
Influence of Preparation Conditions on the Catalytic Performance of Mo/H-ZSM-5 for Methane Dehydroaromatization


Abstract
:1. Introduction
2. Experimental Section
2.1. Catalyst Preparation
2.2. Characterization Methods
2.3. Catalytic Testing
3. Results and Discussions
3.1. Characterization Results
3.2. Catalytic Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gu, F.; Guo, J.; Zhang, W.; Summers, P.A.; Hall, P. From waste plastics to industrial raw materials: A life cycle assessment of mechanical plastic recycling practice based on a real-world case study. Sci. Total Environ. 2017, 601–602, 1192–1207. [Google Scholar] [CrossRef]
- Constantinescu, M.; Bucura, F.; Ionete, E.I.; Ion-Ebrasu, D.; Sandru, C.; Zaharioiu, A.; Marin, F.; Miricioiu, M.G.; Niculescu, V.-C.; Oancea, S.; et al. From Plastic to Fuel—New Challenges. Mater. Plast. 2019, 56, 721–729. [Google Scholar] [CrossRef]
- Constantinescu, M.; Bucura, F.; Ionete, R.-E.; Niculescu, V.-C.; Ionete, E.I.; Zaharioiu, A.; Oancea, S.; Miricioiu, M.G. Comparative Study on Plastic Materials as a New Source of Energy. Mater. Plast. 2019, 56, 41–46. [Google Scholar] [CrossRef]
- Clauser, N.M.; González, G.; Mendieta, C.M.; Kruyeniski, J.; Area, M.C.; Vallejos, M.E. Biomass Waste as Sustainable Raw Material for Energy and Fuels. Sustainability 2021, 13. [Google Scholar] [CrossRef]
- Varma, R.S. Biomass-Derived Renewable Carbonaceous Materials for Sustainable Chemical and Environmental Applications. ACS Sustain. Chem. Eng. 2019, 7, 6458–6470. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratenko, E.V.; Peppel, T.; Seeburg, D.; Kondratenko, V.A.; Kalevaru, N.; Martin, A.; Wohlrab, S. Methane conversion into different hydrocarbons or oxygenates: Current status and future perspectives in catalyst development and reactor operation. Catal. Sci. Technol. 2017, 7, 366–381. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Salvatore, A.; Gabriele, C.; Siglinda, P. Status of Research and Challenges in Converting Natural Gas. In Small-Scale Gas to Liquid Fuel Synthesis; CRC Press: Boca Raton, FL, USA, 2015; pp. 3–50. ISBN 978-1-4665-9938-3. [Google Scholar]
- Perathoner, S.; Gross, S.; Hensen, E.J.M.; Wessel, H.; Chraye, H.; Centi, G. Looking at the Future of Chemical Production through the European Roadmap on Science and Technology of Catalysis the EU Effort for a Long-term Vision. ChemCatChem 2017, 9, 904–909. [Google Scholar] [CrossRef]
- Armor, J.N. Emerging importance of shale gas to both the energy & chemicals landscape. J. Energy Chem. 2013, 22, 21–26. [Google Scholar]
- Zhou, W.; Cheng, K.; Kang, J.; Zhou, C.; Subramanian, V.; Zhang, Q.; Wang, Y. New horizon in C1 chemistry: Breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels. Chem. Soc. Rev. 2019, 48, 3193–3228. [Google Scholar] [CrossRef]
- Roberts, C.B.; Elbashir, N.O. An overview to “advances in C1 chemistry in the year 2002”. Fuel Process. Technol. 2003, 83, 1–9. [Google Scholar] [CrossRef]
