In Vitro and In Silico Screening of 2,4,5-Trisubstituted Imidazole Derivatives as Potential Xanthine Oxidase and Acetylcholinesterase Inhibitors, Antioxidant, and Antiproliferative Agents

 , , ,
, , ,  ,
,  , ,
, ,  , ,
, ,  , and
, and
Abstract
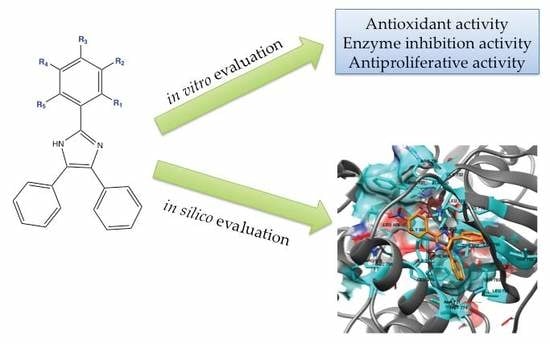
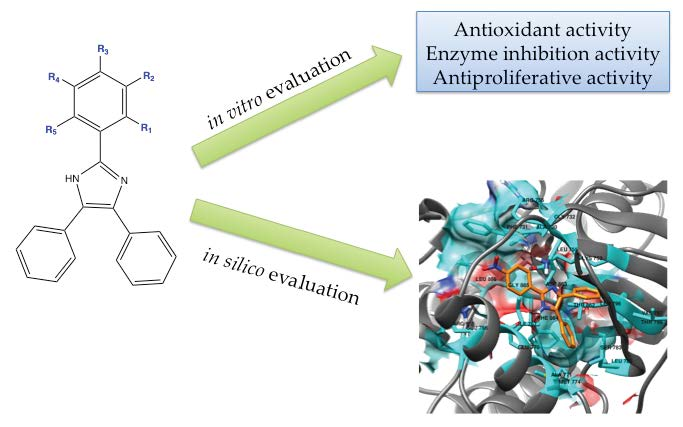
:
1. Introduction
2. Materials and Methods
2.1. General Information
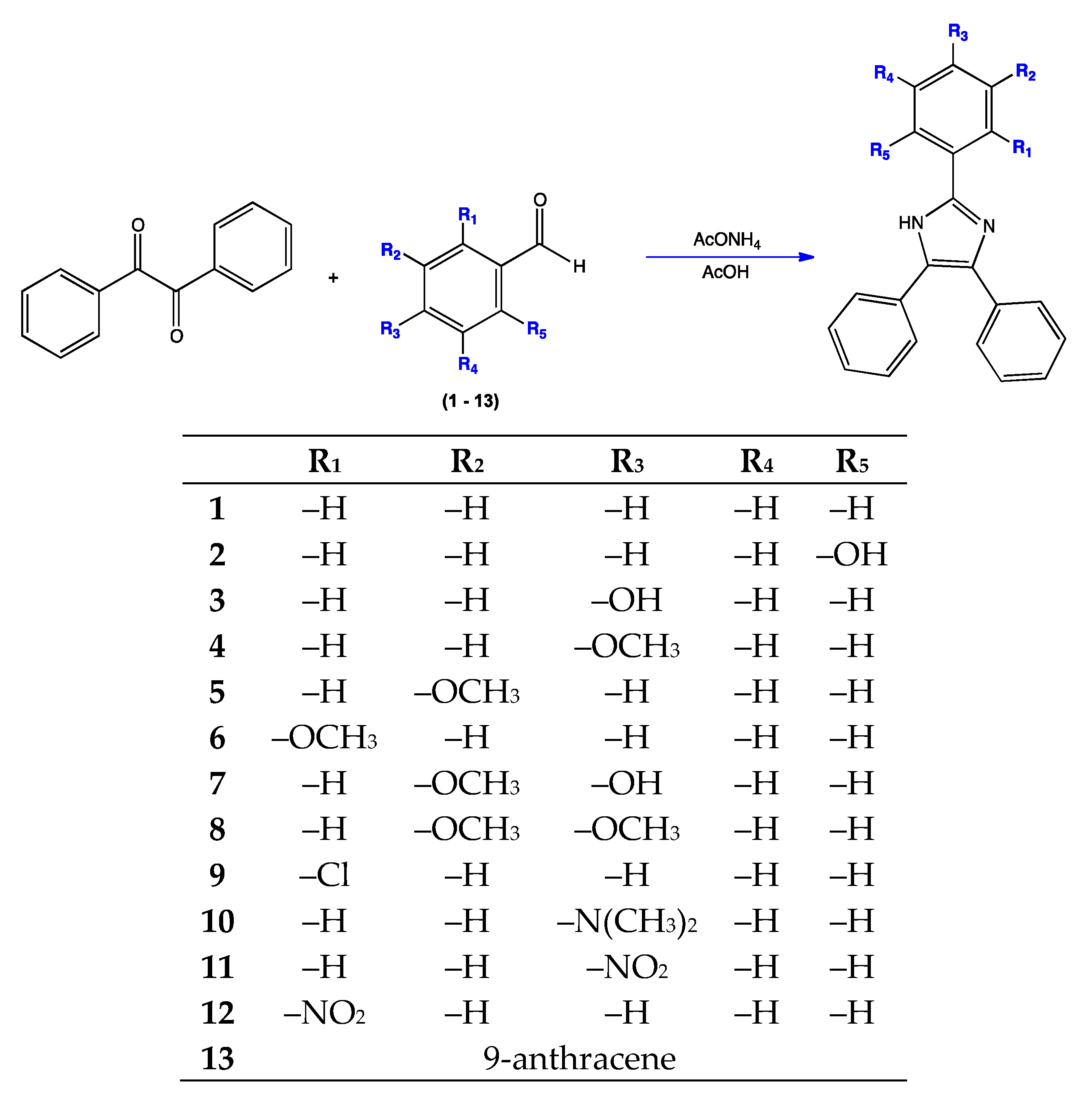
2.2. Synthesis of Triphenyl Imidazole Derivatives
2.3. In Vitro Antioxidant Activity Assay
2.3.1. 1,1-Diphenyl-2-Picrylhydrazyl (DPPH) Radical-Scavenging Assay
2.3.2. ABTS Radical-Scavenging Assay
2.4. In Vitro Acetylcholinesterase Inhibitory Assay
2.5. In Vitro Xanthine Oxidase Inhibitory Assay
2.6. Cell Lines and Culture Conditions
2.7. In vitro Antiproliferative Assay
2.8. Molecular Docking
2.9. In Silico Drug-Likeness Prediction
3. Results and Discussion
3.1. Synthesis of Triphenyl Imidazole Derivatives
3.2. Antioxidant Activity
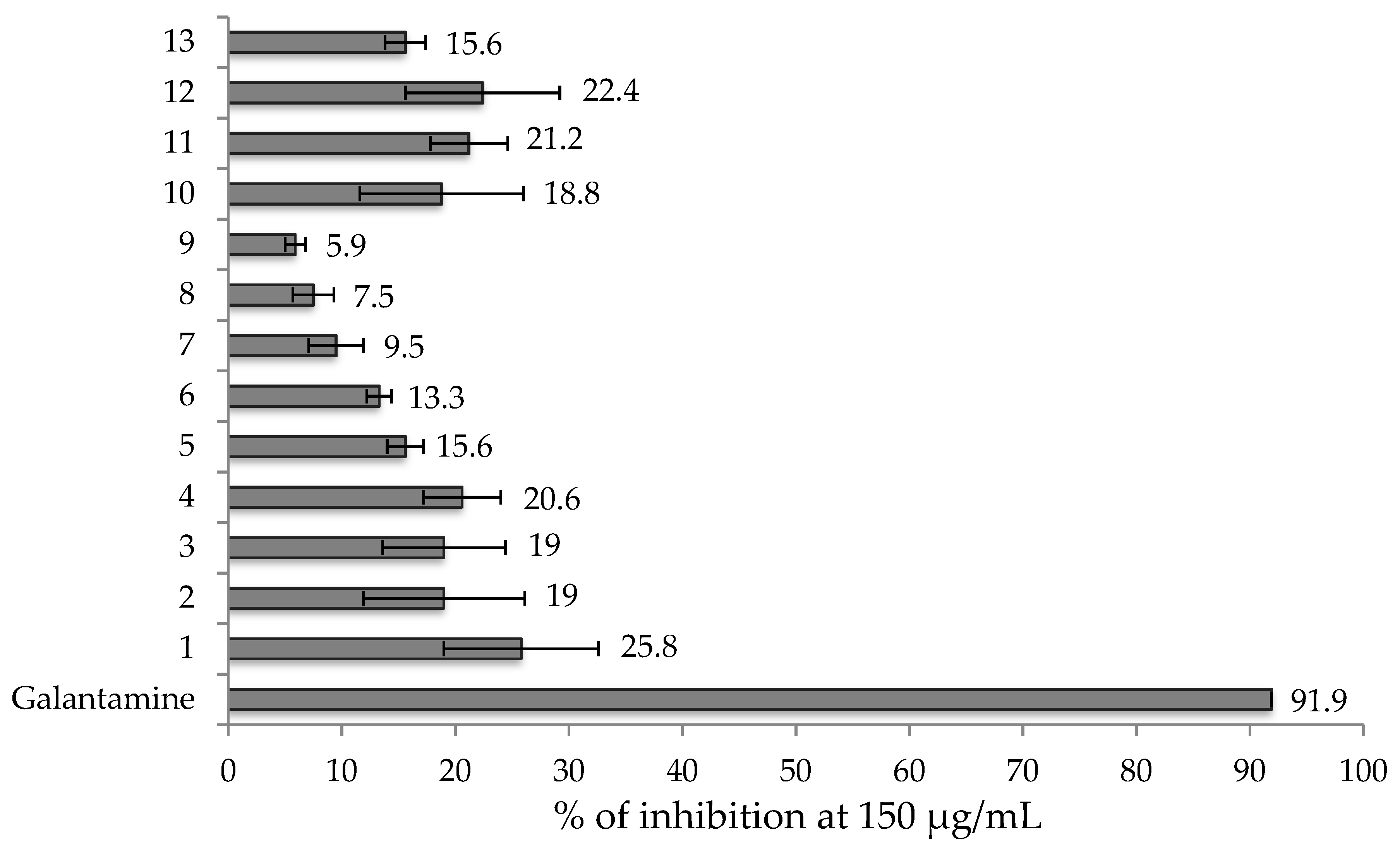
3.3. Acetylcholinesterase Inhibitory Assay
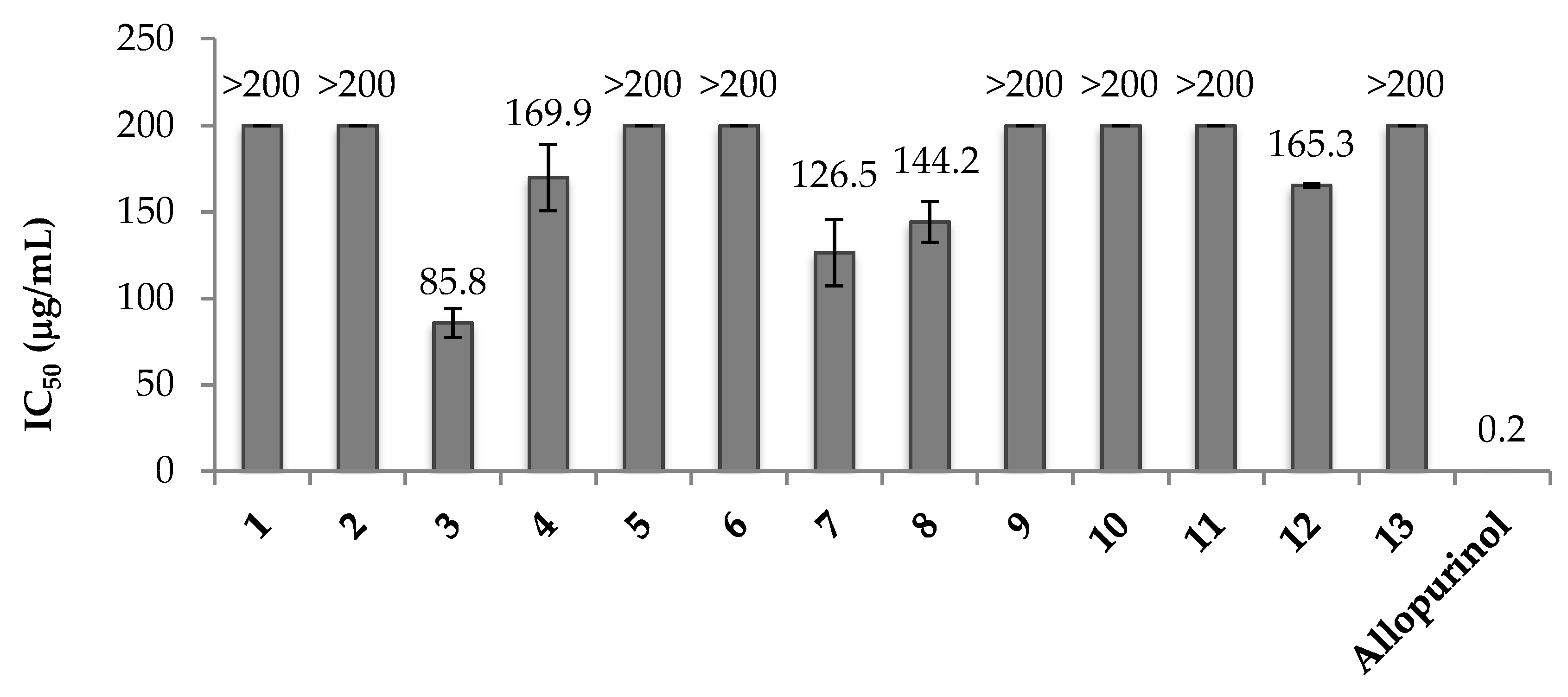
3.4. Xanthine Oxidase Assay
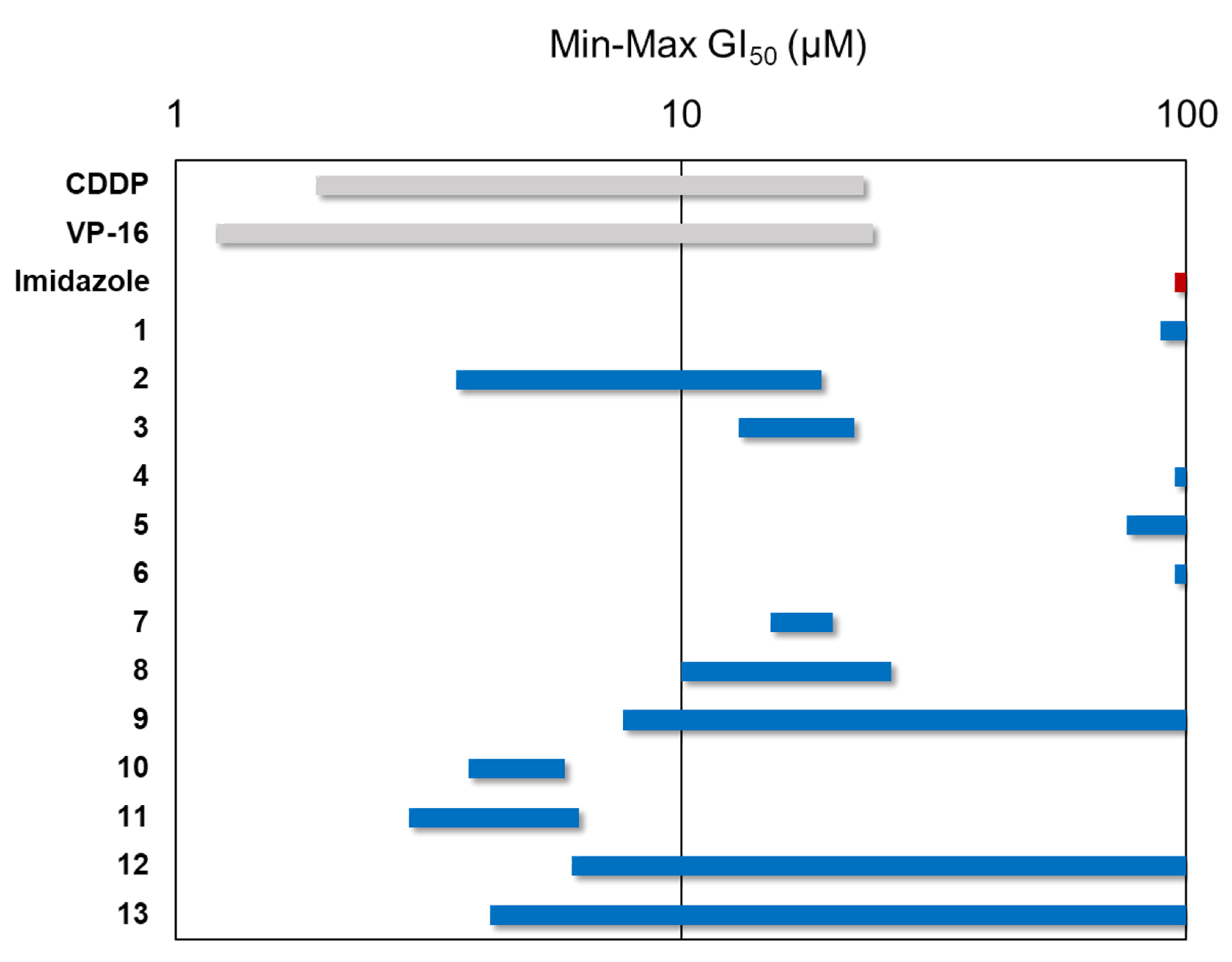
3.5. Antiproliferative Assay
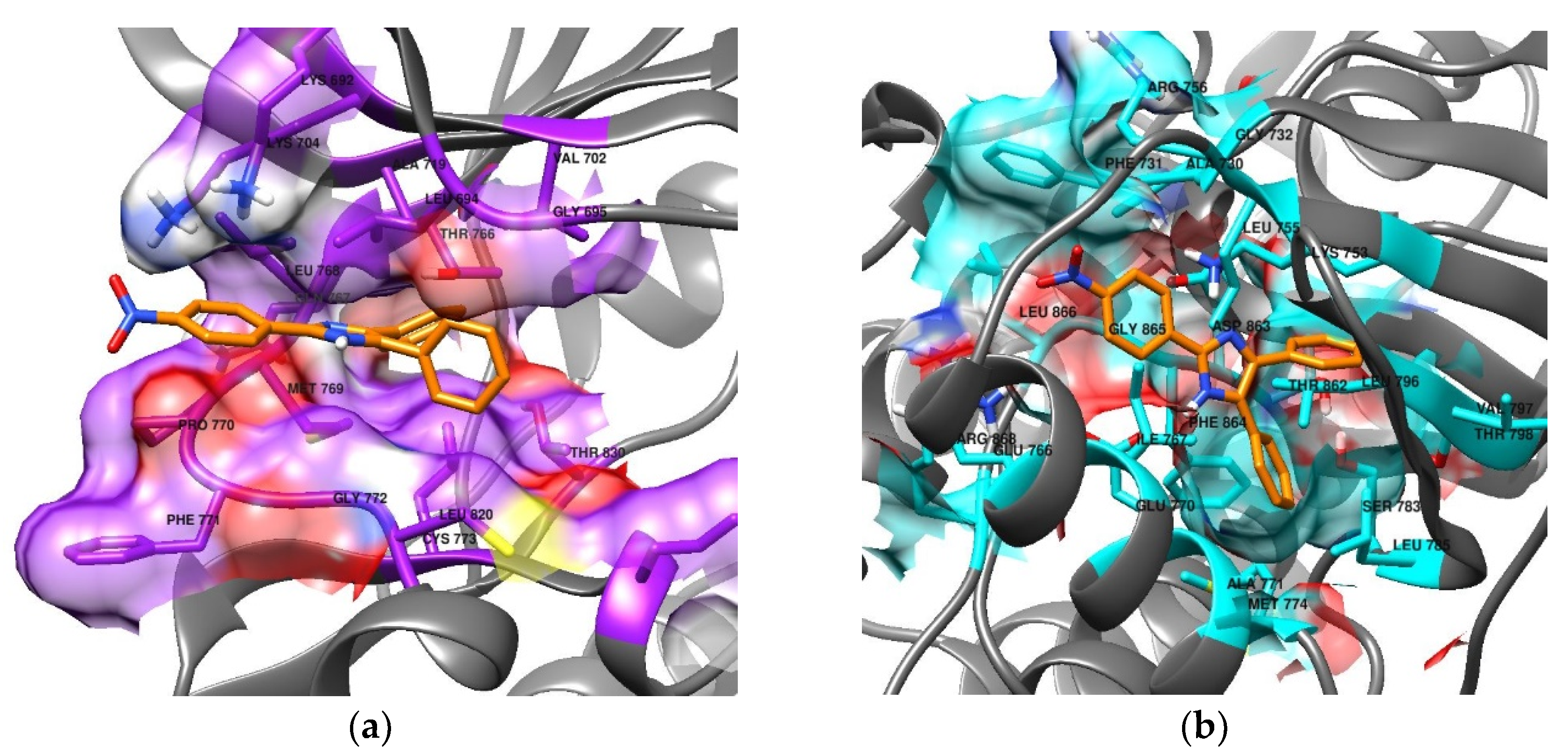
3.6. Molecular Docking
3.7. In Silico Drug-Likeness Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shingalapur, R.V.; Hosamani, K.M.; Keri, R.S. Synthesis and evaluation of in vitro anti-microbial and anti-tubercular activity of 2-styryl benzimidazoles. Eur. J. Med. Chem. 2009, 44, 4244–4248. [Google Scholar] [CrossRef] [PubMed]
- Achar, K.C.; Hosamani, K.M.; Seetharamareddy, H.R. In-vivo analgesic and anti-inflammatory activities of newly synthesized benzimidazole derivatives. Eur. J. Med. Chem. 2010, 45, 2048–2054. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L. Naturally occurring and synthetic imidazoles: Their chemistry and their biological activities. Curr. Med. Chem. 2006, 13, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Daraie, M.; Zadsirjan, V. Current advances in the synthesis and biological potencies of tri- and tetra-substituted 1H-imidazoles. Mol. Divers. 2015, 19, 577–623. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani-Vaghei, R.; Izadkhah, V.; Mahmoodi, J.; Karamian, R.; Ahmadi Khoei, M. The synthesis of imidazoles and evaluation of their antioxidant and antifungal activities. Monatsh. Chem. Chem. Mon. 2018, 149, 1447–1452. [Google Scholar] [CrossRef]
