
Analytic Binary Alloy Volume–Concentration Relations and the Deviation from Zen’s Law



Abstract
:Featured Application
Abstract
1. Introduction
2. Theoretical Background
- 1.
- Continuum approximation. In cases where the field of the disordered solid solution spans throughout the whole composition range, we assume that the atomic volume of the solvent (Ω1(x)) changes linearly with composition from the real value, Ω1, x = 0, to its apparent value, , in the pure solute, x = 1.
- 2.
- Terminal approximation. In cases of limited mutual solubility of the alloy components, it is reasonable to consider the atomic volume of the solvent to be constant and equal to its real value, Ω1(2),
3. Results
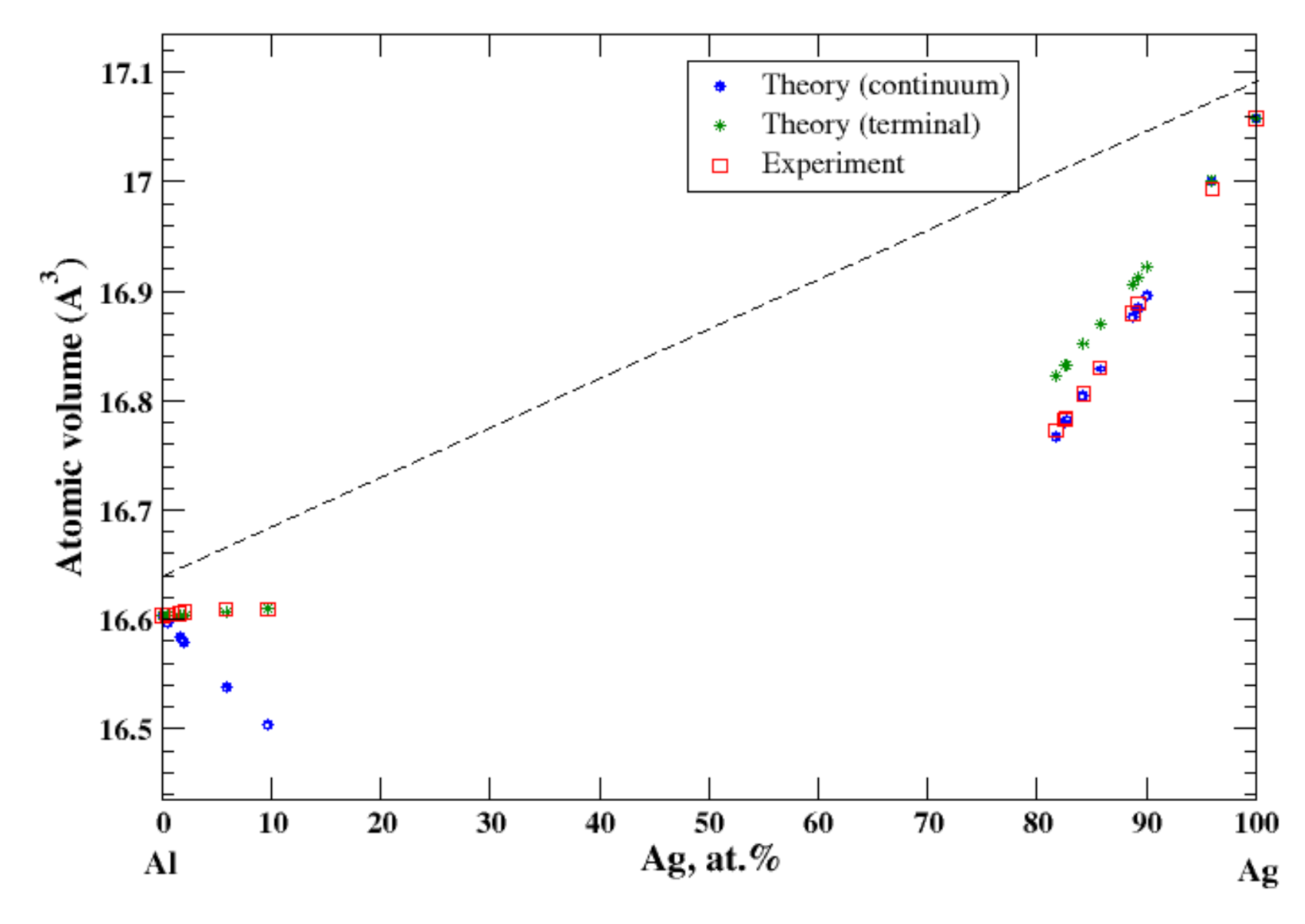
3.1. Al-Ag
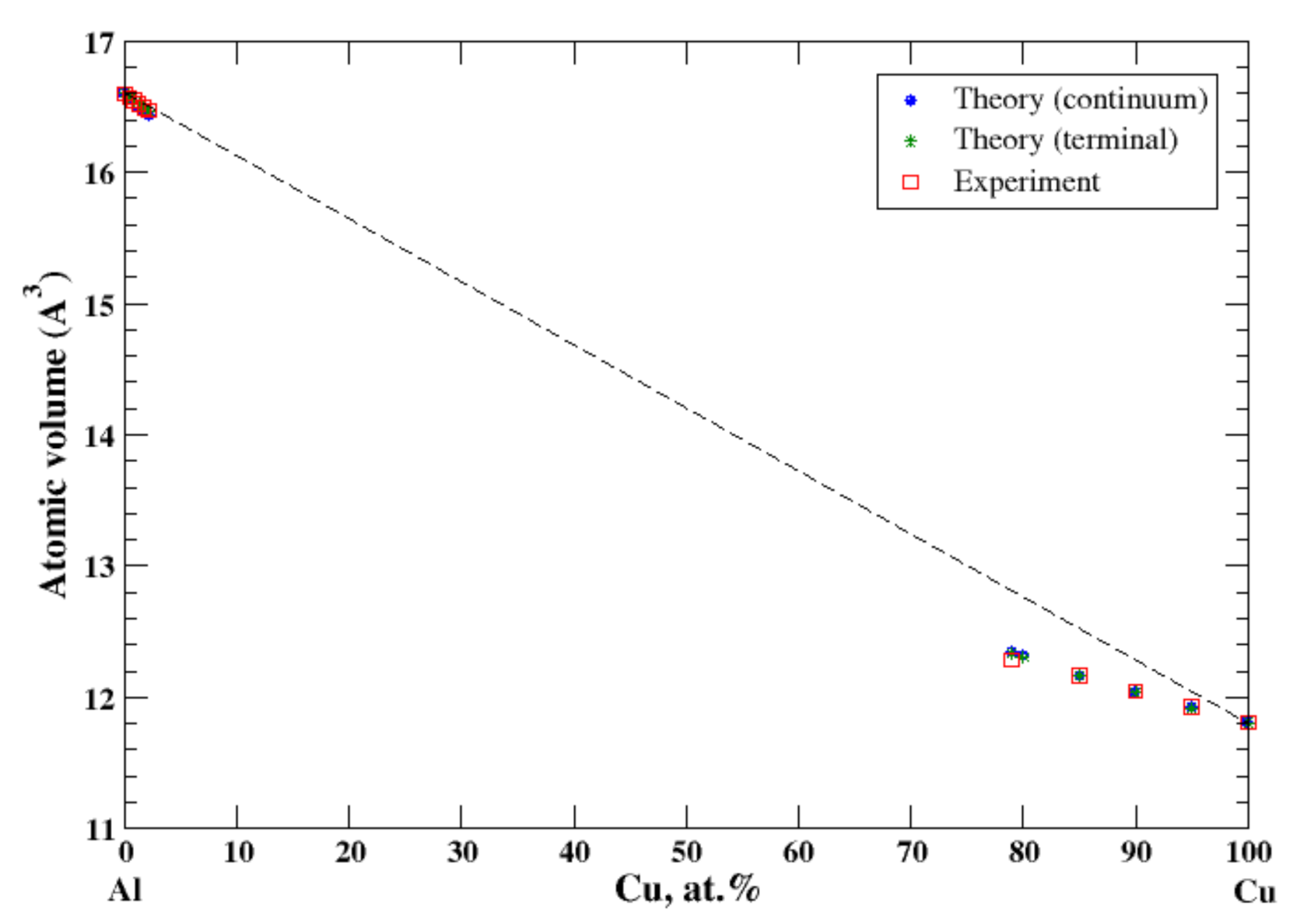
3.2. Al-Cu
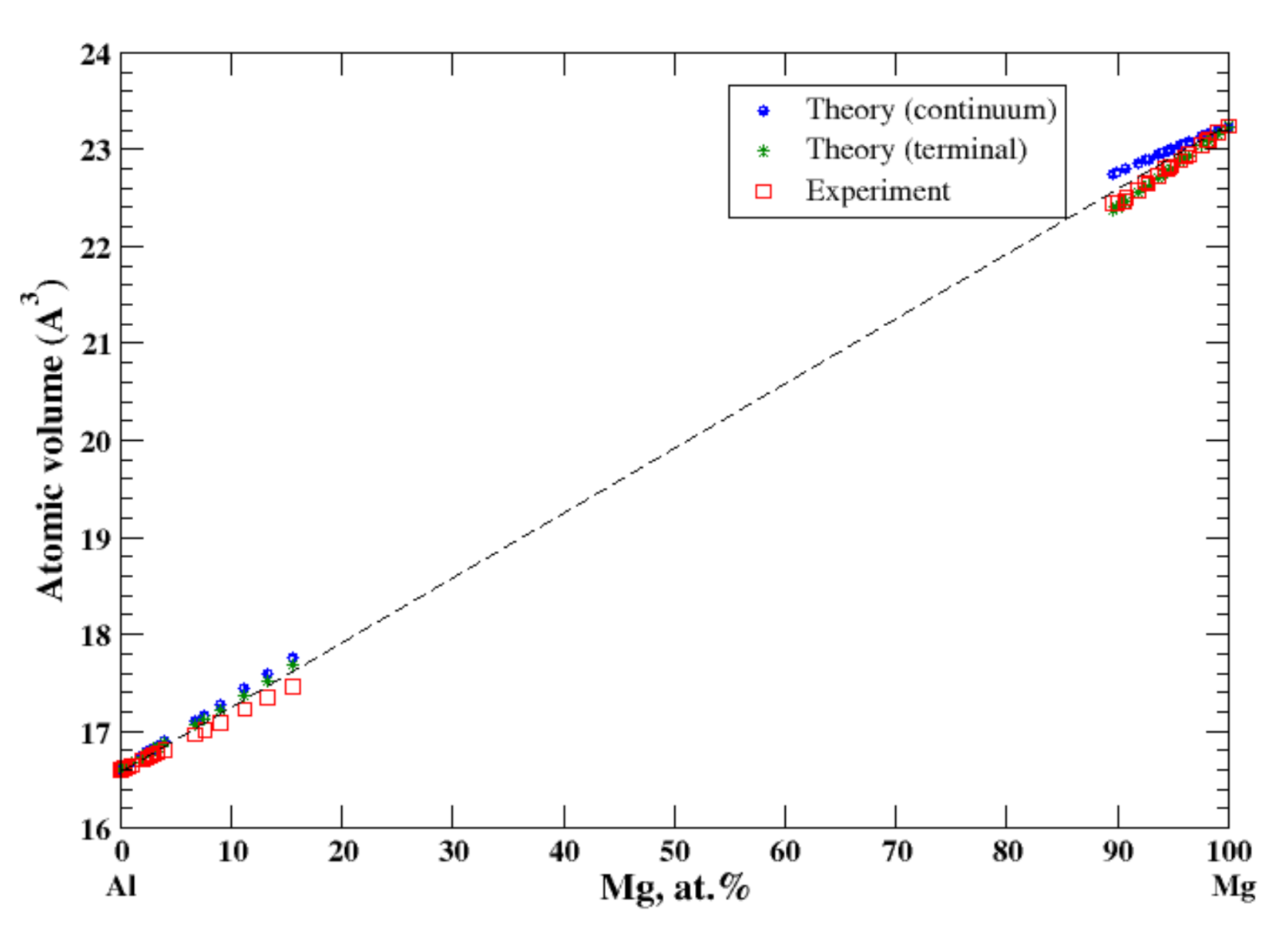
3.3. Al-Mg
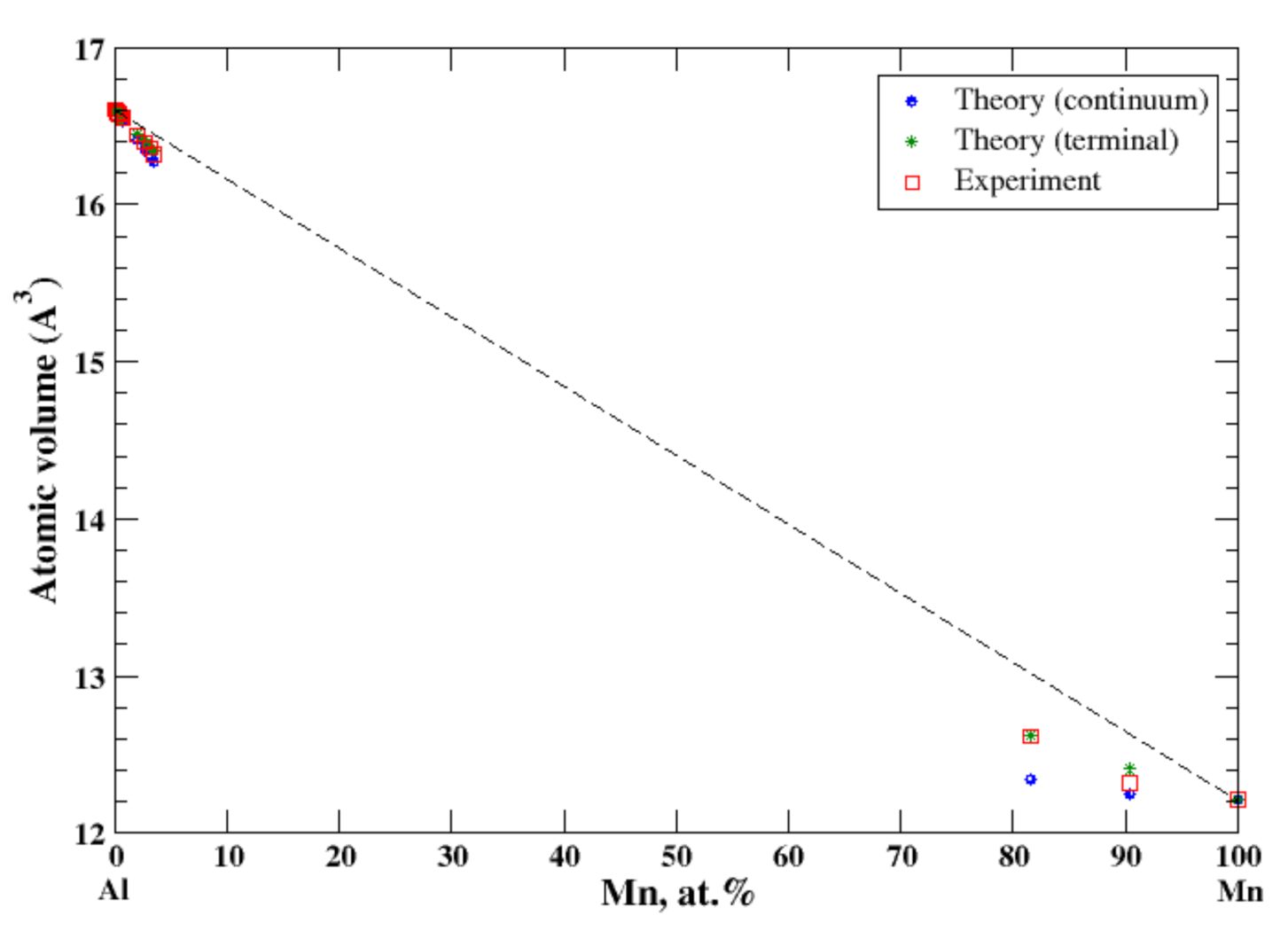
3.4. Al-Mn
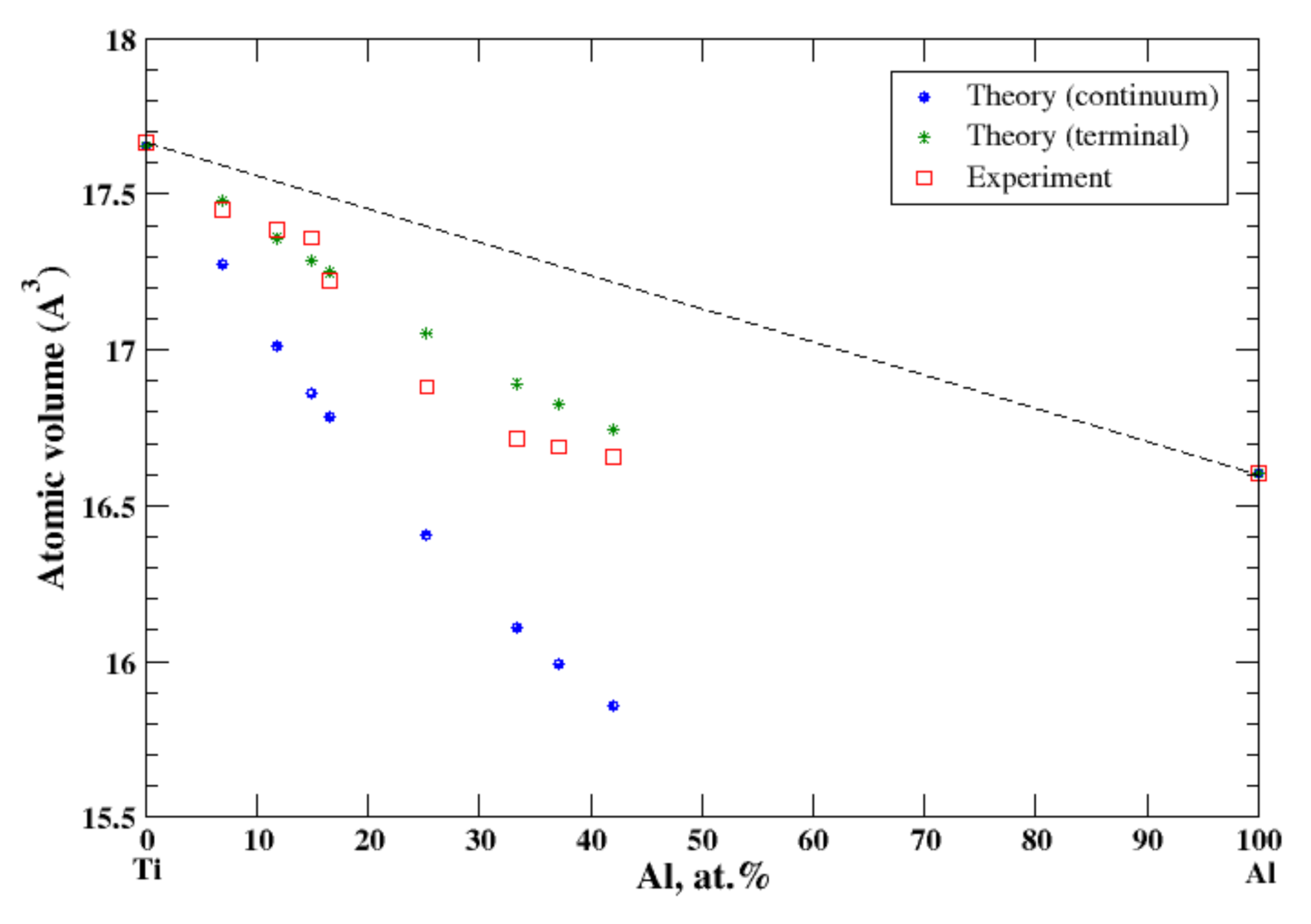
3.5. Al-Ti
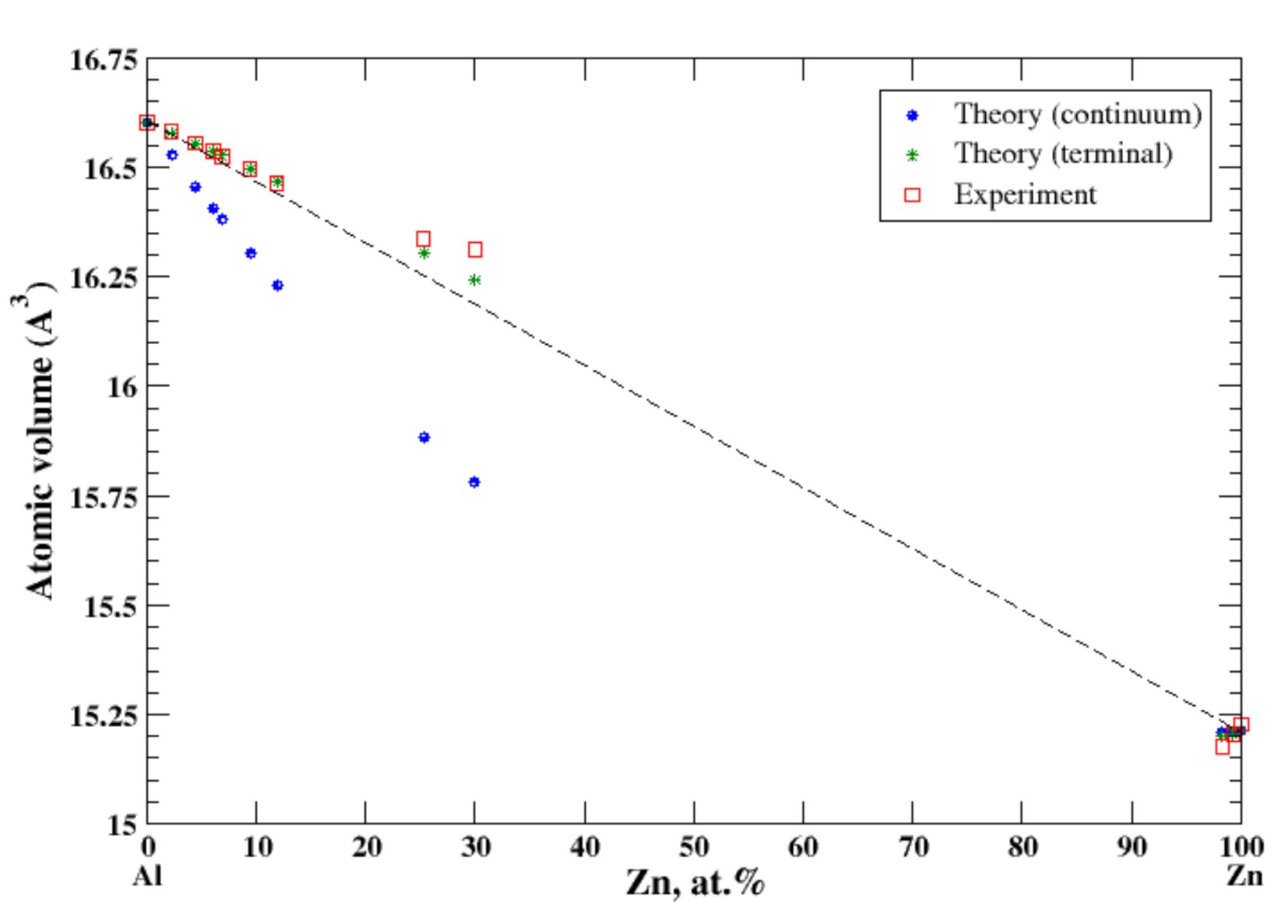
3.6. Al-Zn
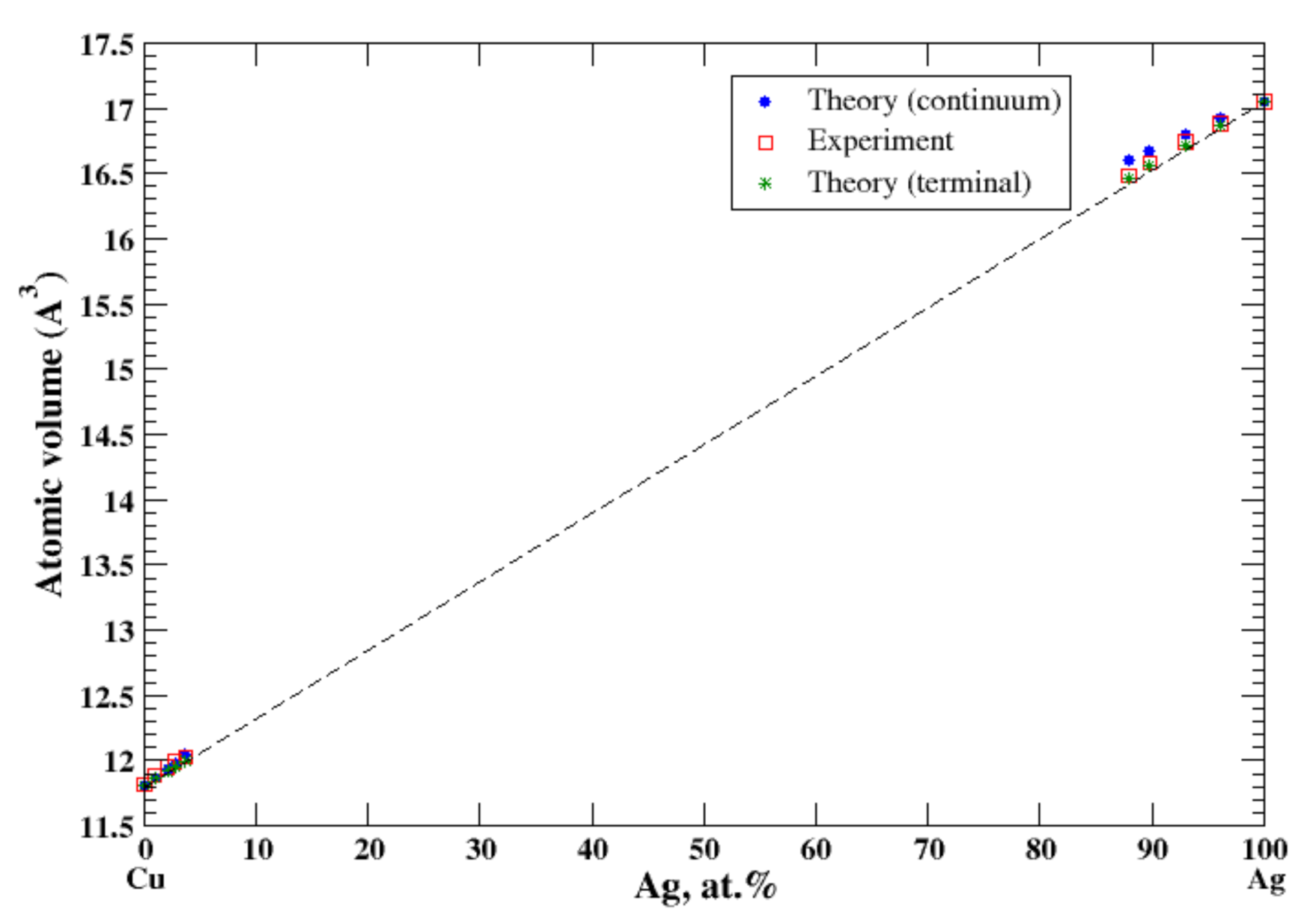
3.7. Ag-Cu
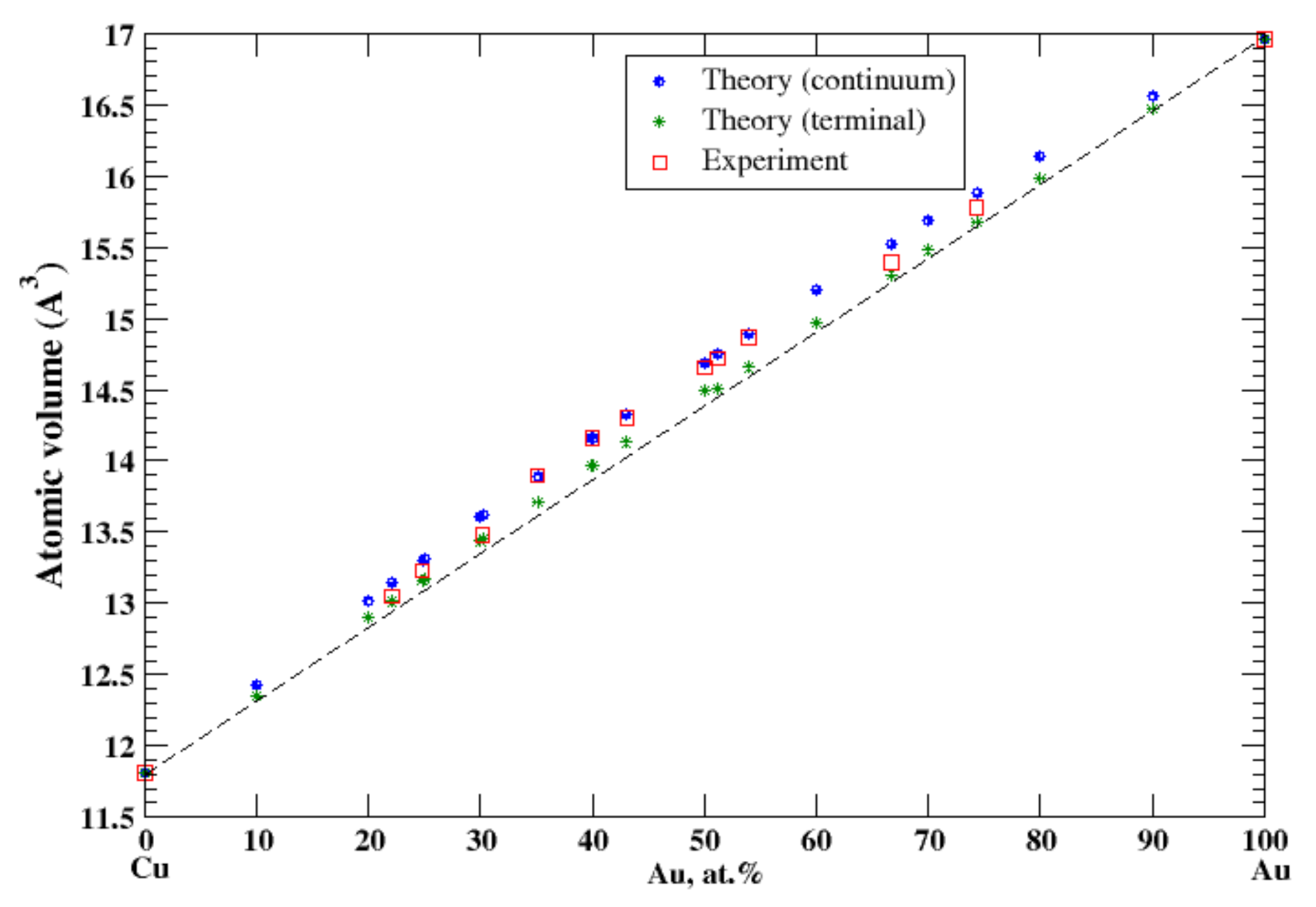
3.8. Cu-Au
3.9. Cu-Fe
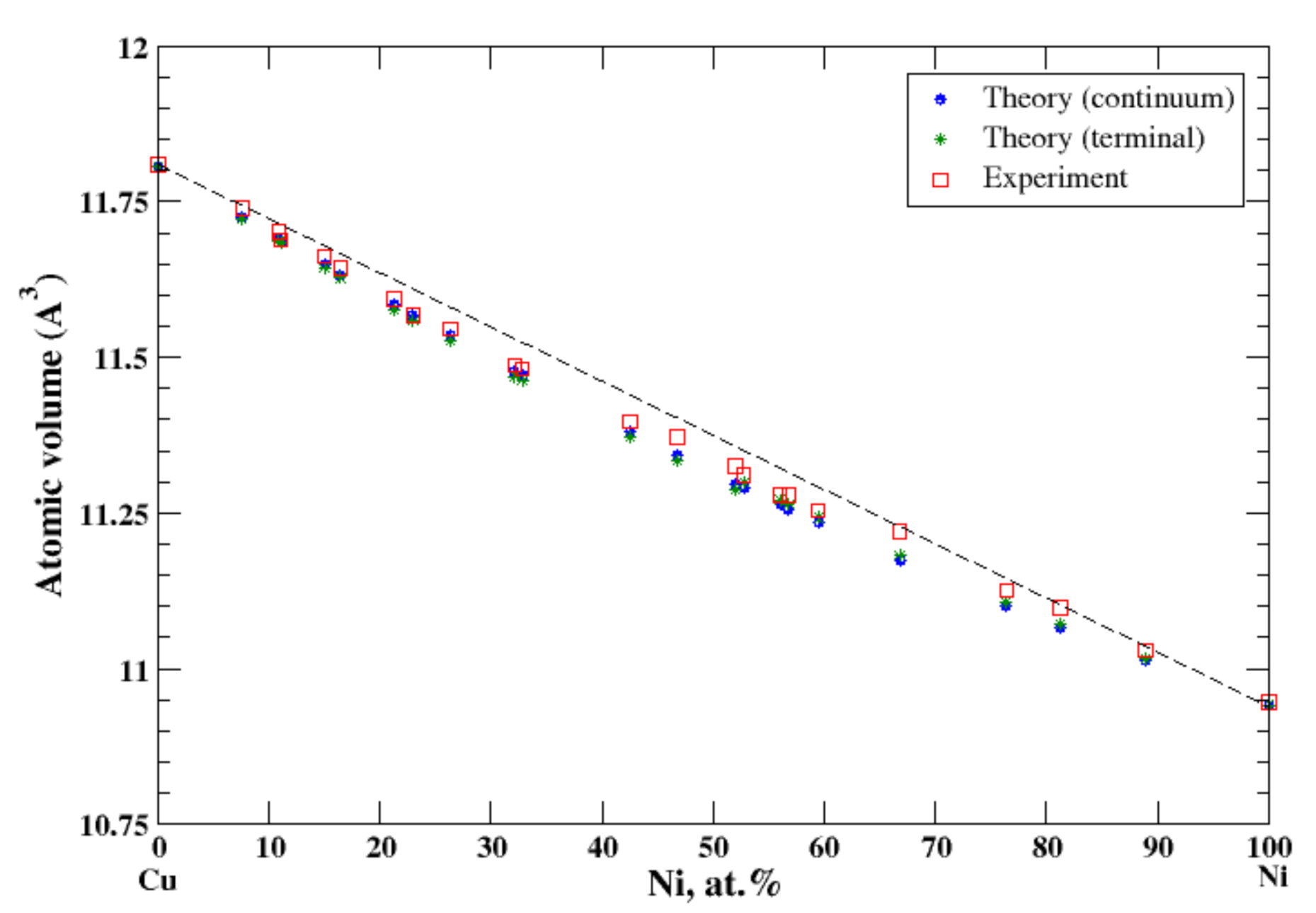
3.10. Cu-Ni
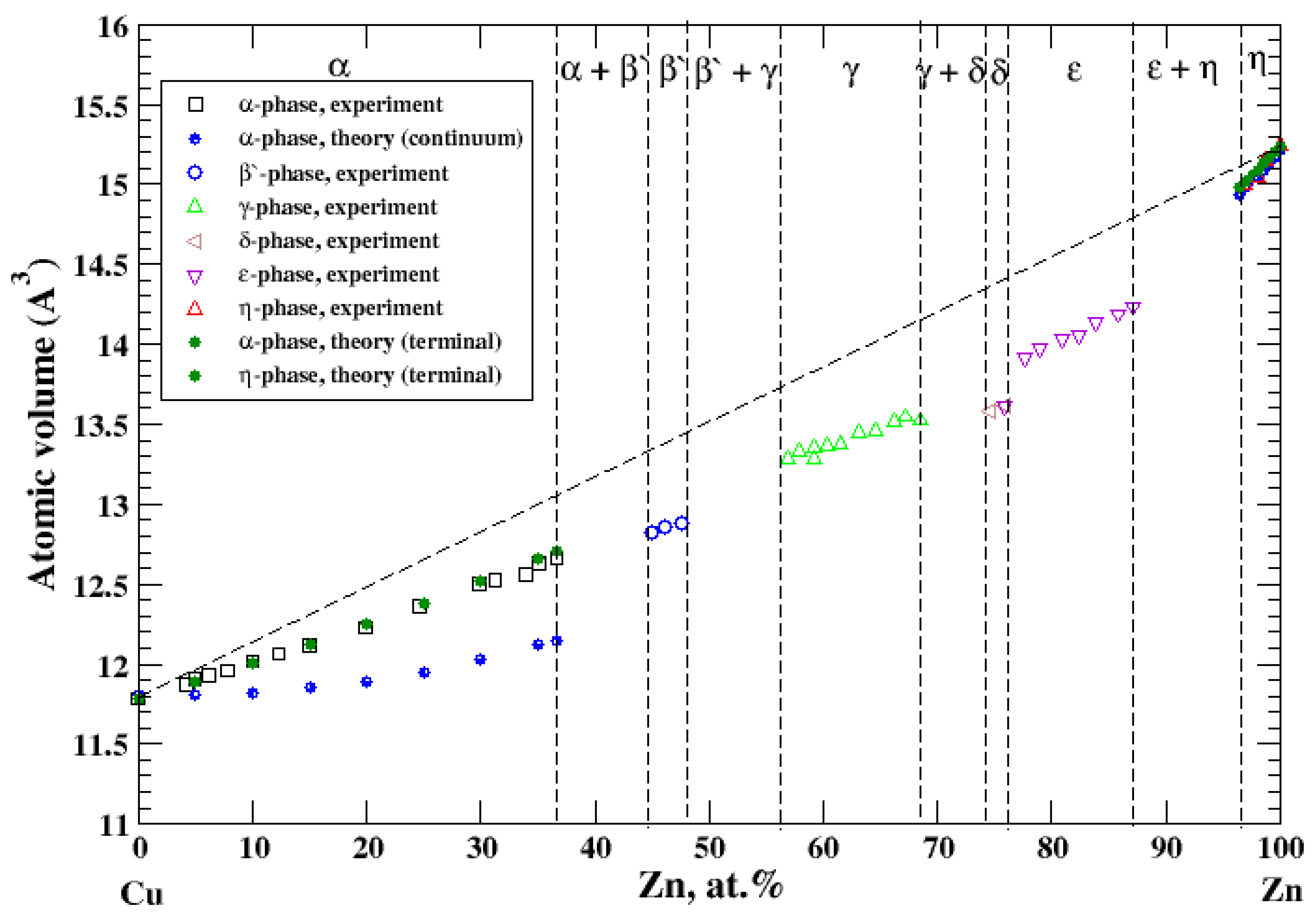
3.11. Cu-Zn
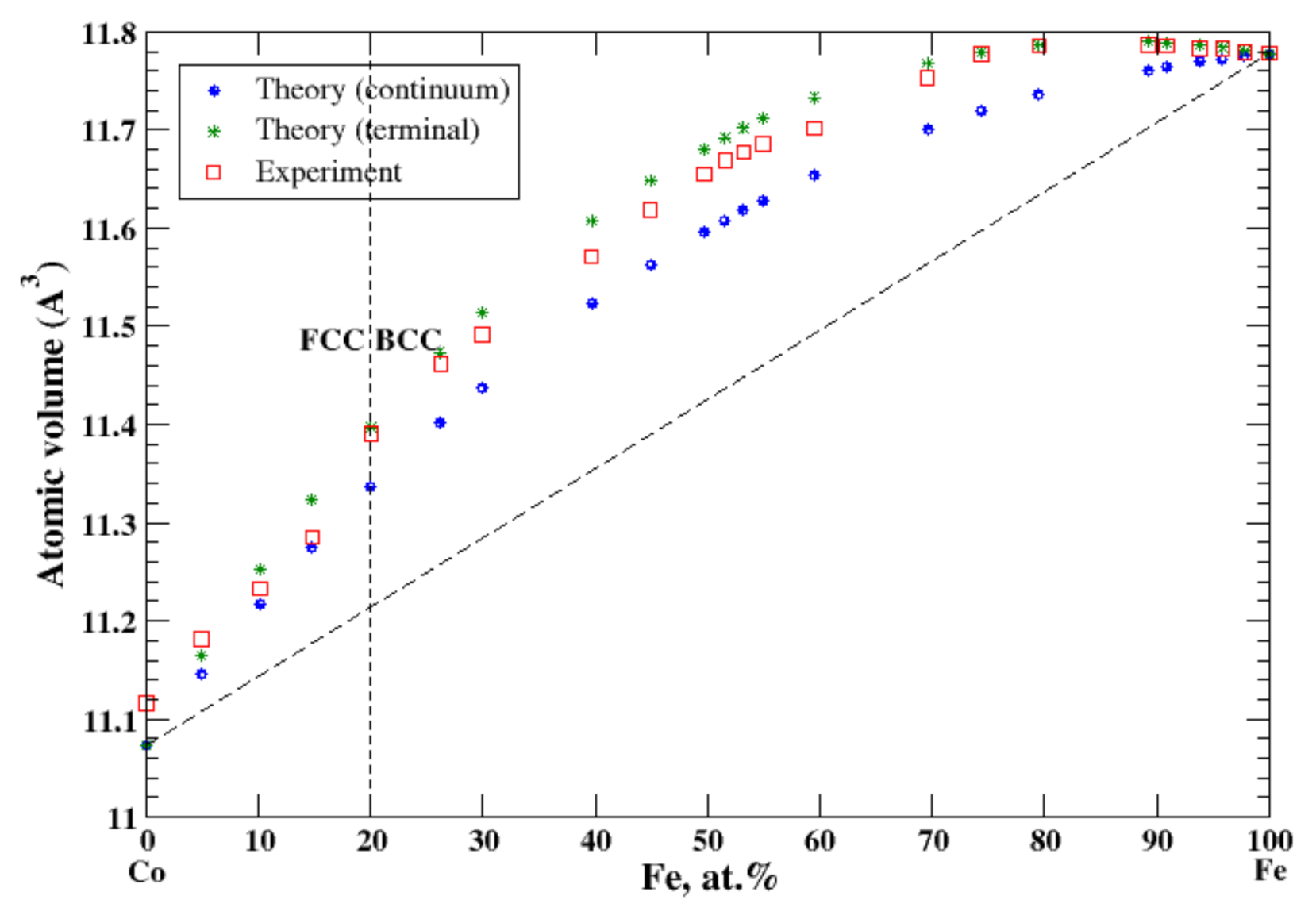
3.12. Co-Fe
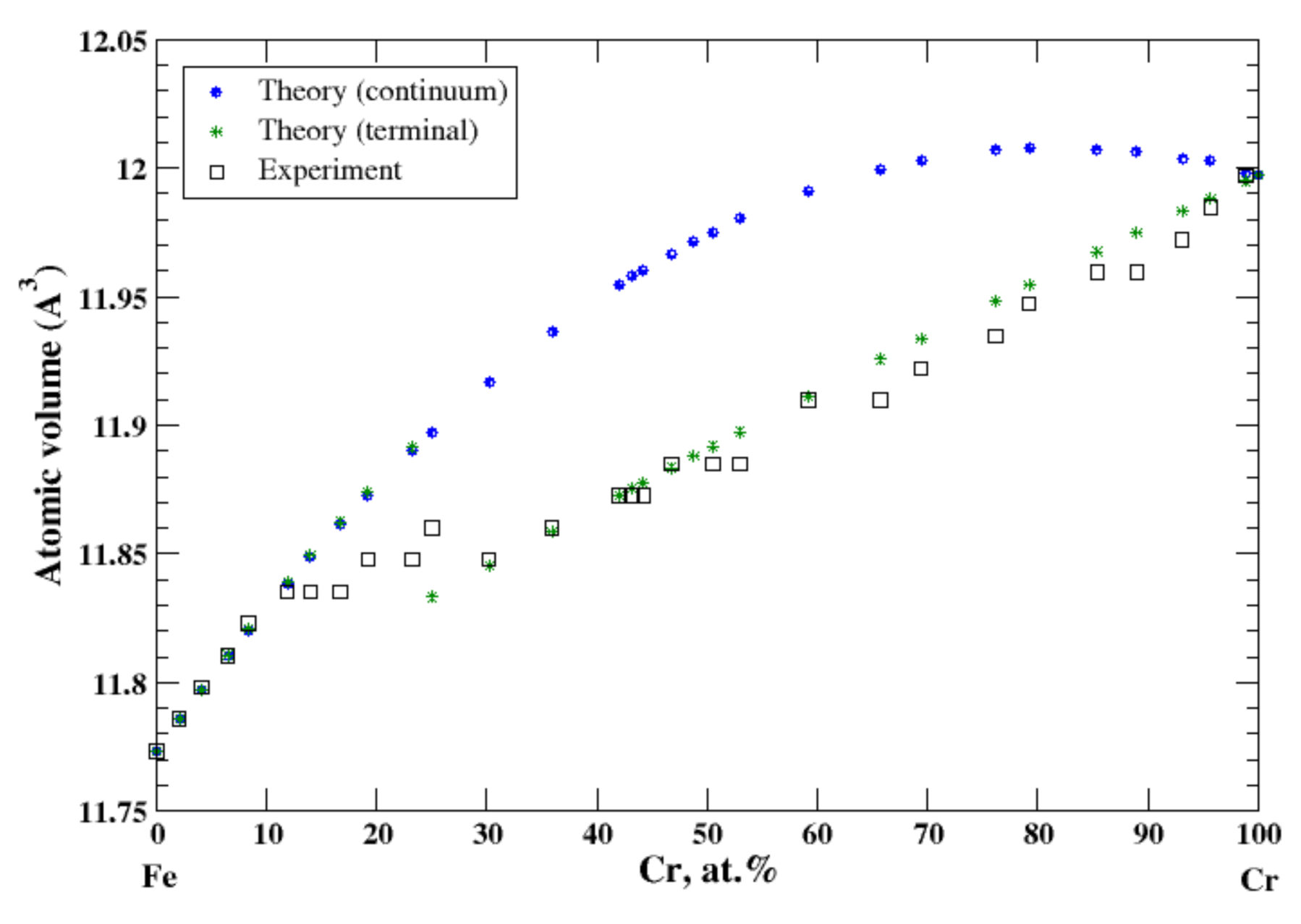
3.13. Fe-Cr
3.14. Fe-V
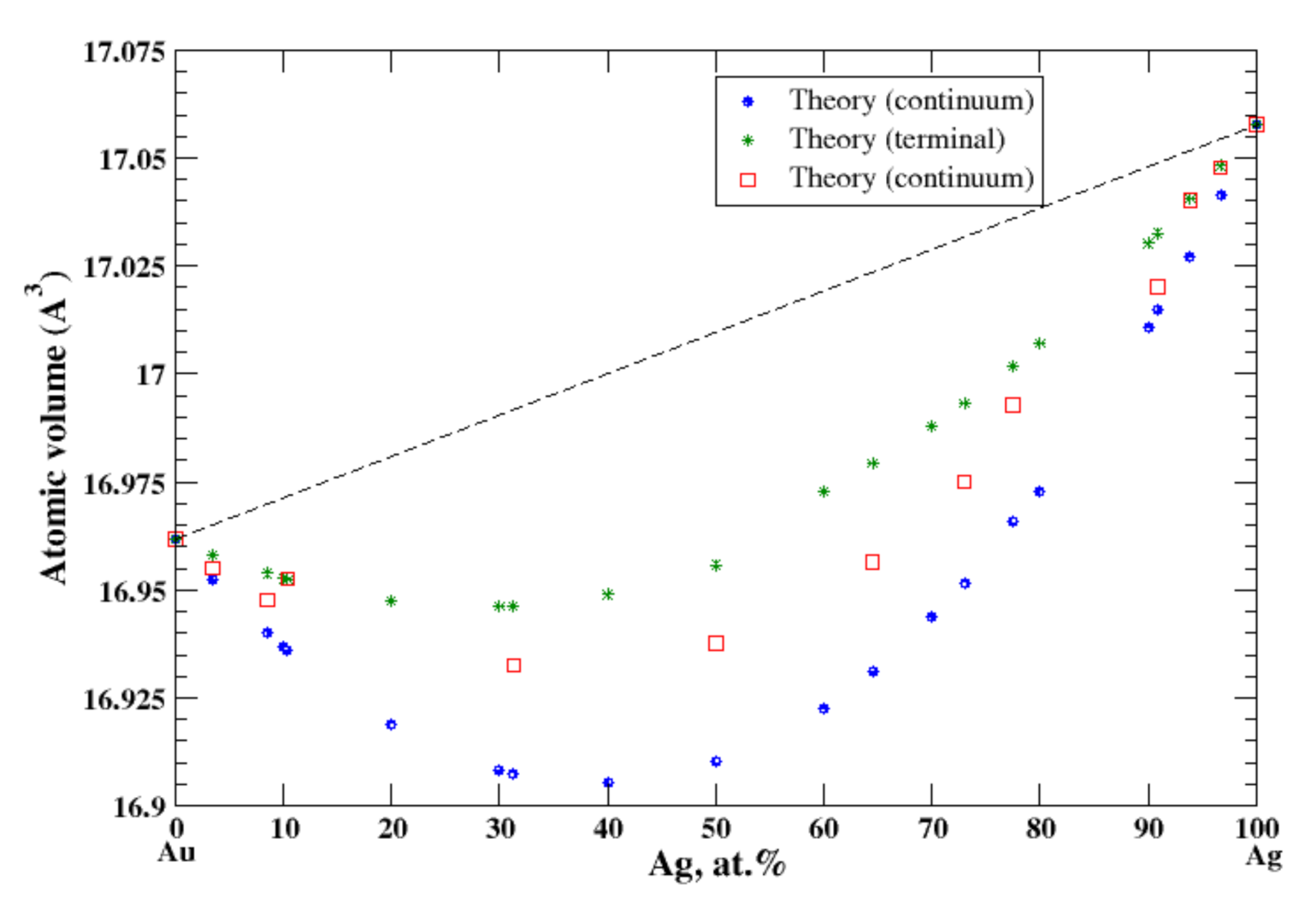
3.15. Ag-Au
3.16. Ag-Mg
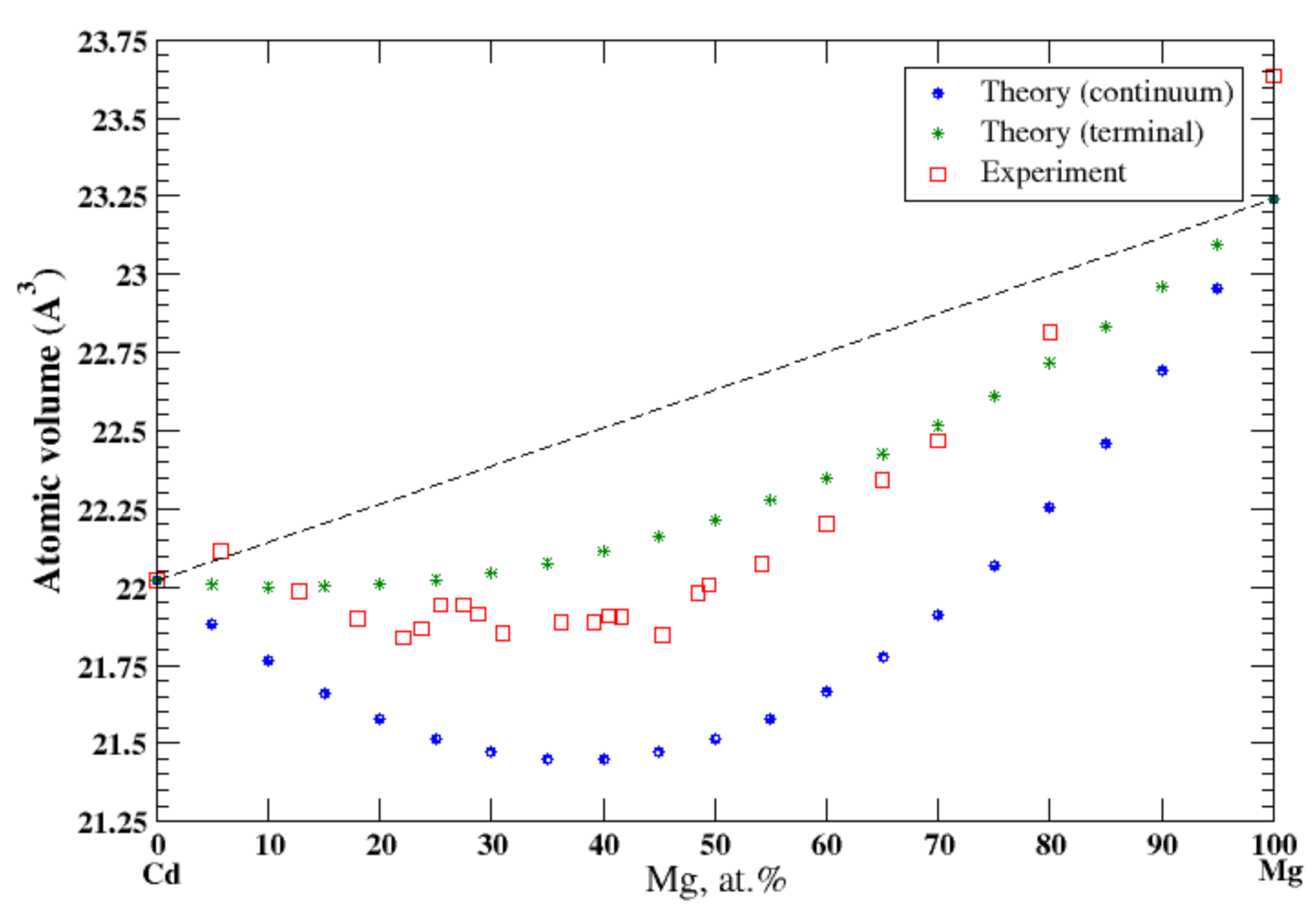
3.17. Cd-Mg
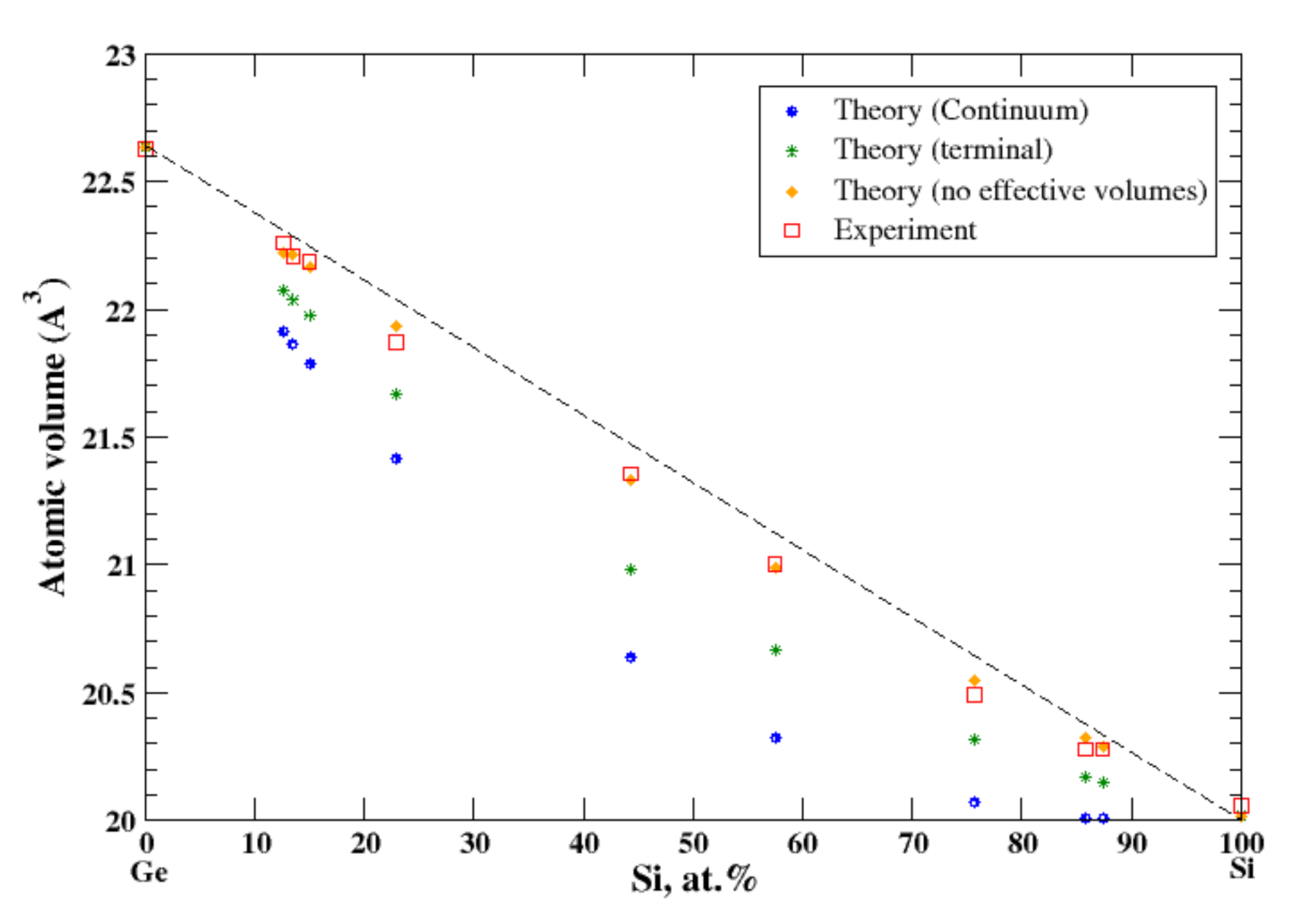
3.18. Ge-Si
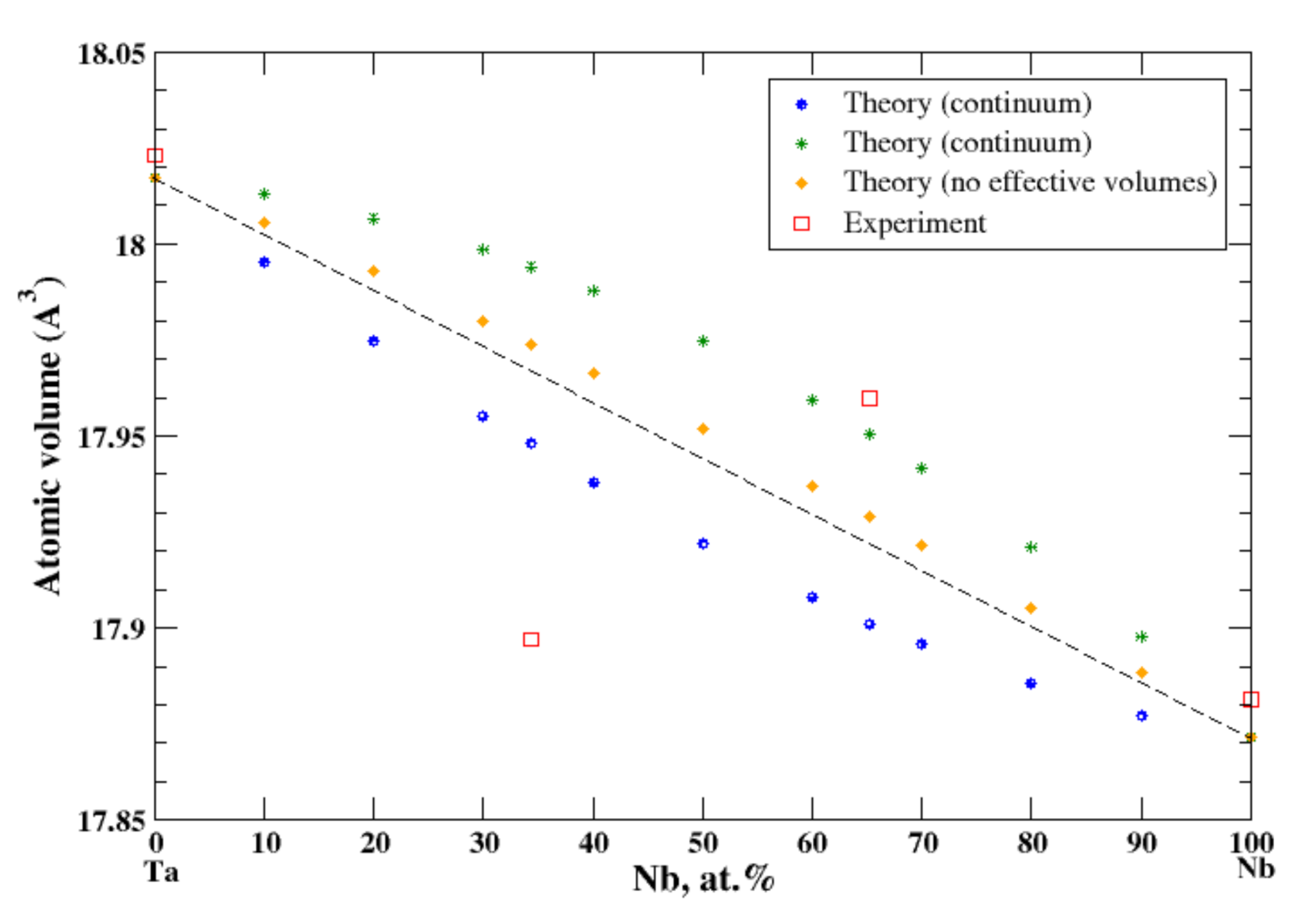
3.19. Nb-Ta
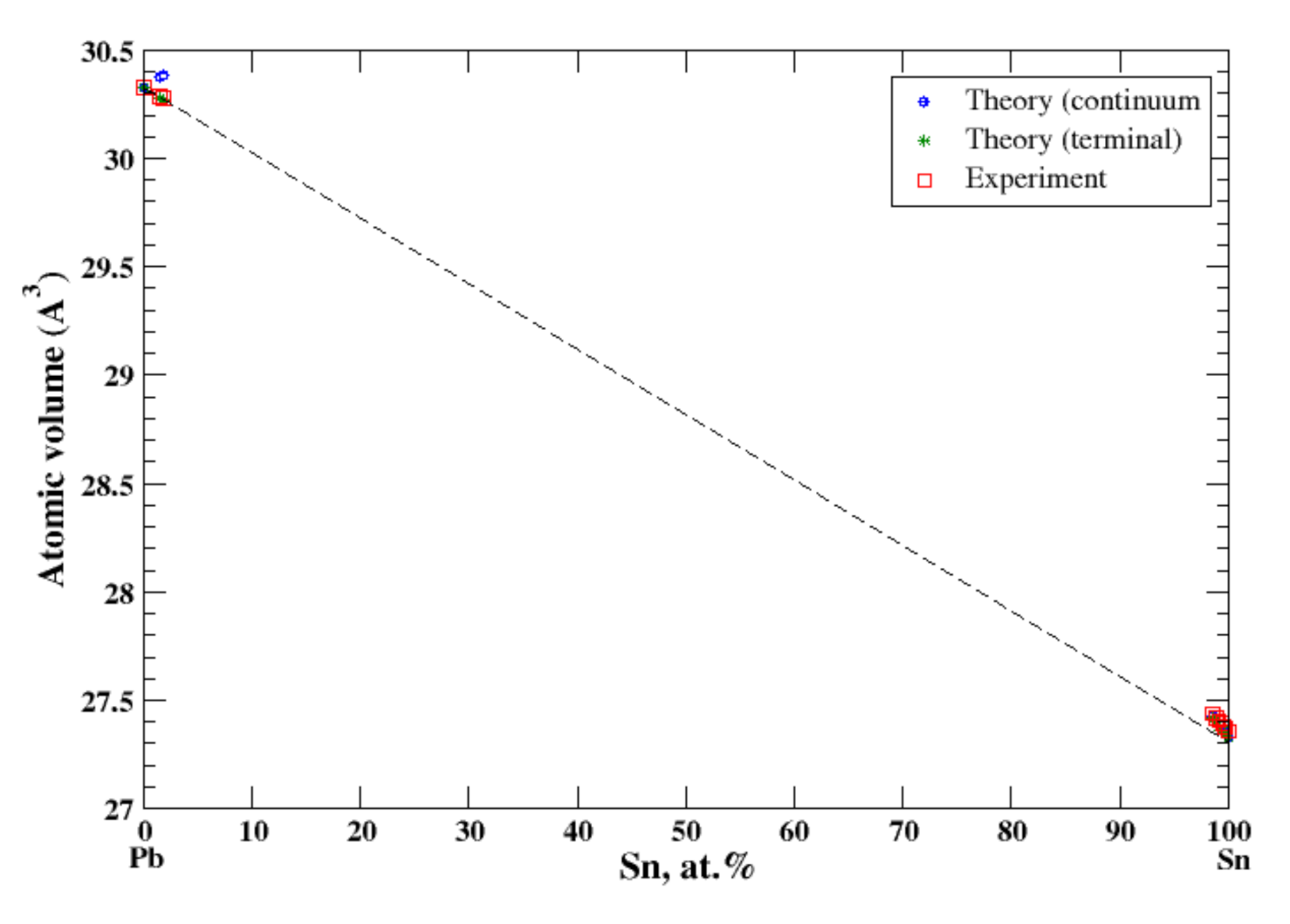
3.20. Pb-Sn
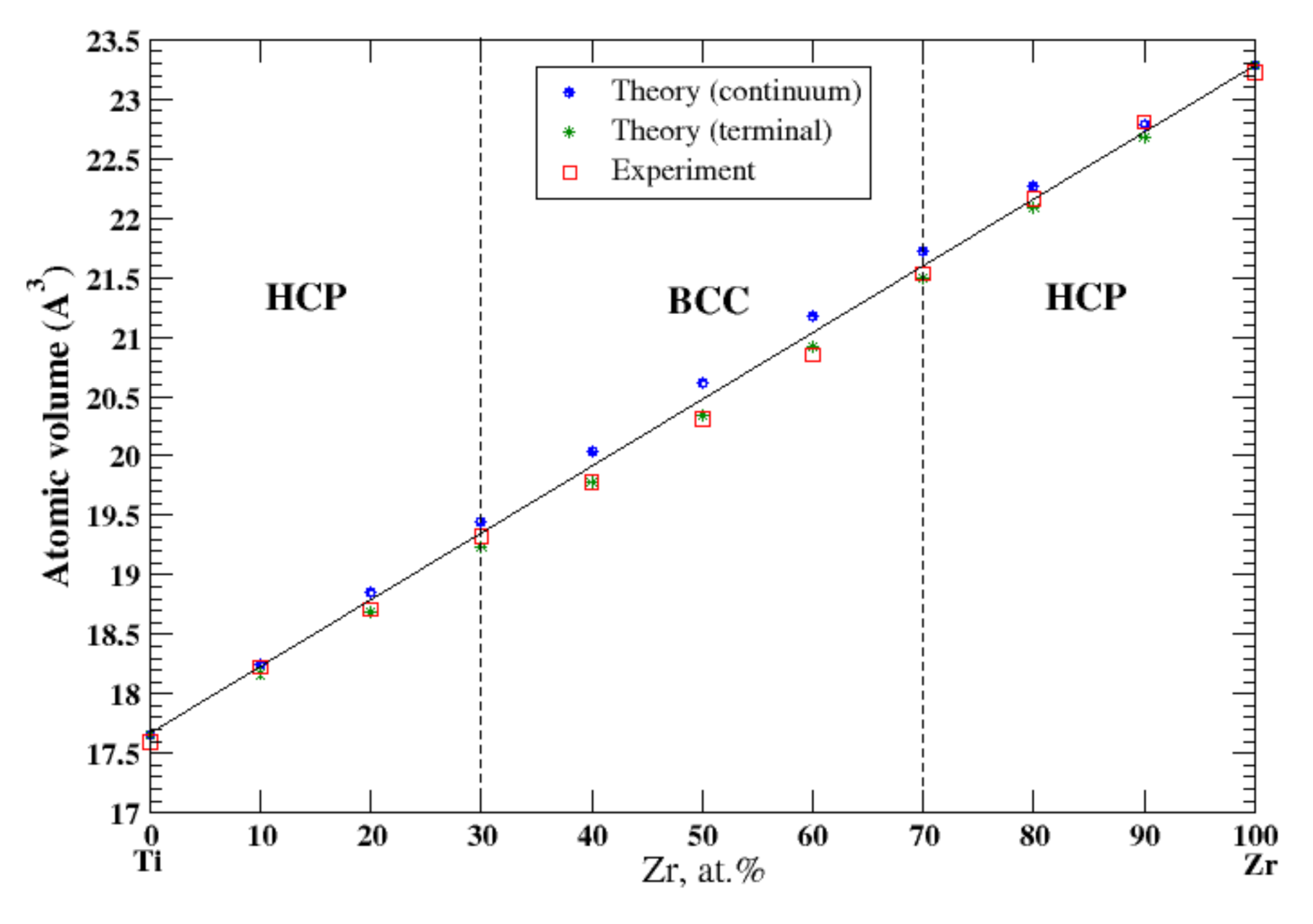