- Wang, S.; Agirrezabal-Telleria, I.; Bhan, A.; Simonetti, D.; Takanabe, K.; Iglesia, E. Catalytic routes to fuels from C1 and oxygenate molecules. Faraday Discuss. 2017, 197, 9–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunsford, J.H. Catalytic conversion of methane to more useful chemicals and fuels: A challenge for the 21st century. Catal. Today 2000, 63, 165–174. [Google Scholar] [CrossRef]
- Al Abdullah, M.; Rodriguez Gomez, A.; Vittenet, J.; Bendjeriou-Sedjerari, A.; Xu, W.; Abba, I.A.; Gascon, J. A Viewpoint on the Refinery of the Future: Catalyst and Process Challenges. ACS Catal. 2020, 10, 8131–8140. [Google Scholar] [CrossRef]
- Caballero, A.; Pérez, P.J. Methane as raw material in synthetic chemistry: The final frontier. Chem. Soc. Rev. 2013, 42, 8809–8820. [Google Scholar] [CrossRef]
- Aasberg-Petersen, K.; Dybkjær, I.; Ovesen, C.V.; Schjødt, N.C.; Sehested, J.; Thomsen, S.G. Natural gas to synthesis gas—Catalysts and catalytic processes. J. Nat. Gas Sci. Eng. 2011, 3, 423–459. [Google Scholar] [CrossRef]
- Baliban, R.C.; Elia, J.A.; Floudas, C.A. Novel Natural Gas to Liquids Processes: Process Synthesis and Global Optimization Strategies. AIChE J. 2013, 59, 505–531. [Google Scholar] [CrossRef]
- Sousa-Aguiar, E.F.; Appel, L.G.; Mota, C. Natural gas chemical transformations: The path to refining in the future. Catal. Today 2005, 101, 3–7. [Google Scholar] [CrossRef]
- Gharibi, M.; Zangeneh, F.T.; Yaripour, F.; Sahebdelfar, S. Nanocatalysts for conversion of natural gas to liquid fuels and petrochemical feedstocks. Appl. Catal. A Gen. 2012, 443–444, 8–26. [Google Scholar] [CrossRef]
- Martínez, A.; Prieto, G.; García-Trenco, A.; Peris, E. Advanced catalysts based on micro- and mesoporous molecular sieves for the conversion of natural gas to fuels and chemicals. In Zeolites and Catalysis, Synthesis, Reactions and Applications; Cejka, J., Corma, A., Zones, S., Eds.; WILEY-VCH: Weinheim, Germany, 2010; p. 649. [Google Scholar]
- Portilla, M.T.; Tempelman, C.H.L.; Martínez, C.; Hensen, E.J.M. New Trends in Catalyst Design for Methane Dehydroaromatization. In Small-Scale Gas to Liquid Fuel Synthesis; CRC Press: Boca Raton, FL, USA, 2015; pp. 263–292. ISBN 978-1-4665-9938-3. [Google Scholar]
- Spivey, J.J.; Hutchings, G. Catalytic aromatization of methane. Chem. Soc. Rev. 2014, 43, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, B.M.; Wang, D.; Rosynek, M.P.; Lunsford, J.H. Conversion of methane to benzene over transition metal ion ZSM-5 zeolites: II. Catalyst characterization by X-ray photoelectron spectroscopy. J. Catal. 1998, 175. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.L.; Li, S.; Yuan, Y.; Zhang, W.X.; Wu, T.H.; Lin, L.W. Aromatization of methane in the absence of oxygen over Mo-based catalysts supported on different types of zeolites. Catal. Lett. 1998, 56, 207–213. [Google Scholar] [CrossRef]
- Shu, Y.; Ma, D.; Xu, L.; Bao, X.; Xu, Y. Effect of pore ring number of zeolites on catalytic performance of Mo/zeolite in methane aromatization under non-oxygen condition. Chin. J. Catal. 2002, 23, 24–28. [Google Scholar]
