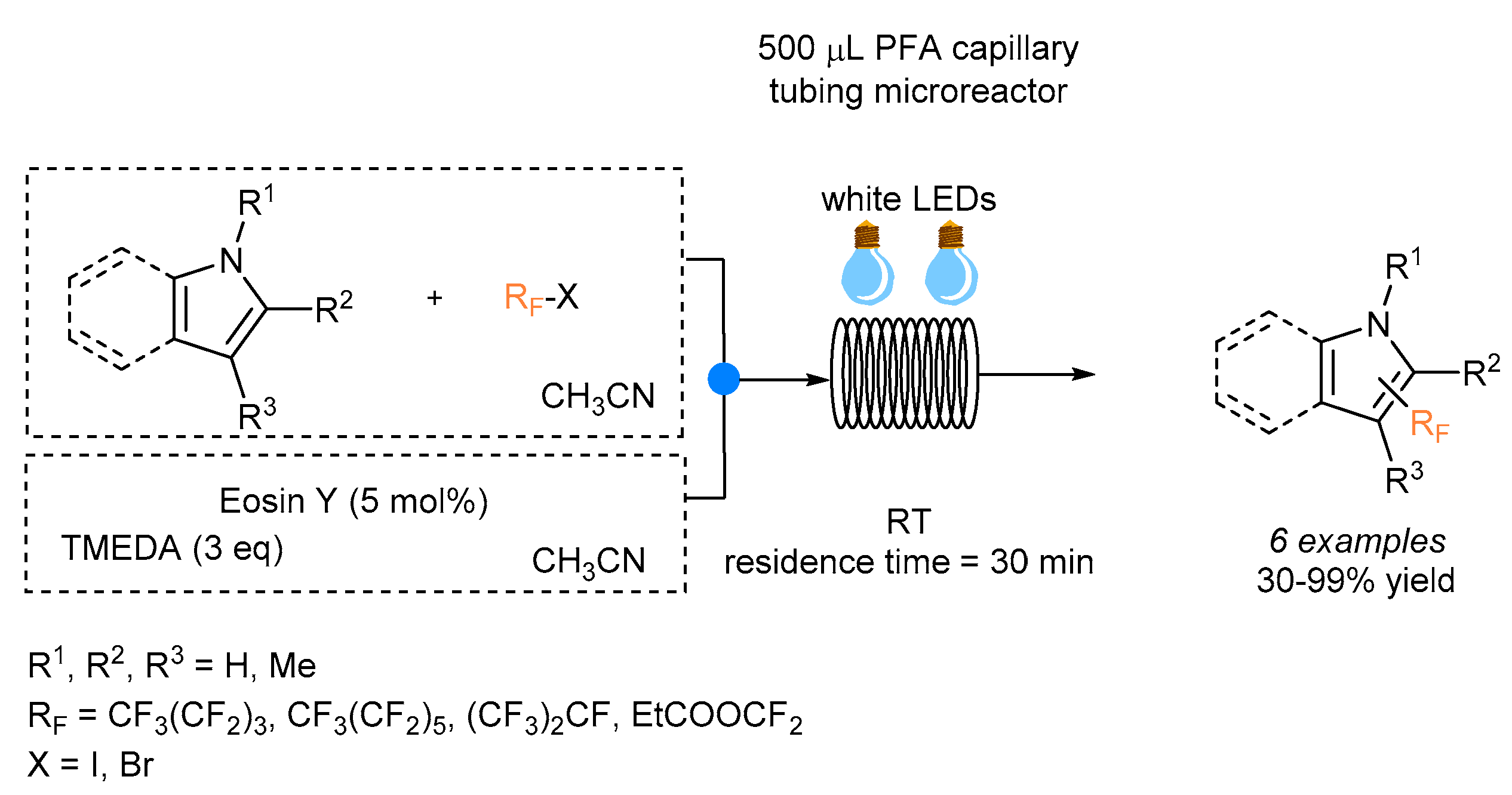
In the same year, Nöel and co-workers reported the photocatalytic perfluoroalkylation of pyrroles and indoles using different perfluoroalkanes in the presence of Eosin Y as a photoredox catalyst [
28]. The microreactor was obtained with transparent perfluoroalkoxy (PFA) capillary tubing (500 μL in volume; internal diameter, 750 μm), and the mixture was irradiated with white LEDs for 30 min as the residence time (
Scheme 6). A solution of Eosin Y (5 mol%) and
N,
N,
N′,
N′-tetramethyl-1,2-ethanediamine (TMEDA) was combined with a solution of the heteroarene premixed with the desired perfluoroalkyl iodides under continuous flow conditions. The presence of TMEDA is necessary to close the catalytic cycle, and the desired products were obtained with yields ranging from 30% to 99%. Lower yields were observed when using perfluoroalkyl bromides, since it is known that they are less effective in promoting this type of transformation.
The same group, after a slight modification of the continuous flow setup, was able to perform the trifluoromethylation of the heteroarenes using dangerous gaseous CF3I. In this case, a mass flow controller was introduced in the system, and N-methylpirrole and 2-methylindole were functionalized with 75% and 47% yields, respectively.