- Sorrenti, V.; Salerno, L.; Di Giacomo, C.; Acquaviva, R.; Siracusa, M.A.; Vanella, A. Imidazole derivatives as antioxidants and selective inhibitors of nNOS. Nitric Oxide 2006, 14, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, B.F.; Awad, G.E.A.; Badria, F.A. Synthesis, antimicrobial, antioxidant, anti-hemolytic and cytotoxic evaluation of new imidazole-based heterocycles. Eur. J. Med. Chem. 2011, 46, 1505–1511. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Tsikas, D.; Rossi, R. Oxidative stress and human diseases: Origin, link, measurement, mechanisms, and biomarkers. Crit. Rev. Clin. Lab. Sci. 2009, 46, 241–281. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef]
- Angoa Pérez, M.; Rivas Arancibia, S. Estrés oxidativo y neurodegeneración: ¿causa o consecuencia? Arch. Neurocien. 2007, 12, 45–54. [Google Scholar]
- Tran, T.D.; Nguyen, T.C.V.; Nguyen, N.S.; Nguyen, D.M.; Nguyen, T.T.H.; Le, M.T.; Thai, K.M. Synthesis of novel chalcones as acetylcholinesterase inhibitors. Appl. Sci. 2016, 6, 198. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s Disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Da Costa, J.S.; Bizarro Lopes, J.P.; Russowsky, D.; Petzhold, C.L.; de Amorim Borges, A.C.; Ceschi, M.A.; Konrath, E.; Batassini, C.; Santana Lunardi, P.; Saraiva Gonçalves, C.A. Synthesis of tacrine-lophine hybrids via one-pot four component reaction and biological evaluation as acetyl- and butyrylcholinesterase inhibitors. Eur. J. Med. Chem. 2013, 62, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šmelcerović, A.; Tomović, K.; Šmelcerović, Ž.; Petronijević, Ž.; Kocić, G.; Tomašič, T.; Jakopin, Ž.; Anderluh, M. Xanthine oxidase inhibitors beyond allopurinol and febuxostat; an overview and selection of potential leads based on in silico calculated physico-chemical properties, predicted pharmacokinetics and toxicity. Eur. J. Med. Chem. 2017, 135, 491–516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lv, Y.; Lei, Y.; Liu, D.; Feng, Y.; Zhao, J.; Chen, S.; Meng, F.; Wang, S. Design, synthesis and biological evaluation of 1-hydroxy-2-phenyl-4-pyridyl-1H-imidazole derivatives as xanthine oxidase