- Wang, L.; Tao, L.; Xie, M.; Xu, G.; Huang, J.; Xu, Y. Dehydrogenation and aromatization of methane under non-oxidizing conditions. Catal. Lett. 1993, 21, 35–41. [Google Scholar] [CrossRef]
- Ismagilov, Z.R.; Matus, E.V.; Kerzhentsev, M.A.; Tsikoza, L.T.; Ismagilov, I.Z.; Dosumov, K.D.; Mustafin, A.G. Methane conversion to valuable chemicals over nanostructured Mo/ZSM-5 catalysts. Pet. Chem. 2011, 51, 174–186. [Google Scholar] [CrossRef]
- Ismagilov, Z.R.; Matus, E.V.; Tsikoza, L.T. Direct conversion of methane on Mo/ZSM-5 catalysts to produce benzene and hydrogen: Achievements and perspectives. Energy Environ. Sci. 2008, 1, 526–541. [Google Scholar] [CrossRef]
- Wei, W. Direct dehydroaromatization of methane. J. Nat. Gas Chem. 2000, 9, 76–86. [Google Scholar]
- Bai, J.; Liu, S.; Xie, S.; Xu, L.; Lin, L. Comparison of 6Mo/MCM-22 and 6Mo/ZSM-5 in the MDA process. React. Kinet. Catal. Lett. 2004, 82, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Shu, Y.; Han, X.; Liu, X.; Xu, Y.; Bao, X. Mo/HMCM-22 catalysts for methane dehydroaromatization: A multinuclear MAS NMR study. J. Phys. Chem. B 2001, 105, 1786–1793. [Google Scholar] [CrossRef]
- Majhi, S.; Mohanty, P.; Wang, H.; Pant, K.K. Direct conversion of natural gas to higher hydrocarbons: A review. J. Energy Chem. 2013, 22, 543–554. [Google Scholar] [CrossRef]
- Shu, Y.; Ma, D.; Xu, L.; Xu, Y.; Bao, X. Methane dehydro-aromatization over Mo/MCM-22 catalysts: A highly selective catalyst for the formation of benzene. Catal. Lett. 2000, 70, 67–73. [Google Scholar] [CrossRef]
- Sobalík, Z.; Tvarůžková, Z.; Wichterlová, B.; Fíla, V.; Špatenka, Š. Acidic and catalytic properties of Mo/MCM-22 in methane aromatization: An FTIR study. Appl. Catal. A Gen. 2003, 253, 271–282. [Google Scholar] [CrossRef]
- Shu, Y.; Ohnishi, R.; Ichikawa, M. A highly selective and coking-resistant catalyst for methane dehydrocondensation. Chem. Lett. 2002, 418–419. [Google Scholar] [CrossRef]
- Smiešková, A.; Hudec, P.; Kumar, N.; Salmi, T.; Murzin, D.Y.; Jorík, V. Aromatization of methane on Mo modified zeolites: Influence of the surface and structural properties of the carriers. Appl. Catal. A Gen. 2010, 377, 83–91. [Google Scholar] [CrossRef]
- Liu, H.; Wu, S.; Guo, Y.; Shang, F.; Yu, X.; Ma, Y.; Xu, C.; Guan, J.; Kan, Q. Synthesis of Mo/IM-5 catalyst and its catalytic behavior in methane non-oxidative aromatization. Fuel 2011, 90, 1515–1521. [Google Scholar] [CrossRef]
- Liu, H.; Hu, J.; Li, Z.; Wu, S.; Liu, L.; Guan, J.; Kan, Q. Synthesis of zeolite IM-5 under rotating and static conditions and the catalytic performance of Mo/H-IM-5 catalyst in methane non-oxidative aromatization. Kinet. Catal. 2013, 54, 443–450. [Google Scholar] [CrossRef]
- Portilla, M.T.; Llopis, F.J.; Martinez, C. Non-oxidative dehydroaromatization of methane: An effective reaction-regeneration cyclic operation for catalyst life extension. Catal. Sci. Technol. 2015, 5, 3806–3821. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, S.; Wu, S.; Shang, F.; Yu, X.; Xu, C.; Guan, J.; Kan, Q. Synthesis of Mo/TNU-9 (TNU-9 Taejon National University No. 9) catalyst and its catalytic performance in methane non-oxidative aromatization. Energy 2011, 36, 1582–1589. [Google Scholar] [CrossRef]