3.21. Ti-Zr
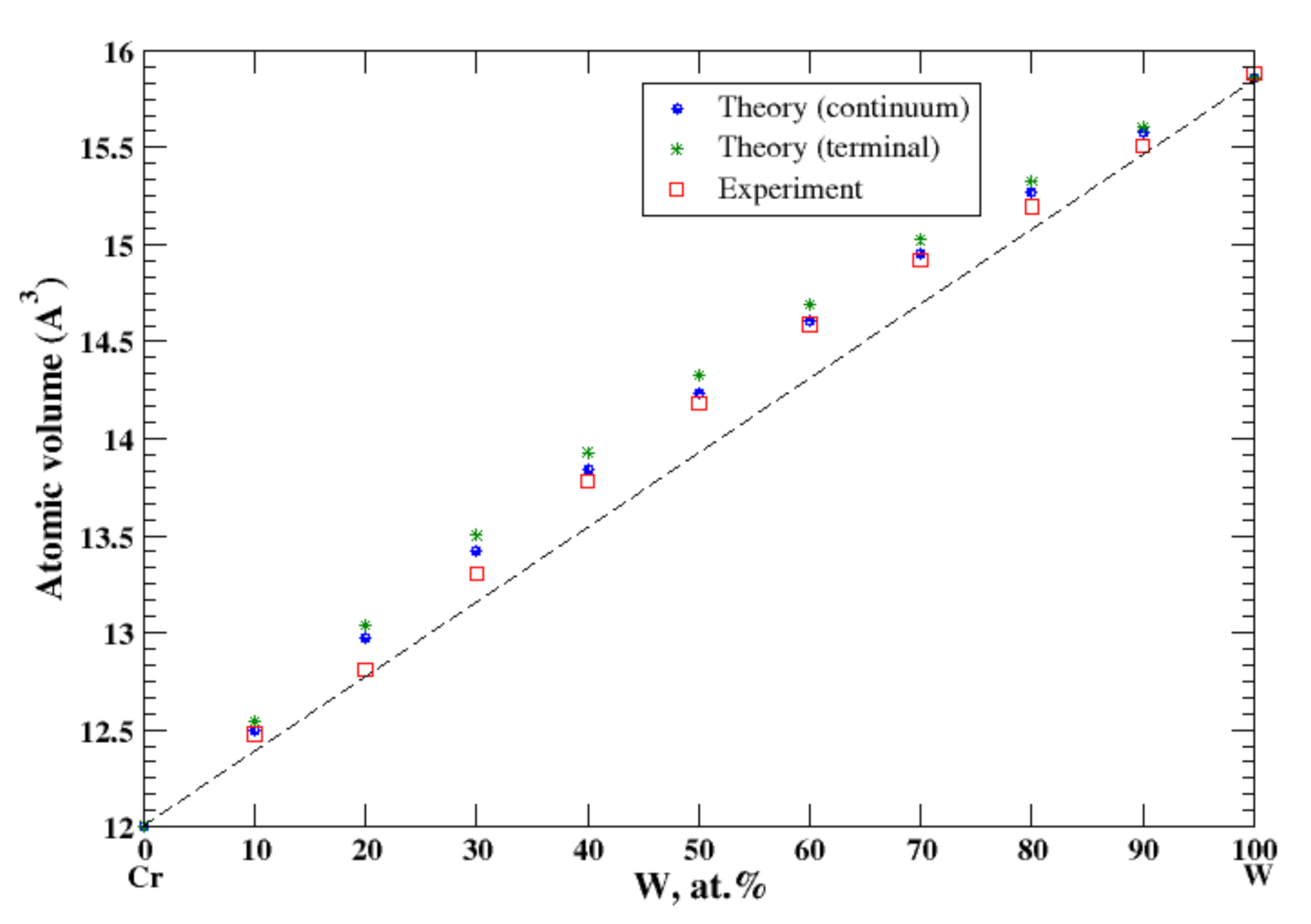
3.22. Cr-W
4. Discussion
4.1. Al-Ag, Al-Cu, Al-Mg, Al-Mn, Al-Ti, and Al-Zn
4.2. Cu-Ag, Cu-Au, Cu-Fe, Cu-Ni, and Cu-Zn
4.3. Co-Fe, Fe-V, and Fe-Cr
4.4. Ag-Au and Ag-Mg
4.5. Nb-Ta, Ti-Zr, and Cr-W
4.6. Ge-Si and Pb-Sn
4.7. Cd-Mg
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mizutani, U. Hume-Rothery Rules for Structurally Complex Alloy Phases; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar] [CrossRef]
- Vegard, L. Die Konstitution der Mischkristallen und die Raumfüllung der Atome. Z. Phys. 1921, 5, 17–26. [Google Scholar] [CrossRef]
- Zen, E. Validity of “Vegard’s Law”. J. Mineral. Soc. Am. 1956, 41, 523–524. [Google Scholar]
- Hume-Rothery, W.; Raynor, G.V. The Structure of Metals and Alloys; Institute of Metals: London, UK, 1962. [Google Scholar]
- Massalski, T.B.; King, H.W. Alloy Phases of the Noble Metal. Prog. Mater. Sci. 1963, 10, 3–78. [Google Scholar] [CrossRef]
- Kozlov, E.V.; Klopotov, A.A.; Solonitsina, N.O.; Tailashev, A.S. Dimensional crystal geometry of binary intermetallic alloys. Russ. Phys. J. 2006, 49, 35–46. [Google Scholar] [CrossRef]
- Kozlov, E.V.; Klopotov, A.A.; Tailashev, A.S.; Solonitsina, N.O. The Ni-Al system: Crystal-geometrical features. Russ. Bull. Acad. Sci. Phys. 2006, 70, 1113–1116. [Google Scholar]
- Klopotov, A.A.; Yasenchuk, Y.F.; Abzaev, Y.A.; Dement’eva, M.G.; Kozlov, E.V.; Potekaev, A.I.; Solonitsyna, N.O. System Ni-Ti. Crystallogeometrical Features. Russ. Phys. J. 2008, 51, 226–239. [Google Scholar] [CrossRef]