inhibitors. Eur. J. Med. Chem. 2018, 146, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Aranda, R.; Granados-Guzmán, G.; Pérez-Meseguer, J.; González, G.M.; Waksman de Torres, N. Activity of Polyphenolic Compounds against Candida glabrata. Molecules 2015, 20, 17903–17912. [Google Scholar] [CrossRef] [PubMed]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Kuskoski, E.M.; Asuero, A.G.; Troncoso, A.M.; Mancini-Filho, J.; Fett, R. Aplicación de diversos métodos químicos para determinar actividad antioxidante en pulpa de frutos. Food Sci. Technol. 2005, 25, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Adewusi, E.A.; Moodley, N.; Steenkamp, V. Antioxidant and acetylcholinesterase inhibitory activity of selected southern African medicinal plants. S. Afr. J. Bot. 2011, 77, 638–644. [Google Scholar] [CrossRef] [Green Version]
- Almada-Taylor, G.; Díaz-Rubio, L.; Salazar-Aranda, R.; Waksman de Torres, N.; Uranga-Solis, C.; Delgadillo-Rodríguez, J.; Ramos, M.A.; Padrón, J.M.; Hernández-Martínez, R.; Córdova-Guerrero, I. Biological Activities of Extracts from Aerial Parts of Salvia pachyphylla Epling Ex Munz. Plants 2018, 7, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Nat. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [CrossRef]
- RSCB PDB Protein Data Bank. Available online: https://www.rcsb.org/ (accessed on 1 August 2019).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puratchikody, A.; Doble, M. Antinociceptive and antiinflammatory activities and QSAR studies on 2-substituted-4,5-diphenyl-1H-imidazoles. Bioorg. Med. Chem. 2007, 15, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Kumar, R.; Tiwari, S.; Khanna, R.S.; Tewari, A.K.; Khanna, H.D. Docking, synthesis and evaluation of antioxidant activity of 2,4,5-triaryl imidazole. Clin. Med. Biochem. Open Access 2015, 1, 105. [Google Scholar] [CrossRef] [Green Version]
- Foti, M.C. Antioxidant properties of phenols. J. Pharm. Pharmacol. 2007, 59, 1673–1685. [Google Scholar] [CrossRef]
- Hemalatha, S.; Naveena, P.A. Synthesis and biological evaluation of substituted diphenyl imidazole derivatives. World J. Pharm. Res. 2015, 4, 1321–1333. [Google Scholar]