- Ma, S.; Guo, X.; Zhao, L.; Scott, S.; Bao, X. Recent progress in methane dehydroaromatization: From laboratory curiosities to promising technology. J. Energy Chem. 2013, 22, 1–20. [Google Scholar] [CrossRef]
- Kosinov, N.; Hensen, E.J.M. Reactivity, Selectivity, and Stability of Zeolite-Based Catalysts for Methane Dehydroaromatization. Adv. Mater. 2020, 32, 2002565. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, I.; Ould-Chikh, S.; Aguilar-Tapia, A.; Li, G.; Pidko, E.; Hazemann, J.-L.; Kapteijn, F.; Gascon, J. Activity Descriptors Derived from Comparison of Mo and Fe as Active Metal for Methane Conversion to Aromatics. J. Am. Chem. Soc. 2019, 141, 18814–18824. [Google Scholar] [CrossRef]
- Çağlayan, M.; Lucini Paioni, A.; Abou-Hamad, E.; Shterk, G.; Pustovarenko, A.; Baldus, M.; Chowdhury, A.D.; Gascon, J. Initial Carbon−Carbon Bond Formation during the Early Stages of Methane Dehydroaromatization. Angew. Chem. Int. Ed. 2020, 59, 16741–16746. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, I.; Gascon, J.; Kapteijn, F.; Yarulina, I.; Olivos Suarez, A.I.; van der Linden, B.; Sneider, Y.G. On the dynamic nature of Mo sites for methane dehydroaromatization. In Proceedings of the Abstracts of Papers, 255th ACS National Meeting & Exposition, New Orleans, LA, USA, 18–22 March 2018; American Chemical Society: Washington, DC, USA, 2018; p. CATL-135. [Google Scholar]
- Li, G.; Vollmer, I.; Liu, C.; Gascon, J.; Pidko, E.A. Structure and Reactivity of the Mo/ZSM-5 Dehydroaromatization Catalyst: An Operando Computational Study. ACS Catal. 2019, 9, 8731–8737. [Google Scholar] [CrossRef] [Green Version]
- Vollmer, I.; Mondal, A.; Yarulina, I.; Abou-hamad, E.; Kapteijn, F.; Gascon, J. Quantifying the impact of dispersion, acidity and porosity of Mo/HZSM-5 on the performance in methane dehydroaromatization. Appl. Catal. A Gen. 2019, 574, 144–150. [Google Scholar] [CrossRef]
- Borry, R.W.; Kim, Y.H.; Huffsmith, A.; Reimer, J.A.; Iglesia, E. Structure and density of Mo and acid sites in Mo-exchanged H-ZSM5 catalysts for nonoxidative methane conversion. J. Phys. Chem. B 1999, 103, 5787–5796. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Shen, W.; Bao, X.; Xu, Y. Identification of Mo active species for methane dehydro-aromatization over Mo/HZSM-5 catalysts in the absence of oxygen: 1H MAS NMR and EPR investigations. J. Mol. Catal. A Chem. 2006, 244, 229–236. [Google Scholar] [CrossRef]
- Liu, H.; Bao, X.; Xu, Y. Methane dehydroaromatization under nonoxidative conditions over Mo/HZSM-5 catalysts: Identification and preparation of the Mo active species. J. Catal. 2006, 239, 441–450. [Google Scholar] [CrossRef]
- Lacheen, H.S.; Iglesia, E. Isothermal activation of Mo2O5 2+-ZSM-5 precursors during methane reactions: Effects of reaction products on structural evolution and catalytic properties. Phys. Chem. Chem. Phys. 2005, 7, 538–547. [Google Scholar] [CrossRef]