- Potekaev, A.I.; Kondratyuk, A.A.; Porobova, S.A.; Klopotov, A.A.; Markova, T.N.; Kukushkun, Y.A.; Klopotov, V.D. Zen’s Law and Features in Liquidus-solidus Curves in Binary State Diagrams Based on Elements VIIIA and IB of the Periodic Table. IOP Conf. Ser. Mater. Sci. Eng. 2016, 156, 012030. [Google Scholar] [CrossRef]
- Potekaev, A.I.; Klopotov, A.A.; Porobova, S.A.; Klopotov, V.D.; Markova, T.N.; Imanaliev, M.I. Binary Phase Diagrams Based on Elements VIIIA and IB Periods of the D.I. Mendeleev’s Table and Features of Crystallographic Parameters. IOP Conf. Ser. Mater. Sci. Eng. 2017, 168, 012071. [Google Scholar] [CrossRef] [Green Version]
- Lubarda, V.A. On the effective lattice parameter of binary alloys. Mech. Mater. 2003, 35, 53–68. [Google Scholar] [CrossRef]
- King, H.W. Quantitative size-factors for metallic solid solutions. J. Mater. Sci. 1966, 1, 79–90. [Google Scholar] [CrossRef]
- Nemat-Nasser, S.; Hori, M. Micromechanics: Overall Properties of Heterogeneous Materials, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Chung, D.-H. Elastic Moduli of Single Crystal and Polycrystalline MgO. Phil. Mag. 1963, 8, 833–841. [Google Scholar] [CrossRef]
- McAlister, A.J. Ag-Al (Silver-Aluminum). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 8–9. [Google Scholar]
- Pearson, W.B. Handbook of Lattice Spacings and Structures of Metals and Alloys; Pergamon Press: London, UK, 1958; Volume 1. [Google Scholar]
- Hultgren, R.; Desai, P.D.; Hawkins, D.T.; Gleiser, M.; Kelley, K.K. Selected Values of Thermodynamic Properties of Binary Alloys; American Society for Metals: Metal Park, OH, USA, 1973. [Google Scholar]
- Murray, J.L. Al-Cu (Aluminum-Copper). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 141–143. [Google Scholar]