- Zheng, L.; Zhao, M.; Xiao, C.; Zhao, Q.; Su, G. Practical problems when using ABTS assay to assess the radical-scavenging activity of peptides: Importance of controlling reaction pH and time. Food Chem. 2016, 192, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.L.; Bosch, P.; Lozano, A.E. Reactivity of Radicals derived from dimethylanilines in acrylic photopolymerization. Macromolecules 1994, 27, 7794–7799. [Google Scholar] [CrossRef]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept®): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef] [Green Version]
- Bartolucci, C.; Perola, E.; Pilger, C.; Fels, G.; Lamba, D. Three-dimensional structure of a complex of galanthamine (Nivalin®) with acetylcholinesterase from Torpedo californica: Implications for the design of new anti-Alzheimer drugs. Proteins 2001, 42, 182–191. [Google Scholar] [CrossRef]
- Zhao, X.-J.; Gong, D.-M.; Jiang, Y.-R.; Guo, D.; Zhu, Y.; Deng, Y.-C. Multipotent AChE and BACE-1 inhibitors for the treatment of Alzheimer’s disease: Design, synthesis and bio-analysis of 7-amino-1,4-dihydro-2H-isoquilin-3-one derivates. Eur. J. Med. Chem. 2017, 138, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Mughal, E.U.; Sadiq, A.; Murtaza, S.; Rafique, H.; Zafar, M.N.; Riaz, T.; Khan, B.A.; Hameed, A.; Khan, K.M. Synthesis, structure-activity relationship and molecular docking of 3-oxoaurones and 3-thioaurones as acetylcholinesterase and butyrylcholinesterase inhibitors. Bioorg. Med. Chem. 2017, 25, 100–106. [Google Scholar] [CrossRef]
- De la Torre, P.; Astudillo Saavedra, L.; Caballero, J.; Quiroga, J.; Alzate-Morales, J.H.; Gutiérrez Cabrera, M.; Trilleras, J. A novel class of selective acetylcholinesterase inhibitors: Synthesis and evaluation of (E)-2-(benzo[d]thiazol-2-yl)-3-heteroarylacrylonitriles. Molecules 2012, 17, 12072–12085. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Eger, B.T.; Nishino, T.; Pai, E.F.; Nishino, T. Mechanism of inhibition of xanthine oxidoreductase by allopurinol: Crystal structure of reduced bovine milk xanthine oxidoreductase bound with oxipurinol. Nucleosides Nucleotides Nucleic Acids 2008, 27, 888–893. [Google Scholar] [CrossRef]