- Ding, W.; Li, S.; Meitzner, G.D.; Iglesia, E. Methane conversion to aromatics on Mo/H-ZSM5: Structure of molybdenum species in working catalysts. J. Phys. Chem. B 2001, 105, 506–513. [Google Scholar] [CrossRef]
- Li, W.; Meitzner, G.D.; Borry, R.W.; Iglesia, E. Raman and X-ray absorption studies of Mo species in Mo/H-ZSM5 catalysts for non-oxidative CH4 reactions. J. Catal. 2000, 191, 373. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Meitzner, G.D.; Iglesia, E. The effects of silanation of external acid sites on the structure and catalytic behavior of Mo/H-ZSM5. J. Catal. 2002, 206, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Martínez, C.; Corma, A. Inorganic molecular sieves: Preparation, modification and industrial application in catalytic processes. Coord. Chem. Rev. 2011, 255, 1558–1580. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Shu, Y.; Liu, S.; Huang, J.; Guo, X. Interaction between ammonium heptamolybdate and NH4ZSM-5 zeolite: The location of Mo species and the acidity of Mo/HZSM-5. Catal. Lett. 1995, 35, 233–243. [Google Scholar] [CrossRef]
- Kim, Y.; Borry, R.W.; Iglesia, E. Genesis of methane activation sites in Mo-exchanged H—ZSM-5 catalysts. Microporous Mesoporous Mater. 2000, 36, 495–509. [Google Scholar] [CrossRef]
- Zheng, H.; Ma, D.; Bao, X.; Jian, Z.H.; Ja, H.K.; Wang, Y.; Peden, C.H.F. Direct observation of the active center for methane dehydroaromatization using an ultrahigh field 95Mo NMR spectroscopy. J. Am. Chem. Soc. 2008, 130, 3722–3723. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, I.; Yarulina, I.; Kapteijn, F.; Gascon, J. Progress in Developing a Structure-Activity Relationship for the Direct Aromatization of Methane. ChemCatChem 2019, 11, 39–52. [Google Scholar] [CrossRef]
- Gao, J.; Zheng, Y.; Jehng, J.-M.; Tang, Y.; Wachs, I.E.; Podkolzin, S.G. Identification of molybdenum oxide nanostructures on zeolites for natural gas conversion. Science 2015, 348, 686–690. [Google Scholar] [CrossRef] [Green Version]
- Kosinov, N.; Coumans, F.J.A.G.; Uslamin, E.A.; Wijpkema, A.S.G.; Mezari, B.; Hensen, E.J.M. Methane Dehydroaromatization by Mo/HZSM-5: Mono-or Bifunctional Catalysis? ACS Catal. 2017, 7, 520–529. [Google Scholar] [CrossRef]
- Liu, S.; Wang, L.; Ohnishi, R.; Ichikawa, M. Bifunctional catalysis of Mo/HZSM-5 in the dehydroaromatization of methane to benzene and naphthalene XAFS/TG/DTA/ MASS/FTIR characterization and supporting effects. J. Catal. 1999, 181, 175–188. [Google Scholar] [CrossRef]
- Su, L.; Xu, Y.; Bao, X. Study on Bifunctionality of Mo/HZSM-5 Catalysts for Methane Dehydro-Aromatization under Non-oxidative Condition. J. Nat. Gas Chem. 2002, 11, 18–27. [Google Scholar]
- Kosinov, N.; Wijpkema, A.S.G.; Uslamin, E.; Rohling, R.; Coumans, F.J.A.G.; Mezari, B.; Parastaev, A.; Poryvaev, A.S.; Fedin, M.V.; Pidko, E.A.; et al. Confined Carbon Mediating Dehydroaromatization of Methane over Mo/ZSM-5. Angew. Chem. Int. Ed. 2018, 57, 1016–1020. [Google Scholar] [CrossRef]