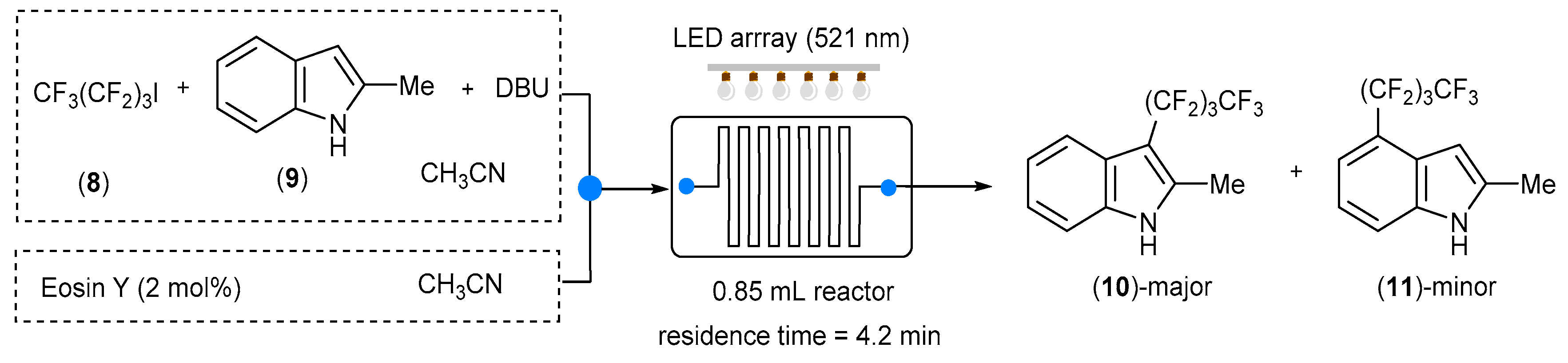
More recently, Tallarek and co-workers reported a technological improvement of this transformation obtained by a systematic study on LED emission bands (350–700 nm) with identical irradiance [
29]. A homemade two-dimensional continuous-flow platform (0.85 mL total volume) equipped with an LED array (12 high-power LEDs, 1.6 W each) was used to perform the addition of nonafluoro-1-iodobutane (8) to 2-methylindole (9) in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and Eosin Y (2 mol%) as a photocatalyst. Under these conditions, the catalyst loading and the reaction time were significantly reduced compared to in Nöel’s work, and the desired product (10) was obtained with 70% yield and 90% selectivity after 4.2 min. Only small amounts of undesired product (11) were detected (
Scheme 7).
The LED array was fixed at the operative wavelength of 521 nm, in order to obtain the largest overlap with the absorption spectrum of Eosin Y. Under these conditions, the reaction time was significantly reduced, with a consequent improvement in terms of productivity. By contrast, no significant absorption of Eosin Y was observed when operating at wavelengths below 450 nm, and the product (10) was synthetized with a lower yield.
2.2. Continuous Flow Photocatalytic Oxidation Mediated by Eosin Y
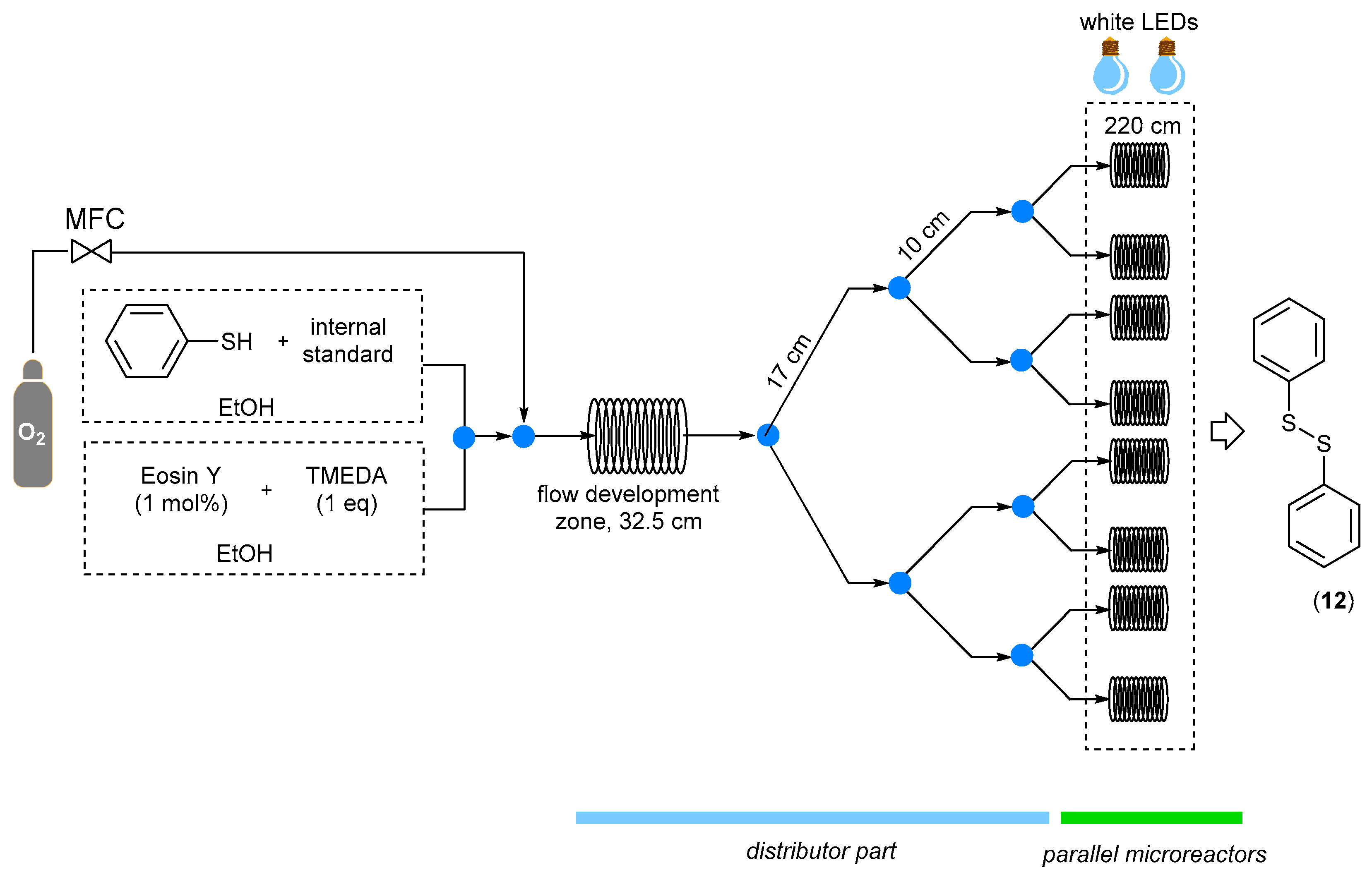
In 2016, it was found that Eosin Y could also operate in aerobic conditions; Su and co-workers demonstrated that thiols could be easily oxidized to disulfides under fluidic conditions by a convenient numbering-up strategy combining up to eight parallel reactors [
30]. Even if this work was focused on the evaluation of the feasibility of the numbering-up approach rather than the chemical application, it was possible to synthetize diphenyl sulfide (12) with a high yield operating with 1 mol% of Eosin Y as a photocatalyst in the presence of tetramethylethylenediamine (TMEDA) and pure oxygen (as oxidant) using a multicapillary photomicroreactor system (
Scheme 8).