- Matsumoto, K.; Okamoto, K.; Ashizawa, N.; Nishino, T. FXY-051: A novel and potent hybrid-type inhibitor of xanthine oxidoreductase. J. Pharmacol. Exp. Ther. 2011, 336, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Zhang, G.; Liao, Y.; Pan, J. Inhibition of chrysin on xanthine oxidase activity and its inhibition mechanism. Int. J. Biol. Macromol. 2015, 81, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tong, T.T.; Yau, L.F.; Chen, C.Y.; Mi, J.N.; Wang, J.R.; Jiang, Z.H. LC-MS based sphingolipidomic study on A549 human lung adenocarcinoma cell line and its taxol-resistant strain. BMC Cancer 2018, 18, 799. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Srinivasa, V.; Rangappa, S.; Mervin, L.; Mohan, S.; Paricharak, S.; Baday, S.; Li, F.; Shanmugam, M.K.; Chinnathambi, A.; et al. Trisubstituted-imidazoles induce apoptosis in human breast cancer cells by targeting the oncogenic PI3K/Akt/mTOR signaling pathway. PLoS ONE 2016, 11, e0153155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dake, S.A.; Kharat, K.R.; Yadav, A.R.; Kendrekar, P.S.; Pawar, R.P. In-vitro antiproliferative activity study of 2,4,5-triphenyl-1H-imidazole derivatives. J. Org. Inorg. Chem. 2017, 3, 5. [Google Scholar] [CrossRef]
- Lopes, M.S.; de Andrade Sena, C.F.; Silva, B.L.; de Souza, C.M.; Ramos, J.P.; Cassali, G.D.; de Souza-Fagundes, E.M.; Alves, R.J.; de Oliveira, M.C.; de Oliveira, R.B. Synthesis of nitroaromatic compounds as potential anticancer agents. Anticancer Agents Med. Chem. 2015, 15, 206–216. [Google Scholar] [CrossRef]
- Galindo-Hernández, O.; Córdova-Guerrero, I.; Díaz-Rubio, L.J.; Pulido-Capiz, A.; Díaz-Villanueva, J.F.; Castañeda-Sánchez, C.Y.; Serafín-Higuera, N.; García-González, V. Protein translation associated to PERK arm is a new target for regulation of metainflammation: A connection with hepatocyte cholesterol. J. Cell. Biochem. 2019, 120, 4158–4171. [Google Scholar] [CrossRef]
- Díaz-Rubio, L.; Hernández-Martínez, R.; Estolano-Cobián, A.; Chávez-Velasco, D.; Salazar-Aranda, R.; Waksman de Torres, N.; Rivero, I.A.; García-González, V.; Ramos, M.A.; Córdova-Guerrero, I. Synthesis, biological evaluation and docking studies of chalcone and flavone analogs as antioxidants and acetylcholinesterase inhibitors. Appl. Sci. 2019, 9, 410. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Aertgeerts, K.; Skene, R.; Yano, J.; Sang, B.-C.; Zou, H.; Snell, G.; Jennings, A.; Iwamoto, K.; Habuka, N.; Hirokawa, A.; et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 2011, 286, 18756–18765. [Google Scholar] [CrossRef] [Green Version]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: Overexpression and therapeutic implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef] [PubMed]