- Kosinov, N.; Uslamin, E.A.; Coumans, F.J.A.G.; Wijpkema, A.S.G.; Rohling, R.Y.; Hensen, E.J.M. Structure and Evolution of Confined Carbon Species during Methane Dehydroaromatization over Mo/ZSM-5. ACS Catal. 2018, 8, 8459–8467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Li, S.; Li, N.; Chen, H.; Zhang, W.; Bao, X.; Lin, B. Structure and acidity of Mo/ZSM-5 synthesized by solid state reaction for methane dehydrogenation and aromatization. Microporous Mesoporous Mater. 2006, 88, 244–253. [Google Scholar] [CrossRef]
- Sedel’nikova, O.V.; Stepanov, A.A.; Zaikovskii, V.I.; Korobitsyna, L.L.; Vosmerikov, A.V. Preparation method effect on the physicochemical and catalytic properties of a methane dehydroaromatization catalyst. Kinet. Catal. 2017, 58, 51–57. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Harkins, W.D.; Jura, G. Surfaces of Solids. XIII. A Vapor Adsorption Method for the Determination of the Area of a Solid without the Assumption of a Molecular Area, and the Areas Occupied by Nitrogen and Other Molecules on the Surface of a Solid. J. Am. Chem. Soc. 1944, 66, 1366–1373. [Google Scholar] [CrossRef]
- Emeis, C.A. Determination of Integrated Molar Extinction Coefficients for Infrared Absorption Bands of Pyridine Adsorbed on Solid Acid Catalysts. J. Catal. 1993, 141, 347–354. [Google Scholar] [CrossRef]
- Martínez, A.; Peris, E.; Derewinski, M.; Burkat-Dulak, A. Improvement of catalyst stability during methane dehydroaromatization (MDA) on Mo/HZSM-5 comprising intracrystalline mesopores. Catal. Today 2011, 169, 75–84. [Google Scholar] [CrossRef]
- Li, W.; Meitzner, G.D.; Kim, Y.H.; Borry, R.W.; Iglesia, E. The location, structure, and role of MoO and MoC species in Mo/H-ZSM5 catalysts for methane aromatization. Stud. Surf. Sci. Catal. 2000, 130 D, 3621–3626. [Google Scholar]
- Song, Y.; Sun, C.; Shen, W.; Lin, L. Hydrothermal post-synthesis of HZSM-5 zeolite to enhance the coke-resistance of Mo/HZSM-5 catalyst for methane dehydroaromatization. Catal. Lett. 2006, 109, 21–24. [Google Scholar] [CrossRef]
- Jin, Z.; Liu, S.; Qin, L.; Liu, Z.; Wang, Y.; Xie, Z.; Wang, X. Methane dehydroaromatization by Mo-supported MFI-type zeolite with core-shell structure. Appl. Catal. A Gen. 2013, 453, 295–301. [Google Scholar] [CrossRef]
- Ding, W.; Meitzner, G.D.; Marler, D.O.; Iglesia, E. Synthesis, structural characterization, and catalytic properties of tungsten-exchanged H-ZSM5. J. Phys. Chem. B 2001, 105, 3928–3936. [Google Scholar] [CrossRef] [Green Version]
- Bijani, P.M.; Sohrabi, M.; Sahebdelfar, S. Thermodynamic Analysis of Nonoxidative Dehydroaromatization of Methane. Chem. Eng. Technol. 2012, 35, 1825–1832. [Google Scholar] [CrossRef]



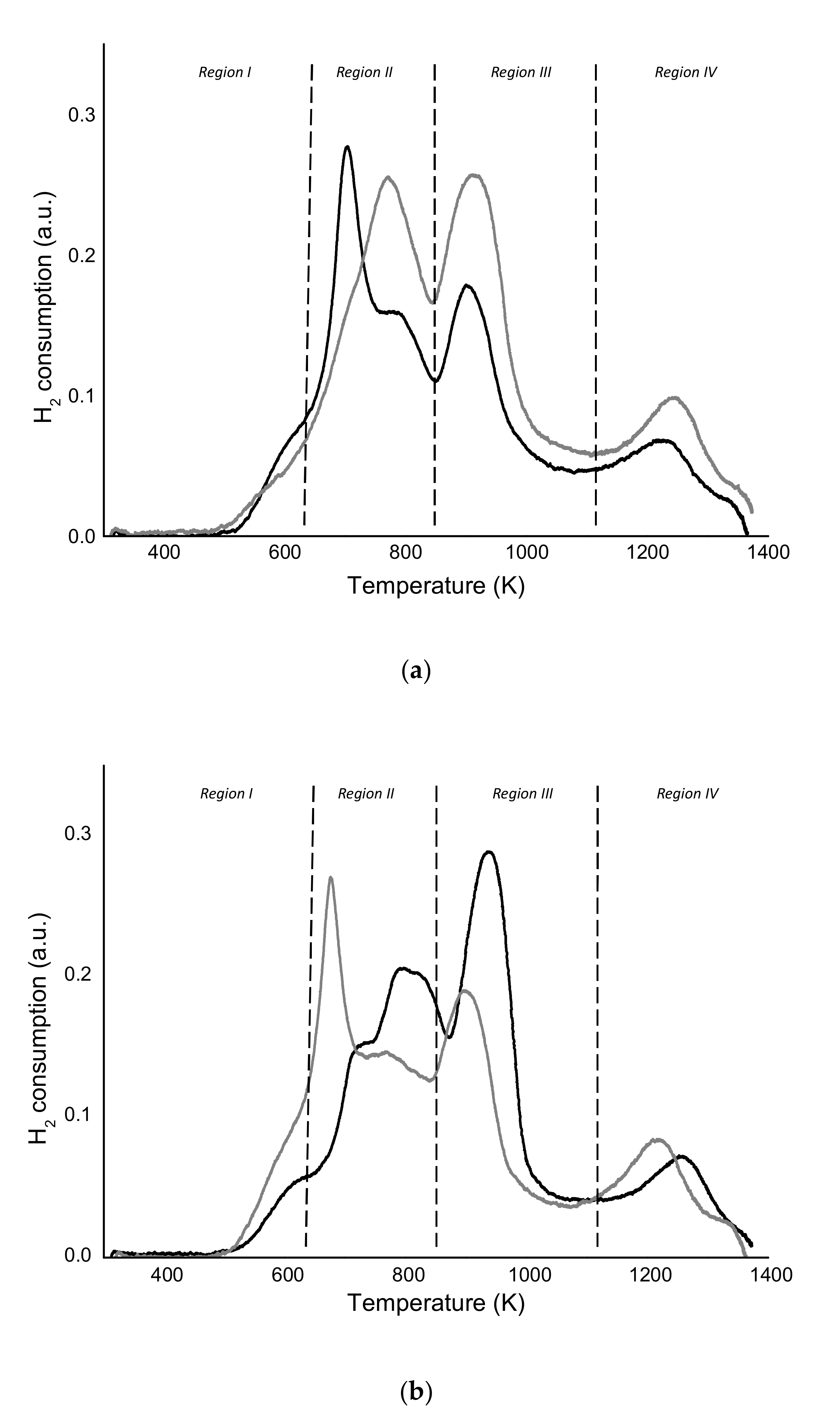
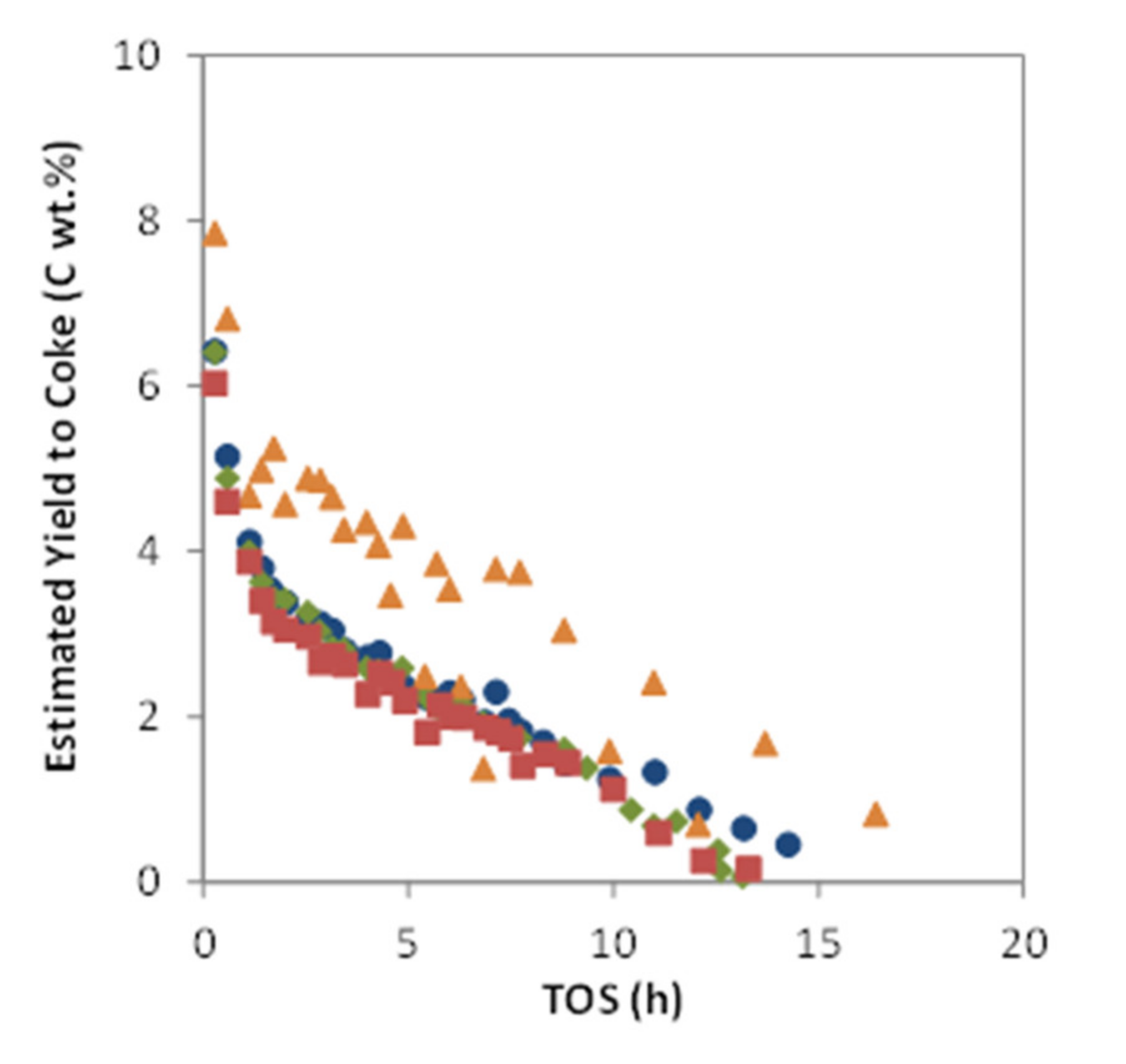
 ), MoZ5-SSR-DC (
), MoZ5-SSR-DC (  ), MoZ5-IW-SC (🔵), and MoZ5-IW-DC (
), MoZ5-IW-SC (🔵), and MoZ5-IW-DC (  ), at 973 K, atmospheric pressure and w/F = 16 g·h/mol.