The multicapillary microreactor system employed by Su was assembled starting from commercially available parts, and it is composed of two units: the first one guarantees the equal distribution of reagents to all parallel microreactors, whereas the second module consists of eight micro-photoreactors, each one irradiated by white LEDs. The substrate solution and the catalyst were filled into the device using a series of T-micromixers and combined with pure oxygen controlled by a gas mass flow controller, resulting in a two-phase flow. The flow rate ratio between the oxygen and the liquid phase was kept at 3:1, and different flow rates were investigated. The best results were obtained with 0.3 mL/min, leading to the formation of the desired product with up to 90% yield. Even if a small standard deviation in the flow distribution performance was observed (SD < 10%), the global yield of the numbered-up photomicroreactors was comparable to the yield obtained when a single fluidic device was used.
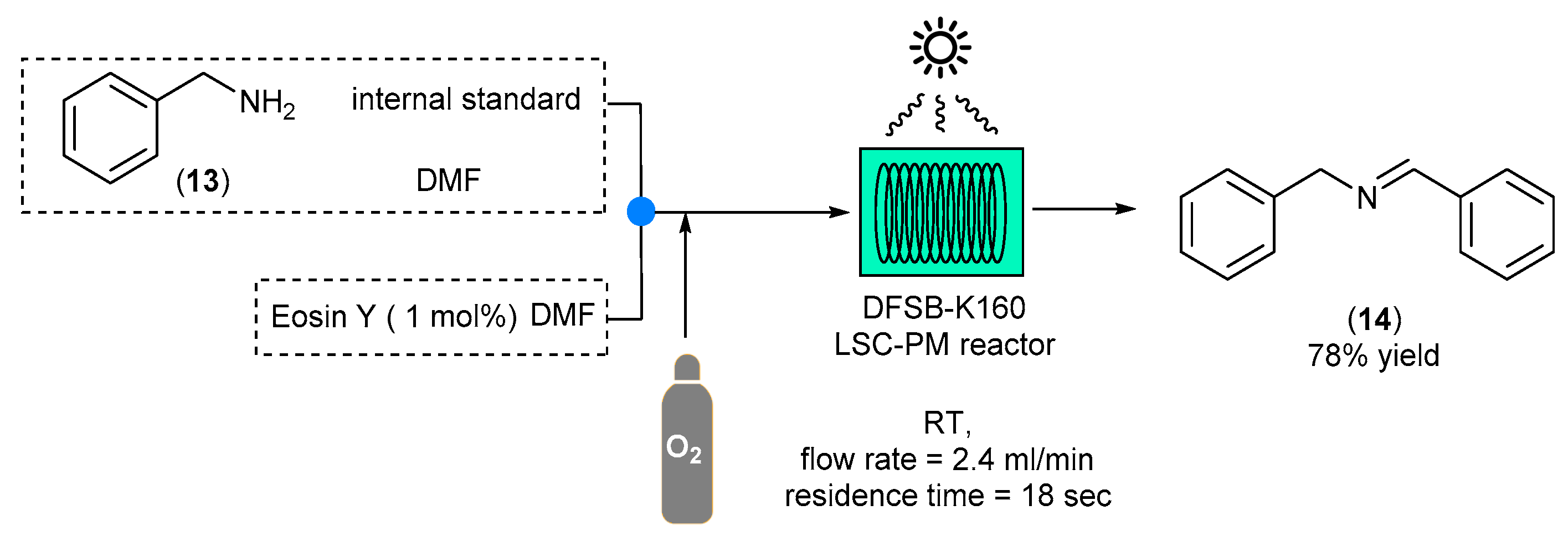
Another photocatalytic oxidation mediated by Eosin Y was reported in 2019 by Noel and co-workers [
22]. In this example, the authors reported the fabrication of a new fluidic device based on the combination of luminescent solar concentrators (LSC) and a photomicroreactor (PM). LCS are inexpensive slabs of luminophore-doped polymeric materials that are able to harvest and concentrate photons. LSC-PM devices couple sunlight modulation/concentration with microflow chemistry; under this system, the solar photons reaching the device are absorbed by the fluorescent dye and re-emitted. However, due to the nature of the system, emitted photons remain trapped in the polymeric material, which acts as a waveguide for the delivery of the down-converted photon to the reaction channels. In this example, perfluoroalkoxy alkane capillaries containing DFSB-K160 dye (emitting at 530 nm) were embedded in poly(methyl methacrylate) lightguides and were employed in the photocatalytic oxidation of benzylamine (13) to dibenzylimine (14) mediated by the presence of Eosin Y (
Scheme 9).
As the results, the desired product was obtained with a 78% yield after 18 s only (productivity: 30 mmol m2 h−1). The Eosin Y solution and the benzylamine solution were fed into the a 20 × 20 cm2 LSC-PM device using a syringe pump and mixed with pure oxygen with a 5:1 gas/liquid ratio with a flowrate of 2.02 mL/min for O2 and 405 μL/min for the liquid stream. The mixture was then irradiated under solar-simulated conditions, and the presence of the desired product was investigated by gas chromatography (GC).
3. Solid-Supported Eosin Y
As shown in the previous chapter, the possibility of operating under continuous flow conditions can overcome the intrinsic problems of the
Beer–Lambert–Bouguer Law related to the non-uniform irradiation of the solution. Using microfluidic devices, the surface-to-volume ratio can be easily increased by several orders of magnitude compared to that in the same process performed under the traditional batch protocol. Another way to overcome the problem consists of operating under heterogeneous conditions by anchoring the photocatalyst to a supporting material. This approach has been investigated by tethering Eosin Y to the solid phase in a covalent or ionic manner or by incorporating Eosin Y into the backbone of the material. An advantage of this approach consists of the easy workup isolation, and purification procedures from the catalytic species when the reaction is complete and, possibly, the reuse of the material in a second chemical transformation [
31]. In this section, organocatalytic photoredox reactions involving the use of “supported” Eosin Y will be discussed.