- Guda, R.; Kumar, G.; Korra, R.; Balaji, S.; Dayakar, G.; Palabindela, R.; Myadaraveni, P.; Yellu, N.R.; Kasula, M. EGFR, HER2 target based molecular docking analysis, in vitro screening of 2,4,5-trisubstituted imidazole derivatives as potential anti-oxidant and cytotoxic agents. J. Photochem. Photobiol. B Biol. 2017, 176, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.T.; Luque, F.J.; Barril, X. Toward accurate relative energy predictions of the bioactive conformation of drugs. J. Comput. Chem. 2009, 30, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, F.; Valabrega, G.; Aglietta, M. Lapatinib: A dual inhibitor of EGFR and HER2 tyrosine kinase activity. Expert Opin. Biol. Ther. 2007, 7, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Sulimov, A.V.; Kutov, D.C.; Katkova, E.V.; Sulimov, V.B. Combined docking with classical force field and quantum chemical semiempirical method PM7. Adv. Bioinform. 2017, 2017, 7167691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Antioxidant Activity (EC50, mg/mL) | |
|---|---|---|
| DPPH | ABTS | |
| 1 | 3.25 ± 0.137 | 34.312 ± 0.245 |
| 2 | 1.389 ± 0.631 | 0.188 ± 0.011 |
| 3 | 0.141 ± 0.094 | 0.168 ± 0.046 |
| 4 | 16.74 ± 0.003 | 1.644 ± 0.584 |
| 5 | 16.89 ± 0.636 | 37.223 ± 2.629 |
| 6 | 7.12 ± 1.916 | 15.643 ± 0.324 |
| 7 | 0.341 ± 0.101 | 0.199 ± 0.001 |
| 8 | 12.23 ± 3.042 | 1.964 ± 0.37 |
| 9 | 5.62 ± 1.752 | ND |
| 10 | 0.174 ± 0.041 | 0.162 ± 0.006 |
| 11 | ND | 8.025 ± 0.771 |
| 12 | ND | 42.158 ± 2.697 |
| 13 | 4.00 ± 0.135 | 0.449 ± 0.03 |
| Imidazole | >15 | >10 |
| * Quercetin | 0.052 ± 0.037 | 0.075 ± 0.002 |
| Compound | Substituent | Cell Lines (GI50, μM) | |||||
|---|---|---|---|---|---|---|---|
| A549 | HBL-100 | HeLa | SW1573 | T-47D | WiDr | ||
| 1b | -H | >100 | >100 | >100 | 89 | >100 | >100 |
| 2 | o-OH | 11 ± 5.5 | 7.0 ± 2.0 | 4.3 ± 0.6 | 3.6 ± 0.2 | 18 ± 0.3 | 19 ± 0.5 |
| 3 | p-OH | 19 ± 4.0 | 16 ± 0.4 | 13 ± 2.5 | 15 ± 2.3 | 20 ± 1.5 | 22 ± 0.7 |
| 4 | p-OMe | >100 | >100 | >100 | >100 | >100 | >100 |
| 5b | m-OMe | >100 | >100 | >100 | 76 | >100 | >100 |
| 6 | o-OMe | >100 | >100 | >100 | >100 | >100 | >100 |