- Okamoto, H. Al-Mg (Aluminum-Magnesium). J. Phase Equilib. 1998, 19, 598. [Google Scholar] [CrossRef]
- Liu, X.J.; Ohnuma, I.; Kainuma, R.; Ishida, K. Thermodynamic Assessment of the Aluminum-Manganese (Al-Mn) Binary Phase Diagram. J. Phase Equilib. 1999, 20, 45–56. [Google Scholar] [CrossRef]
- Schuster, J.C.; Palm, M. Reassessment of the Binary Aluminum-Titanium Phase Diagram. J. Phase Equilib. Diffus. 2006, 27, 255–277. [Google Scholar] [CrossRef]
- Okamoto, H. Al-Zn (Aluminum-Zinc). J. Phase Equilib. 1995, 16, 281–282. [Google Scholar] [CrossRef]
- Subrumanian, P.R.; Perepezko, J.H. The Ag-Cu (Silver-Copper) System. J. Phase Equilib. 1993, 14, 62–75. [Google Scholar] [CrossRef]
- Okamoto, H.; Chakrabarti, D.J.; Laughlin, D.E.; Massalski, T.B. Au-Cu (Gold-Copper). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 358–362. [Google Scholar]
- Amara, S.E.; Belhadj, A.; Kesri, R.; Hamar-Thibault, S. Stable and Metastable Equilibria in the Binary Fe-Cu and Ternary Fe-Cu-C systems. Z. Metallkd. 1999, 90, 116–123. [Google Scholar]
- Chakrabarti, D.J.; Laughlin, D.E.; Chen, S.W.; Chang, Y.A. Cu-Ni (Copper-Nickel). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 1442–1446. [Google Scholar]
- Miodownik, A.P. Cu-Zn (Copper-Zinc). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 1508–1510. [Google Scholar]
- Nishizawa, T.; Ishida, K. Co-Fe (Cobalt-Iron). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 1186–1187. [Google Scholar]
- Okamoto, H. Cr-Fe (Chromium-Iron). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 1271–1273. [Google Scholar]