3.1. Functionalized Eosin Y Materials in Photocatalytic Applications
A straightforward way of supporting Eosin Y is the exploitation of the fact that it readily generates an anion. The
EY3 and
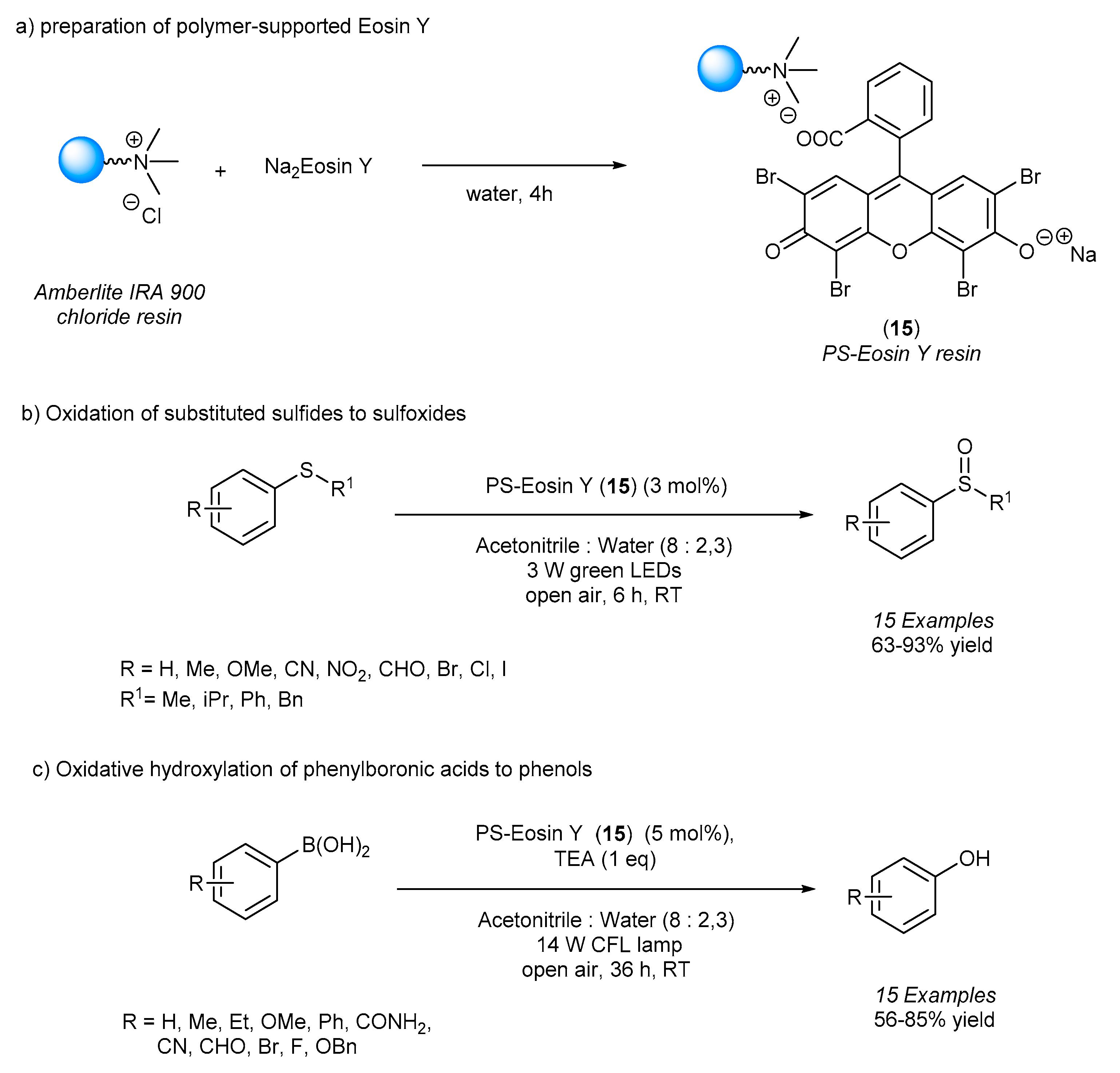
EY4 anionic forms of Eosin Y can be tethered by ionic interactions to a positively charged ion-exchange resin. In this sense, very recently, Selvaraj and co-workers reported the realization of a polystyrene-supported Eosin Y (PS-eosin Y,
15) through an anion-exchange reaction (
Scheme 10a) as a reusable photocatalyst for visible light-mediated organic transformations such as the oxidation of thioethers to sulfoxides and phenylboronic acids to phenols under open atmospheric air [
32].
In the first case, sulfoxides are generally obtained with modest-to-good yields; thioanisole bearing both electron-donating groups and electron-withdrawing groups underwent selective oxidation, leading to the formation of the corresponding sulfoxides with 64–88% yield when treated with 3 mol% of PS-eosin Y upon irradiation with 3 W green LEDs for 6 h at room temperature. Higher results were obtained when the alkyl chain of the sulfide was increased (
Scheme 10b). In the same manner, substituted phenylboronic acids irradiated using a 14 W household CFL lamp in the presence of 5 mol% of PS-eosin Y and triethylamine (TEA) as a sacrificial electron donor gave the desired phenols with 56–87% yield after 36 h (
Scheme 10c).
In both cases, the reaction requires only atmospheric air as an oxidant, and the functionalized resin can be recycled and reused up to six times without exhibiting a drop in chemical efficiency.