| 7 | m-OMe, p-OH | 17 ± 1.3 | 17 ± 0.7 | 15 ± 1.7 | 15 ± 0.5 | 20 ± 2.0 | 17 ± 1.2 |
| 8 | m-OMe, p-OMe | 26 ± 1.0 | 15 ± 2.0 | 10 ± 0.4 | 15 ± 3.1 | 16 ± 1.2 | 13 ± 6.8 |
| 9b | o-Cl | >100 | >100 | 7.7 | 17 | >100 | >100 |
| 10 | p-N(CH3)2 | 3.8 | 5.9 ± 3.1 | 4.5 ± 1.2 | 4.4 ± 2.5 | 5.3 ± 1.9 | 4.5 ± 0.8 |
| 11 | p-NO2 | 6.3 ± 3.4 | 3.3 ± 1.6 | 3.0 ± 0.9 | 2.9 ± 0.4 | 5.5 ± 0.2 | 4.6 ± 0.2 |
| 12b | o-NO2 | >100 | >100 | 6.1 | 66 | >100 | >100 |
| 13 | 9-anthracene | 45 ± 14 | 23 ± 10 | 12 ± 1.5 | 4.2 ± 1.4 | >100 | >100 |
| Imidazole | - | >100 | >100 | >100 | >100 | >100 | >100 |
| CDDP | - | 4.9 ± 0.3 | 1.9 ± 0.2 | 1.9 ± 0.4 | 2.7 ± 0.4 | 17 ± 2.3 | 23 ± 4.3 |
| VP-16 | - | 1.5 ± 0.3 | 1.2 ± 0.3 | 2.4 ± 0.9 | 15 ± 1.5 | 18 ± 4.4 | 24 ± 2.6 |
| Compound | Binding Energy (kcal/mol) | |
|---|---|---|
| EGFR | HER2 | |
| 1 | −8.32 | −8.59 |
| 2 | −7.92 | −8.92 |
| 3 | −8.24 | −8.1 |
| 4 | −7.89 | −8.4 |
| 5 | −8.87 | −9.13 |
| 6 | −8.2 | −8.58 |
| 7 | −8.68 | −8.99 |
| 8 | −7.63 | −8.99 |
| 9 | −8.49 | −9.1 |
| 10 | −8.28 | −8.98 |
| 11 | −9.11 | −9.19 |
| 12 | −9.88 | −9.31 |
| 13 | −8.37 | −8.23 |
| Imidazole | −2.89 | −3.21 |
| Lapatinib | −10.48 | −9.88 |
| Descriptors | Compound | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | |
| MW (g/mol) | 296.37 | 312.36 | 312.36 | 326.39 | 326.39 | 326.39 | 342.39 | 356.42 | 330.81 | 339.43 | 341.36 | 341.36 | 396.48 |
| #H−bond acceptors | 1 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 1 | 1 | 3 | 3 | 1 |
| #H−bond donors | 1 | 2 | 2 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 |
| TPSA (Å2) | 28.68 | 48.91 | 48.91 | 37.91 | 37.91 | 37.91 | 58.14 | 47.14 | 28.68 | 31.92 | 74.5 | 74.5 | 28.68 |
| Consensus Log P | 4.59 | 4.26 | 4.17 | 4.56 | 4.55 | 4.52 | 4.27 | 4.54 | 5.15 | 4.6 | 4 | 3.95 | 6.36 |
| ESOL Log S | −5.4 | −5.24 | −5.24 | −5.44 | −5.44 | −5.44 | −5.29 | −5.49 | −5.97 | −5.59 | −5.42 | −5.42 | −7.6 |
| ESOL Class | MS | MS | MS | MS | MS | MS | MS | MS | MS | MS | MS | MS | PS |
| GI absorption | High | High | High | High | High | High | High | High | High | High | High | High | Low |
| BBB permeant | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | No | No | No |
| P−gp substrate | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | No | No |