- Smith, J.F. Fe-V (Iron-Vanadium). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 1787–1791. [Google Scholar]
- Okamoto, H.; Massalski, T.B. Ag-Au (Silver-Gold). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 12–13. [Google Scholar]
- Nayeb Hashemi, A.A.; Clark, J.B. Ag-Mg (Silver-Magnesium). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 55–57. [Google Scholar]
- Moser, Z.; Gasior, W.; Wypartowicz, J.; Zabdyr, L. Cd-Mg (Cadmium-Magnesium). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 995–996. [Google Scholar]
- Olesinski, R.W.; Abbaschian, G.J. Ge-Si (Germanium-Silicon). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 2000–2001. [Google Scholar]
- Krishnan, R.; Garg, S.P.; Krishnamurthy, N. Nb-Ta (Niobium-Tantalum). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 2772–2773. [Google Scholar]
- Fetch, H.J.; Perepezko, J.H. Metastable Phase Equilibria in the Lead-Tin Alloy System: Part I. Experimental. Metall. Trans. A 1989, 20, 785–794. [Google Scholar] [CrossRef]
- Saunders, N.; Argent, B.B. On the α->β transformation in Ti-Zr alloys. J. Less Common Met. 1986, 125, L11–L13. [Google Scholar] [CrossRef]
- Naidu, S.V.N.; Sriramamurthy, A.M.; Rama Rao, P. Cr-W (Chromium-Tungsten). In Binary Alloy Phase Diagrams, 2nd ed.; Massalski, T.B., Ed.; ASM International: Materials Park, OH, USA, 1990; Volume 1, pp. 1353–1354. [Google Scholar]
- Ponomareva, A.V.; Ruban, A.V.; Vekilova, O.; Yu Simak, S.I.; Abrikosov, I.A. Effect of Pressure on Phase Stability in Fe-Cr Alloys. Phys. Rev. B 2011, 84, 094422. [Google Scholar] [CrossRef] [Green Version]