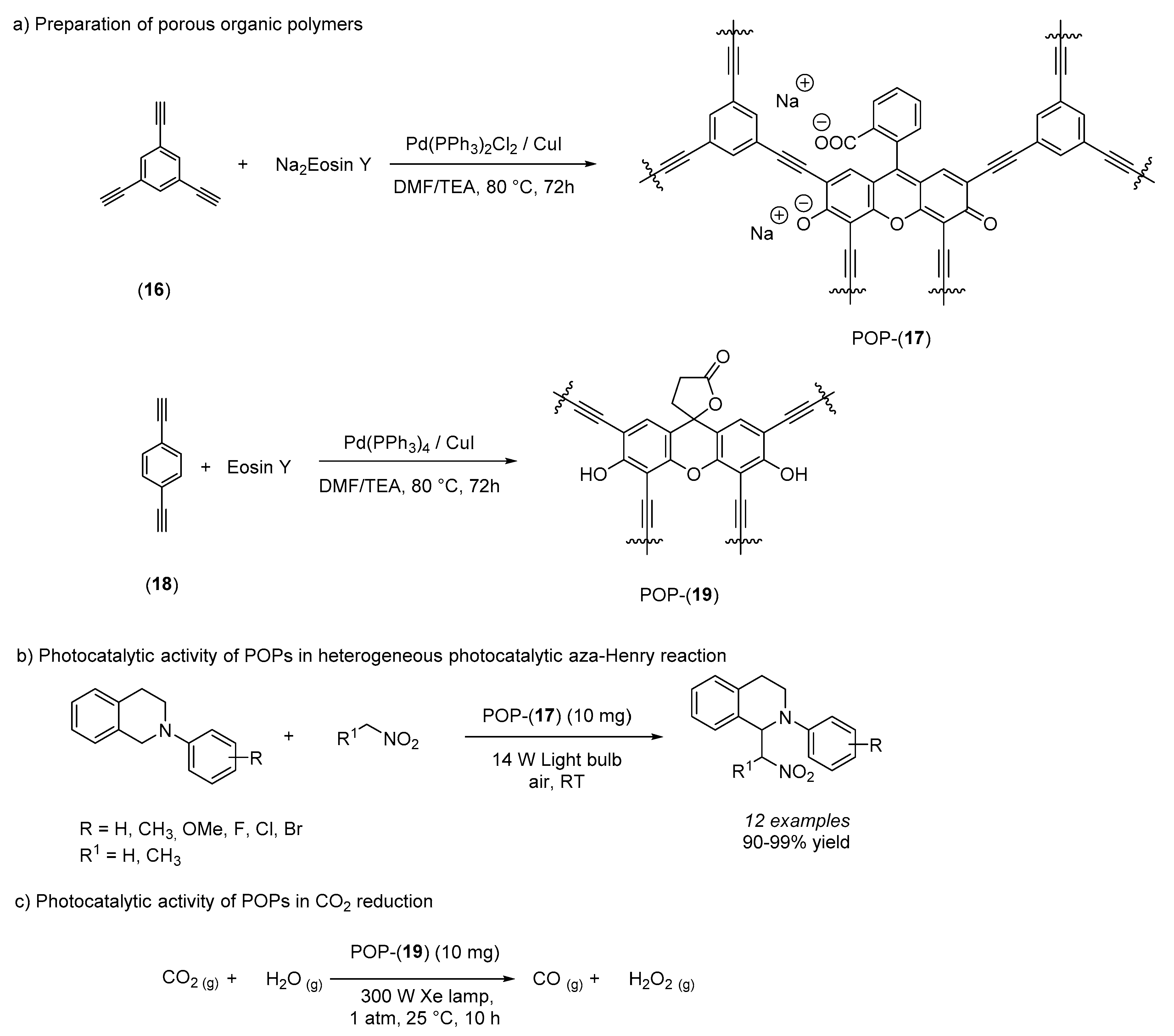
In a completely different approach, Wang and Han [
33] and Yang and Liu [
34] used the four bromine atoms of the Eosin Y backbone to create different porous organic polymers characterized by an extensive π-network, which have been employed in dehydrogenative aza-Henry reactions and in the photoreduction of CO
2 under light activation. Many porous organic polymers were synthetized, and high levels of efficiency were observed when POP-(
17) and POP-(
19) materials were used. These functionalized polymers were, respectively, synthetized by a palladium-catalyzed Sonogashira–Hagihara cross-coupling reaction between Eosin Y and 1,3,5-triethynylbenzene (
16) and 1,4-diethynylbenzene (
18) (
Scheme 11a). Morphological investigations confirmed that these polymers are microporous materials in which the Eosin Y monomer skeleton is perfectly embedded into the polymer network. POP-(
17) material has been successfully employed in the photocatalytic aza-Henry reaction of tetrahydroisoquinolines with nitroalkanes, leading to the formation of desired products with good-to-excellent yields after 24 h of irradiation with a 14 W fluorescent lamp. After reaction completion, the material was recovered and recycled twelve times without any significant loss of catalytic efficiency (
Scheme 11b). POP-(
19) material consists of a dark-red powder, which was employed to promote the photoreduction of CO
2 to CO in the presence of gaseous H
2O under visible-light irradiation under ambient conditions (
Scheme 11c). CO was produced with a production rate of 33 μmol g
−1 h
−1 and selectivity of 92%, which decreased to 73.5% after five runs.
In 2019, taking advantage of magnetic nanoparticle properties, Li and Wang reported the use of Eosin Y-functionalized nanoparticles to promote C-C and C-P coupling reactions under irradiation with green light [
35]. As illustrated in
Scheme 12, 15 nm magnetite silica core-shell nanoparticles were chemically modified with a benzyl linker bearing a quaternary ammonium-ion salt to ionically tether Eosin Y to the magnetic nanoparticle. The catalytic nanoparticles (NPs) Eosin Y-(
20) were then used in the photoredox catalytic generation of an iminium ion derived from different tetrahydroisoquinolines, which undergoes the nucleophilic addition of different nucleophiles such as nitroalkanes, malononitrile, dimethylmalonate and phosphites. The corresponding products were obtained with good-to-excellent yields, and the supported Eosin Y nanoparticles were removed by means of an external permanent magnet. The recovered nanoparticles where then used in subsequent runs up to eight times without Eosin Y leaching or without the chemical efficiency being affected; only a slight aggregation was observed in TEM imaging.