| CYP1A2 inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No |
| CYP2C19 inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| CYP2C9 inhibitor | No | No | No | No | No | No | No | Yes | No | No | No | No | No |
| CYP2D6 inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | No |
| CYP3A4 inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | No | No |
| Lipinski #violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noriega-Iribe, E.; Díaz-Rubio, L.; Estolano-Cobián, A.; Barajas-Carrillo, V.W.; Padrón, J.M.; Salazar-Aranda, R.; Díaz-Molina, R.; García-González, V.; Chávez-Santoscoy, R.A.; Chávez, D.; et al. In Vitro and In Silico Screening of 2,4,5-Trisubstituted Imidazole Derivatives as Potential Xanthine Oxidase and Acetylcholinesterase Inhibitors, Antioxidant, and Antiproliferative Agents. Appl. Sci. 2020, 10, 2889. https://doi.org/10.3390/app10082889
Noriega-Iribe E, Díaz-Rubio L, Estolano-Cobián A, Barajas-Carrillo VW, Padrón JM, Salazar-Aranda R, Díaz-Molina R, García-González V, Chávez-Santoscoy RA, Chávez D, et al. In Vitro and In Silico Screening of 2,4,5-Trisubstituted Imidazole Derivatives as Potential Xanthine Oxidase and Acetylcholinesterase Inhibitors, Antioxidant, and Antiproliferative Agents. Applied Sciences. 2020; 10(8):2889. https://doi.org/10.3390/app10082889
Chicago/Turabian StyleNoriega-Iribe, Eduardo, Laura Díaz-Rubio, Arturo Estolano-Cobián, Victor Wagner Barajas-Carrillo, José M. Padrón, Ricardo Salazar-Aranda, Raúl Díaz-Molina, Victor García-González, Rocio Alejandra Chávez-Santoscoy, Daniel Chávez, and et al. 2020. "In Vitro and In Silico Screening of 2,4,5-Trisubstituted Imidazole Derivatives as Potential Xanthine Oxidase and Acetylcholinesterase Inhibitors, Antioxidant, and Antiproliferative Agents" Applied Sciences 10, no. 8: 2889. https://doi.org/10.3390/app10082889
APA StyleNoriega-Iribe, E., Díaz-Rubio, L., Estolano-Cobián, A., Barajas-Carrillo, V. W., Padrón, J. M., Salazar-Aranda, R., Díaz-Molina, R., García-González, V., Chávez-Santoscoy, R. A., Chávez, D., & Córdova-Guerrero, I. (2020). In Vitro and In Silico Screening of 2,4,5-Trisubstituted Imidazole Derivatives as Potential Xanthine Oxidase and Acetylcholinesterase Inhibitors, Antioxidant, and Antiproliferative Agents. Applied Sciences, 10(8), 2889. https://doi.org/10.3390/app10082889