- Hume-Rothery, W.; Raynor, G.V. The Equilibrium and Lattice-Spacing Relations in the System Magnesium-Cadmium. Proc. R. Soc. 1940, A174, 471–486. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.H.; Scott, M.J.; Young, W.H. Thermodynamic Properties of Solid Alloys: Application to MgxCd1-x. J. Phys. F Metal. Phys. 1976, 6, 1039–1051. [Google Scholar] [CrossRef]
- Hasegawa, M.; Young, W.H. Pseudopotential Theory of the Solid-Liquid Transition in Binary Alloys: Applications to CdxMg1-x and NaxK1-x. J. Phys. F Metal. Phys. 1977, 7, 2271–2283. [Google Scholar] [CrossRef]
- Hasegawa, M.; Young, W.H. Entropy-Volume Correlation for Some Homovalent Disordered solid Alloys. J. Phys. F Metal. Phys. 1978, 8, 1105–1117. [Google Scholar] [CrossRef]
- Landa, A.I.; Psahie, S.G.; Panin, V.E.; Zhorovkov, M.F. Vegard’s Rule for the Equilibrium Volume of Solid Solutions. Izv. Vyssh. Uchebnykh Zaved. Fiz. 1981, 24, 118–120. [Google Scholar]
- Landa, A.I.; Yuryev, A.A.; Ruban, A.V.; Gurskaya, E.G.; Kovnersityi, Y.K.; Vatolin, N.A. Pseudopotential Calculation of Thermodynamic Properties and Glass Transition Temperatures of Binary Ni-Al Alloys. J. Phys. Condens. Matter 1991, 3, 9229–9243. [Google Scholar] [CrossRef]
- Jacob, K.T.; Raj, S.; Rannesh, L. Vegard’s Law: A Fundamental Relation or an Approximation? Int. J. Mater. Res. 2007, 98, 776–779. [Google Scholar] [CrossRef] [Green Version]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Ω (Å3) | K (GPa) | μ (GPa) |
|---|---|---|---|
| Mg | 23.2396 | 35.6 | 17.3 |
| Cd | 22.0210 | 46.8 | 19.1 |
| Al | 16.6036 | 72.6 | 26.0 |
| Si | 20.0182 | 97.6 | 66.2 |
| Ti | 17.6542 | 108.2 | 45.6 |
| V | 13.8256 | 157.9 | 46.7 |
| Cr | 12.0064 | 160.0 | 115.1 |
| Mn | 12.2199 | 98.0 | 39.0 |
| Fe | 11.7771 | 169.6 | 81.4 |