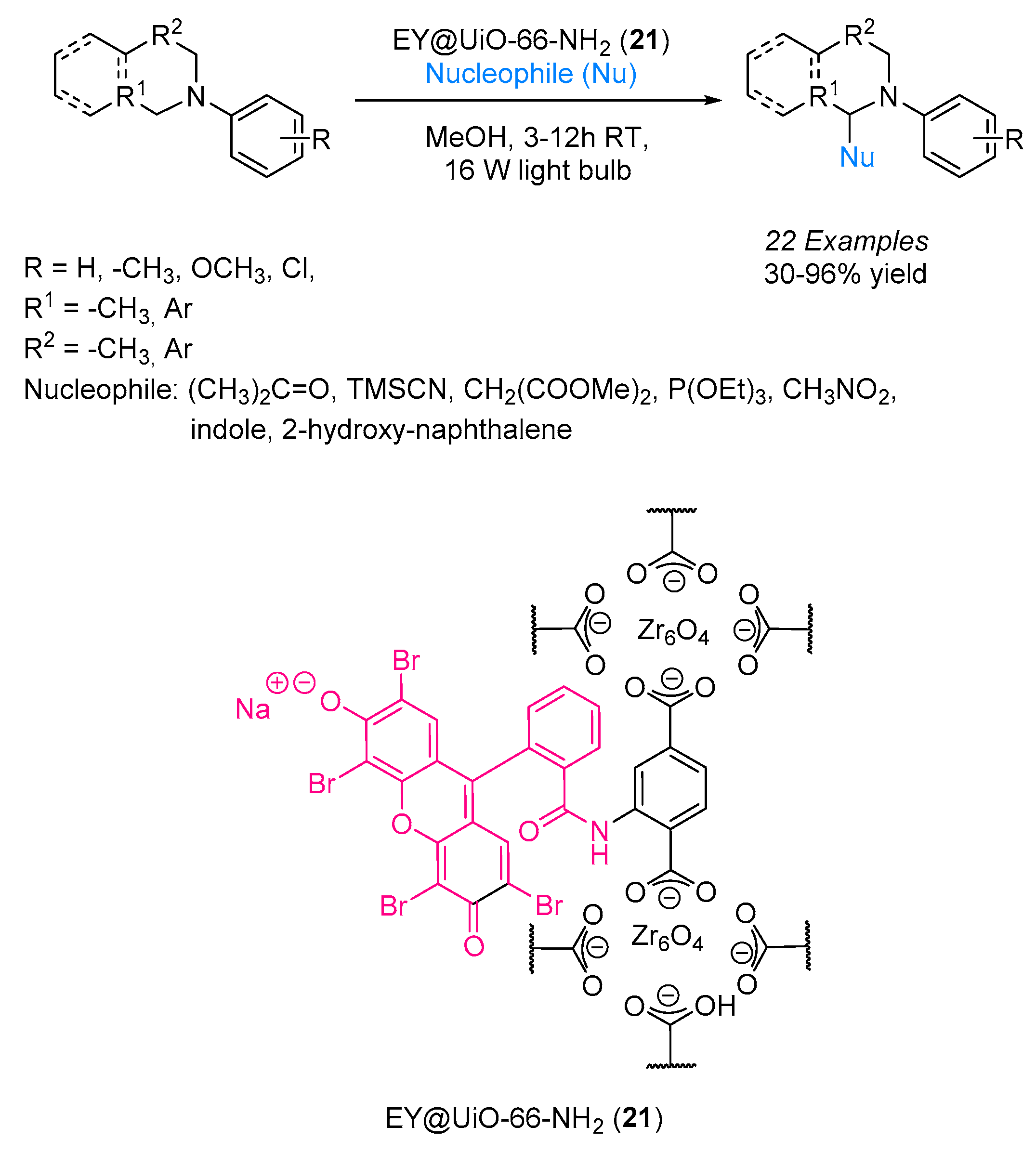
To further explore the versatility of Eosin Y anchored to different materials, Neogi and Khan exploited the well-defined dimension of metal–organic framework (MOF) pores as an anchor site for Eosin Y. A Zr(IV) framework was dye-functionalized and employed as a visible light heterogeneous photocatalyst in the functionalization of tertiary amines [
36]. The application of MOF as catalysts for the synthesis of fine chemicals is not new [
37], but in this application, the organic dye was covalently attached to the MOF by means of free amino groups available on the MOF surface. For this purpose, MOF UiO-66-NH
2 was selected and Eosin Y was anchored via an amide bond formation (
Scheme 13). The newly created material EY@UiO-66-NH
2 (
21) was able to efficiently catalyze the oxidation of tertiary amines to their respective iminium ions, which undergo a subsequent nucleophilic addition by several nucleophiles under the activation of visible green light. The photocatalytic cyanation of tertiary amines—as well as the addition of nitroalkanes, dimethylmalonate and triethyl phosphite to
N-aryl tetrahydroisoquinolines—was accomplished with good results, showing that the catalytic efficiency of the heterogeneous photocatalyst was superior to that observed for unsupported Eosin Y. In addition, a recycling test showed that this new material can be recycled up to 12 times without exhibiting a decrement in terms of reaction yield.