), MoZ5-SSR-DC ( ), MoZ5-IW-SC (🔵), and MoZ5-IW-DC ( ), at 973 K, atmospheric pressure and w/F = 16 g·h/mol.
), at 973 K, atmospheric pressure and w/F = 16 g·h/mol.
), MoZ5-SSR-DC ( ), MoZ5-IW-SC (🔵), and MoZ5-IW-DC ( ), at 973 K, atmospheric pressure and w/F = 16 g·h/mol. ), MoZ5-SSR-DC ( ), MoZ5-IW-SC (🔵), and MoZ5-IW-DC ( ), at 973K, atmospheric pressure and w/F = 16 g·h/mol.
), MoZ5-SSR-DC ( ), MoZ5-IW-SC (🔵), and MoZ5-IW-DC ( ), at 973K, atmospheric pressure and w/F = 16 g·h/mol.
), MoZ5-SSR-DC ( ), MoZ5-IW-SC (🔵), and MoZ5-IW-DC ( ), at 973K, atmospheric pressure and w/F = 16 g·h/mol.
), MoZ5-SSR-DC ( ), MoZ5-IW-SC (🔵), and MoZ5-IW-DC ( ), at 973K, atmospheric pressure and w/F = 16 g·h/mol. ), MoZ5-SSR-SC-N2 (
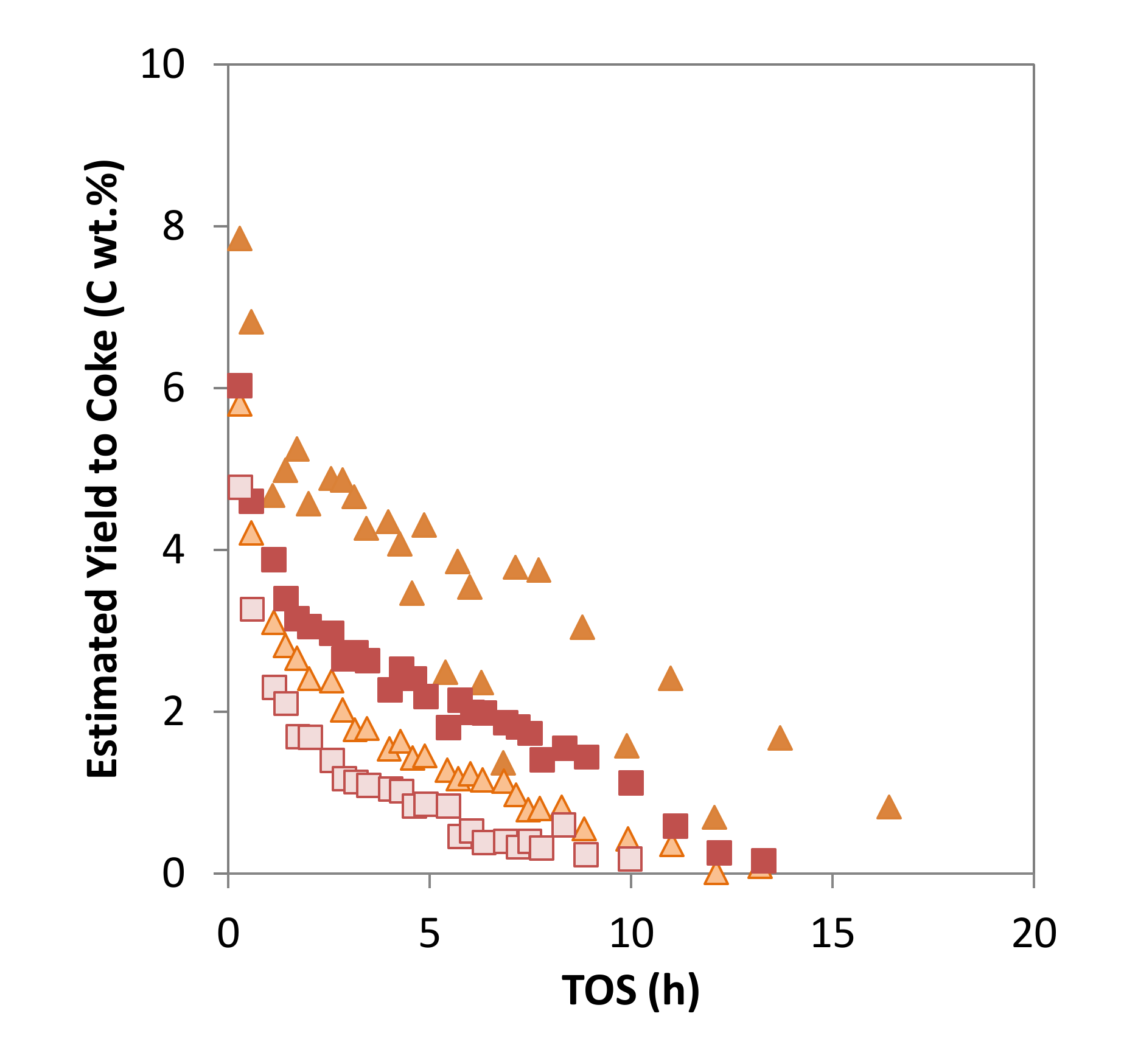
), MoZ5-SSR-SC-N2 (  ), MoZ5-SSR-DC ( ) and MoZ5-SSR-DC-N2 (
), MoZ5-SSR-DC ( ) and MoZ5-SSR-DC-N2 (  ), at 973 K and atmospheric pressure (w/F = 16 g·h/mol).