| Co | 11.0732 | 82.3 | 88.8 |
| Ni | 10.9415 | 183.0 | 80.0 |
| Cu | 11.8072 | 136.4 | 46.8 |
| Zn | 15.2123 | 69.6 | 41.9 |
| Ge | 22.6345 | 75.0 | 54.9 |
| Zr | 23.2790 | 94.0 | 30.0 |
| Nb | 17.8715 | 170.3 | 37.5 |
| Mo | 15.5834 | 261.3 | 125.5 |
| Ag | 17.0578 | 103.4 | 30.3 |
| Sn | 27.3255 | 58.2 | 18.4 |
| Ta | 18.0173 | 196.5 | 69.0 |
| W | 15.8566 | 311.0 | 160.6 |
| Au | 16.9618 | 170.7 | 27.5 |
| Pb | 30.3246 | 45.9 | 5.6 |
| Alloy | ω1 | ω2 |
|---|---|---|
| Al-Ag | −0.0918 | +0.0012 |
| Al-Cu | +0.2000 | −0.3780 |
| Al-Mg | −0.3580 | +0.4082 |
| Al-Mn | +0.1620 | −0.4681 |
| Al-Ti | −0.2009 | −0.1506 |
| Al-Zn | −0.0625 | −0.0574 |
| Cu-Ag | −0.2775 | +0.4352 |
| Cu-Au | −0.2781 | +0.4759 |
| Cu-Fe | +0.1753 | +0.0457 |
| Cu-Ni | +0.0718 | −0.0845 |
| Cu-Zn | −0.5457 | +0.1710 |
| Fe-Co | +0.0524 | +0.0154 |
| Fe-Cr | −0.0207 | +0.0436 |
| Fe-V | −0.1886 | +0.1051 |
| Ag-Au | −0.0064 | −0.0178 |
| Ag-Mg | −0.6342 | +0.0713 |
| Mg-Cd | −0.0160 | −0.2108 |
| Si-Ge | −0.2065 | +0.0468 |
| Nb-Ta | −0.0023 | −0.0026 |
| Pb-Sn | +0.2905 | −0.0825 |
| Ti-Zr | −0.2233 | +0.3008 |
| Cr-W | −0.2173 | +0.3735 |
| Alloy | Ω1 (Å3) | Ω2 (Å3) | ||
|---|---|---|---|---|
| Al-Ag | 16.6036 | 17.0578 | 15.3642 | 16.6193 |
| Al-Cu | 16.6036 | 11.8072 | 15.0820 | 11.8247 |
| Al-Mg | 16.6036 | 23.2396 | 17.2056 | 27.4045 |
| Al-Mn | 16.6036 | 12.2129 | 14.5684 | 10.4510 |
| Al-Ti | 16.6036 | 17.6542 | 13.8034 | 14.4677 |
| Al-Zn | 16.6036 | 15.2123 | 14.2961 | 15.6559 |
| Cu-Ag | 11.8072 | 17.0578 | 13.0147 | 18.4090 |
| Cu-Au | 11.8072 | 16.9618 | 12.4972 | 17.8913 |
| Cu-Fe | 11.8072 | 11.7771 | 14.1683 | 12.3236 |