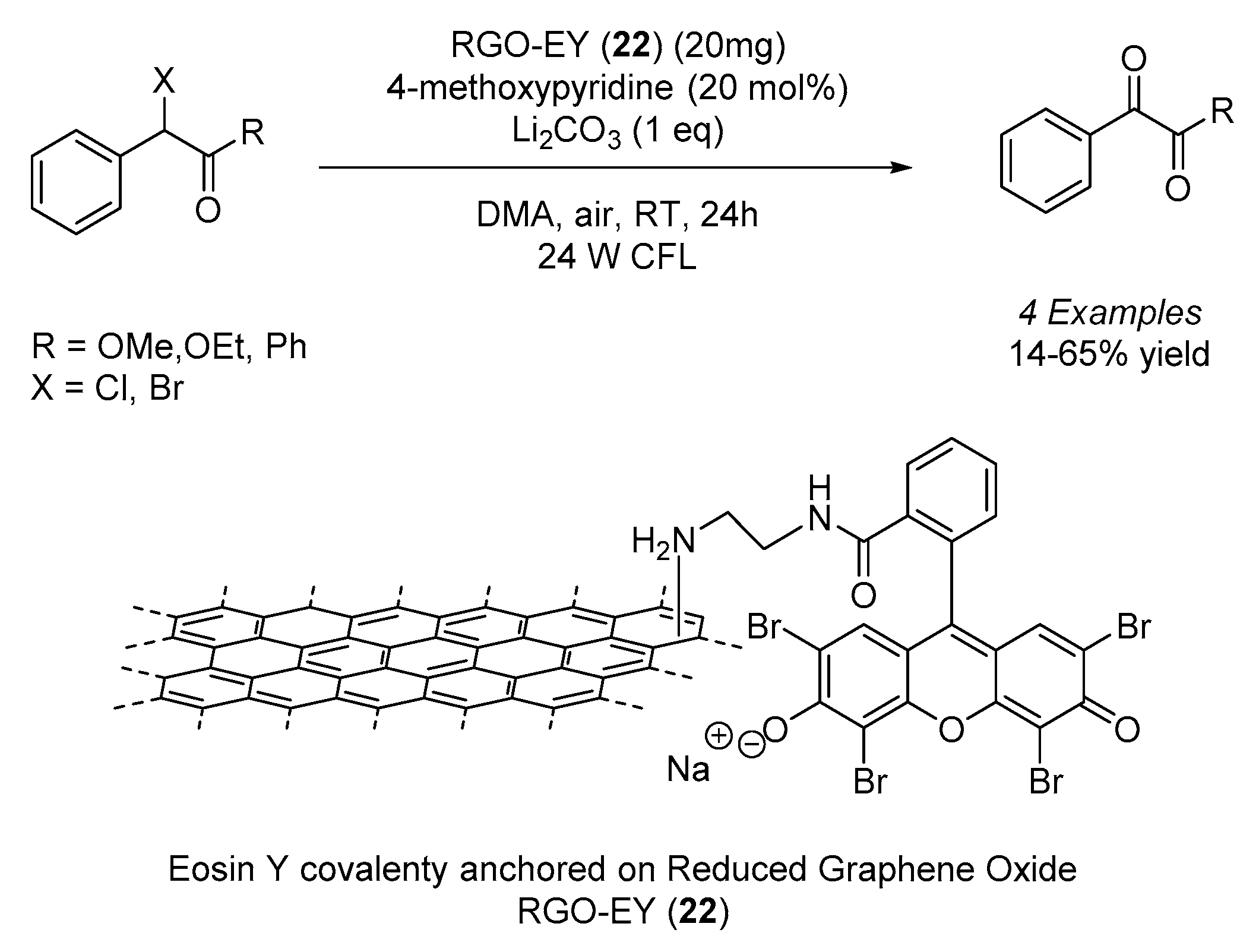
Eosin Y was also covalently anchored to other backbones, such as graphene. Over the last decades, this interesting material has attracted increasing attention due to its outstanding mechanical strength and electronic and optical properties. In this sense, Fan and co-workers used a reduced graphene oxide functionalized with a diethyleneamine linker to covalently tether Eosin Y to the surface, creating a new material of unique photocatalytic properties (
Scheme 14) [
38]. This new material (
22) was then applied in the photocatalytic aerobic oxidation of α-halo carbonyl compounds upon irradiation with a 24 W compact fluorescent lamp, leading to the formation of desired compounds with moderate yields. In addition, the recycling and reusing of the same material up to five times induced no significant drop in terms of its catalytic activity.
3.2. Functionalized Eosin Y Materials in Biomedical Imaging
High interest of scientists in the synthesis of materials that can provide high selectivity for tumor cells and low toxicity to normal cells has led to major breakthroughs in the field of biomedical imaging. Many types of materials have been studied for biomedical imaging applications; for example, it is known that Eosin Y can be used as photoinitiator both in the photo-crosslinking of hydrogels, which act as matrices for tissue engineering and biosensing [
39], and in the realization of new promising shear thinning/self-healing hydrogels for drug delivery purposes [
40].
Other studies have indicated significant applications of metal nanoparticles as a suitable material for the early disclosure of diseases [
41]. As shown before, Eosin Y has been supported on iron nanoparticles, but examples of Eosin Y-functionalized gold nanoparticles are also known [
42]. Unfortunately, one of the major issues of the limited use of nanoparticles in biomedicine relates to their sizes, which have been proved to play a critical role in the exploitation of their biological activity [
43]. Unfunctionalized nanoparticles exhibit hydrophobic surfaces and tend to agglomerate and form large clusters, which are not suitable for biological purposes.