), MoZ5-SSR-SC-N2 ( ), MoZ5-SSR-DC ( ) and MoZ5-SSR-DC-N2 ( ), at 973 K and atmospheric pressure (w/F = 16 g·h/mol).
), at 973 K and atmospheric pressure (w/F = 16 g·h/mol).
), MoZ5-SSR-SC-N2 ( ), MoZ5-SSR-DC ( ) and MoZ5-SSR-DC-N2 ( ), at 973 K and atmospheric pressure (w/F = 16 g·h/mol). ), MoZ5-SSR-SC-N2 ( ), MoZ5-SSR-DC ( ) and MoZ5-SSR-DC-N2 ( ), at 973 K and atmospheric pressure (w/F = 16 g·h/mol).
), MoZ5-SSR-SC-N2 ( ), MoZ5-SSR-DC ( ) and MoZ5-SSR-DC-N2 ( ), at 973 K and atmospheric pressure (w/F = 16 g·h/mol).
), MoZ5-SSR-SC-N2 ( ), MoZ5-SSR-DC ( ) and MoZ5-SSR-DC-N2 ( ), at 973 K and atmospheric pressure (w/F = 16 g·h/mol).
), MoZ5-SSR-SC-N2 ( ), MoZ5-SSR-DC ( ) and MoZ5-SSR-DC-N2 ( ), at 973 K and atmospheric pressure (w/F = 16 g·h/mol).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ICP | S BET (m2/g) | S micro (m2/g) | V micro (cm3/g) | ||
|---|---|---|---|---|---|---|
| Si/Al | Mo/Al | Mo (wt. %) | ||||
| Z5 (TZP302A) | 10.0 | --- | --- | 368 | 355 | 0.17 |
| MoZ5-IW-SC | 10.5 | 0.5 | 6.0 | 270 | 257 | 0.12 |
| MoZ5-IW-DC | 9.9 | 0.5 | 6.1 | 263 | 246 | 0.12 |
| MoZ5-SSR-SC | 10.3 | 0.5 | 6.4 | 292 | 275 | 0.13 |
| MoZ5-SSR-DC | 10.2 | 0.5 | 6.3 | 276 | 260 | 0.13 |
| MoZ5-SSR-SC-N2 | 10.3 | 0.5 | 6.4 | 254 | 241 | 0.12 |
| MoZ5-SSR-DC-N2 | 10.2 | 0.5 | 6.3 | 237 | 221 | 0.11 |
| Sample | Brønsted Acidity (mol Py/g) | Lewis Acidity (mol Py/g) | |||||
|---|---|---|---|---|---|---|---|
| B523 | B623 | B673 | B673/B523 | L523 | L623 | L673 | |
| Z5 (TZP302A) | 564 | 368 | 241 | 0.43 | 7 | 0 | 0 |
| MoZ5-IW-SC | 213 | 122 | 107 | 0.50 | 23 | 14 | 3 |
| MoZ5-IW-DC | 199 | 150 | 65 | 0.32 | 21 | 13 | 0 |
| MoZ5-SSR-SC | 295 | 184 | 116 | 0.39 | 31 | 19 | 9 |
| MoZ5-SSR-DC | 250 | 138 | 124 | 0.50 | 13 | 5 | 5 |
| MoZ5-SSR-SC-N2 | 237 | 132 | 95 | 0.40 | 37 | 25 | 18 |
| MoZ5-SSR-DC-N2 | 236 | 131 | 100 | 0.42 | 32 | 21 | 19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portilla, M.T.; Llopis, F.J.; Moliner, M.; Martinez, C. Influence of Preparation Conditions on the Catalytic Performance of Mo/H-ZSM-5 for Methane Dehydroaromatization. Appl. Sci. 2021, 11, 5465. https://doi.org/10.3390/app11125465
Portilla MT, Llopis FJ, Moliner M, Martinez C. Influence of Preparation Conditions on the Catalytic Performance of Mo/H-ZSM-5 for Methane Dehydroaromatization. Applied Sciences. 2021; 11(12):5465. https://doi.org/10.3390/app11125465
Chicago/Turabian StylePortilla, Maria Teresa, Francisco J. Llopis, Manuel Moliner, and Cristina Martinez. 2021. "Influence of Preparation Conditions on the Catalytic Performance of Mo/H-ZSM-5 for Methane Dehydroaromatization" Applied Sciences 11, no. 12: 5465. https://doi.org/10.3390/app11125465
APA StylePortilla, M. T., Llopis, F. J., Moliner, M., & Martinez, C. (2021). Influence of Preparation Conditions on the Catalytic Performance of Mo/H-ZSM-5 for Methane Dehydroaromatization. Applied Sciences, 11(12), 5465. https://doi.org/10.3390/app11125465