| Cu-Ni | 11.8072 | 10.9415 | 11.8423 | 10.9082 |
| Cu-Zn | 11.8072 | 15.2123 | 9.5991 | 14.6320 |
| Fe-Co | 11.7771 | 11.0732 | 11.4817 | 12.0343 |
| Fe-Cr | 11.7771 | 12.0064 | 11.7664 | 12.3105 |
| Fe-V | 11.7771 | 13.8256 | 11.4252 | 13.0978 |
| Ag-Au | 17.0578 | 16.9618 | 16.8408 | 16.7868 |
| Ag-Mg | 17.0578 | 23.2396 | 13.9318 | 18.9916 |
| Mg-Cd | 23.2396 | 22.0210 | 21.6319 | 19.4024 |
| Si-Ge | 20.0182 | 22.6345 | 18.7414 | 21.1106 |
| Nb-Ta | 17.8715 | 18.0173 | 17.9742 | 17.8253 |
| Pb-Sn | 30.3246 | 27.3255 | 36.8260 | 27.9621 |
| Ti-Zr | 17.6542 | 23.2790 | 18.6324 | 23.8674 |
| Cr-W | 12.0064 | 15.8566 | 11.5458 | 15.7591 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landa, A.; Klepeis, J.E.; Rudd, R.E.; Caspersen, K.J.; Young, D.A. Analytic Binary Alloy Volume–Concentration Relations and the Deviation from Zen’s Law. Appl. Sci. 2021, 11, 6231. https://doi.org/10.3390/app11136231
Landa A, Klepeis JE, Rudd RE, Caspersen KJ, Young DA. Analytic Binary Alloy Volume–Concentration Relations and the Deviation from Zen’s Law. Applied Sciences. 2021; 11(13):6231. https://doi.org/10.3390/app11136231
Chicago/Turabian StyleLanda, Alexander, John E. Klepeis, Robert E. Rudd, Kyle J. Caspersen, and David A. Young. 2021. "Analytic Binary Alloy Volume–Concentration Relations and the Deviation from Zen’s Law" Applied Sciences 11, no. 13: 6231. https://doi.org/10.3390/app11136231
APA StyleLanda, A., Klepeis, J. E., Rudd, R. E., Caspersen, K. J., & Young, D. A. (2021). Analytic Binary Alloy Volume–Concentration Relations and the Deviation from Zen’s Law. Applied Sciences, 11(13), 6231. https://doi.org/10.3390/app11136231