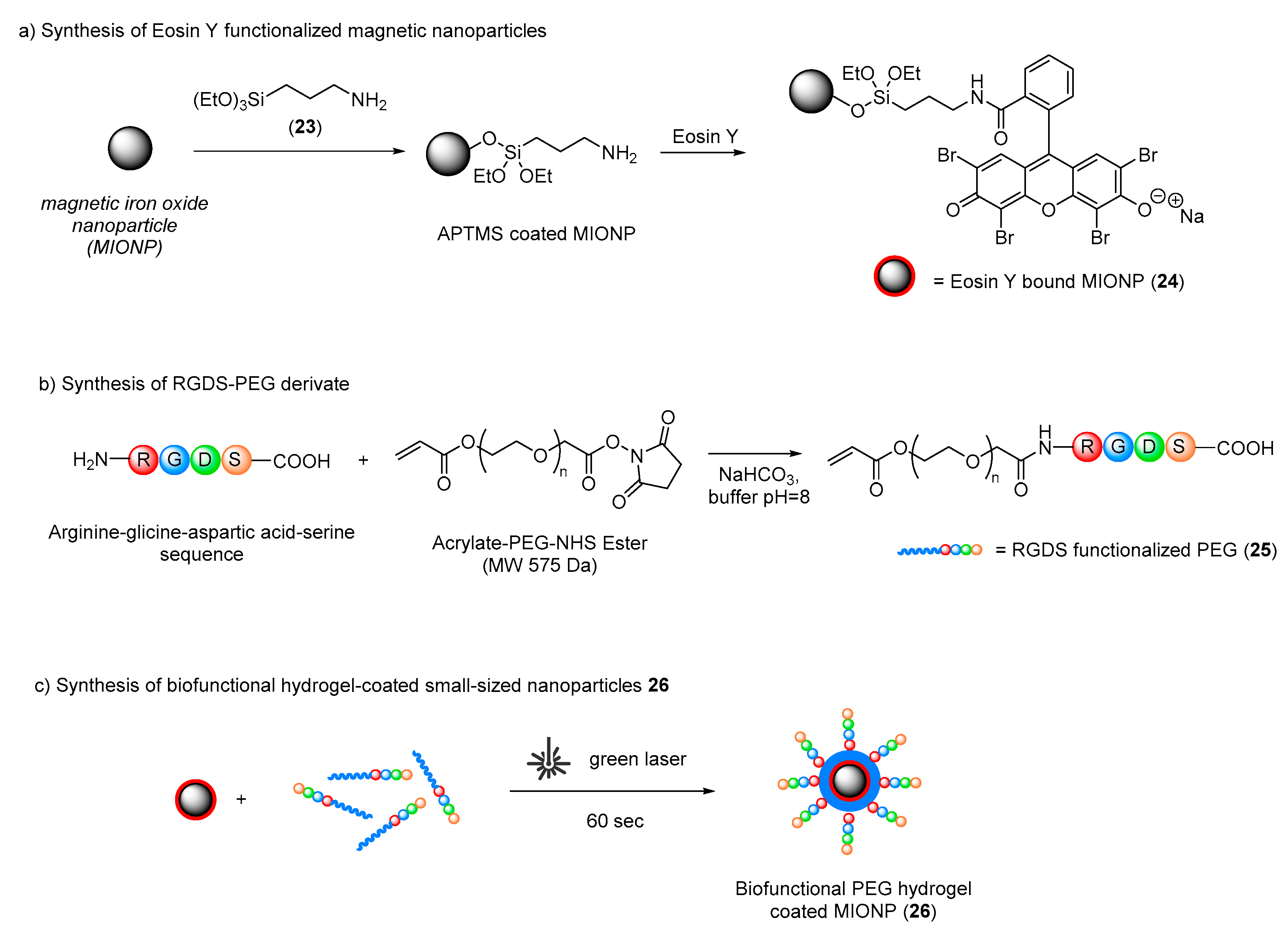
In order to develop more stable, specific and biocompatible derivatized materials, Nazli and co-workers investigated the possibility of combining the advantages of nanoparticles and hydrogels for the development of thin, biocompatible and biofunctional hydrogel-coated small-sized nanoparticles for specific cell targeting in biomedical imaging and cancer therapy [
43]. In this particular approach, magnetic iron oxide nanoparticles were first derivatized with Eosin Y to form nanoparticles (
24) and then coated with a biofunctional polyethylene glycol (PEG) hydrogel by photopolymerization to generate (
26). Biological activity was ensured by the presence of the peptide sequence arginine–glycine–aspartic acid–serine (RGDS sequence), which showed great ability to bind HeLa cancer cell receptors (
Scheme 15). Nazli, for the first time, employed the surface-initiated photopolymerization technique to encapsulate a single magnetic nanoparticle (or its small cluster) within a biofunctional polyethylene glycol (PEG) hydrogel. Eosin Y, as a photosensitizer, had a great impact on the coating of the biofunctional PEG hydrogel through a photopolymerization process on the nanoparticle surface.
Nanoparticles were first coated with 3-aminopropyltrimethoxysilane (23) (APTMS); then, the free amino groups introduced were used to covalently bond Eosin Y onto the nanoparticle core. A solution of these nanoparticles was then mixed with the RGDS–PEG derivative (25) and irradiated under green light in order to create the hydrogel shell around the nanoparticles. Different exposure times were evaluated in order to tune the hydrogel thickness around the magnetic core, and the best results were obtained with 60 s of green laser illumination. After biological investigations, it was shown that RGDS-functionalized polyethylene glycol hydrogel-coated magnetic iron oxide nanoparticles enhanced specific intracellular uptake by HeLa cells and increased the viability of cells compared to the uptake and viability measured in control groups.
Although more studies are needed, it could be stated that the use of nanoparticles as an inorganic material with organic molecules can serve as a platform for preparing biomedical building blocks that can enhance stability in physiological media and transportation to target cells. The formation of biologically acceptable PEG hydrogel nanocomposites via the photopolymerization technique is a good way to perfect the biocompatibility of the molecules used in biomedical imaging.
4. Outlook and Perspectives
The direct use of light in photochemistry is extremely appealing, and in the last few years, the demand for new energetically efficient chemical processes, which minimize their environmental and economic impacts, has led to the development of many photochemical transformations in which the use of more sustainable organic dyes are involved. In this context, it is not surprising that Eosin Y—thanks to its non-toxicity, lower cost, easy manageability and eco-friendly characteristics—became a landmark in photochemical applications. Small amounts of Eosin Y (generally less than 1 mol%) are able to promote many chemical transformations with good yields and short reaction times, making these organocatalytic processes comparable (and, in some cases, also more competitive) to their corresponding metal-based counterparts.
Organic dyes capable of capturing light and transforming it into chemical energy are now an attractive alternative to active metal complexes, and their immobilization on different supports (which have been proven to maintain excellent catalytic performance) to generate different photocatalytic materials expands their applications to unconventional fields, enabling transformations that are notoriously challenging.
In addition, photochemical processes promoted under fluidic conditions are gaining popularity due to the many advantages of this enabling technology compared to traditional processes. Unfortunately, in this case, their spread is being slowed down by some technical limitations: on one hand, there is the impossibility of using solids under flow conditions, and on the other hand, the variable radiance of light sources (which decreases when the usage time increases) makes these processes still unripe for industrial applications.
Despite this, Eosin Y remains an appealing and green photocatalyst for organic synthesis, and we believe that in this emerging field, there are still numerous possibilities to be explored, especially in combination with chiral organocatalysts for the development of stereoselective photoredox transformations, producing impactful research for years to come.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














