1. Introduction
Neuroimaging has improved the understanding, diagnosis, and management of several pediatric cerebellar degenerative diseases.
The accurate observation of structural imaging studies with MRI in search of atrophic changes and subtle signal abnormalities due to neuronal loss, gliosis, changes in the water content of the nervous tissue is the best possible way to reach a neuroradiological diagnosis.
Advanced MR techniques such as MR spectroscopy [
1,
2] and voxel-based morphometry (VBM) have been applied for years in adults and children with sporadic and genetic ataxia, but nowadays play a minor role. Other techniques, such as and diffusion tensor imaging (DTI) [
3,
4], have more application in selected cases.
Functional MRI (fMRI) using the BOLD (blood oxygenation level-dependent) effect has also been applied more recently to the investigation of pediatric patients with neurodegeneration and is still a matter of research.
In this paper, we shall concentrate on MRI, pointing out the characteristic MRI findings of some paediatric cerebellar degenerative diseases: pontocerebellar hypoplasia, (PCH 1–13), infantile neuroaxonal dystrophy (INAD), autosomal recessive spastic ataxia of Charlevoix–Saguenay (ARSACS), inherited defects in glycosylation (GDC), Cockayne Syndrome, GM2-gangliosidosis and neuronal ceroid lipofuscinoses (NCLs), Progressive encephalopathy, edema, hypsarrhythmia, and optic atrophy (PEHO), Ataxia-telangiectasia (AT), ataxias with oculomotor apraxia (AOAs, AOA1, AOA2), mitochondrial diseases and spinocerebellar ataxia (SCAs). Furthermore, we only mention two leukodystrophies with hypomyelination and cerebellar atrophy, 4H-syndrome, and H-ABC.
1.1. Pontocerebellar Hypoplasia
Pontocerebellar hypoplasia (PCH) is a group of neurodegenerative disorders with autosomal recessive inheritance and prenatal onset, mainly affecting the survival and growth of neurons in the cerebellar cortex, dentate nuclei, ventral pontine, and inferior olivary nuclei. Microcephaly, neocortical atrophy, and ventriculomegaly are also usually observed. The common MRI findings include cerebellar hypoplasia and a variable degree of cerebellar and ventral pons atrophy [
5].
To date, there have been identified 19 genes implied in 13 PCH subtypes (OMIM). Like the genetic spinal muscular atrophies, Type 1 is characterized by the loss of motor neurons in the spinal cord [
6]. Prenatal ultrasounds are reported to show arthrogryposis and polyhydramnios, while postnatally neonates present with hypotonia, primary hypoventilation, impaired swallowing with consequent feeding difficulties, and progressive microcephaly [
7]. In PCH type 2, which represents the most frequent PCH and the best-studied subtype, the distinctive features are dystonia and dyskinesia, severe microcephaly, and, more rarely, pure spasticity [
8,
9]. Prenatal ultrasounds are usually unremarkable and pregnancies uneventful. Affected neonates present usually, but not always, with dysphagia, respiratory and feeding difficulties, while extrapyramidal dyskinesia with mixed spasticity develops later. PCH2 is often fatal in early childhood. Affected children develop progressive microcephaly, visual impairment, seizures, and severe cognitive and motor delays. Impaired motor development with a lack of voluntary hand control is not uncommon. PCH type 4, former known as olivopontocerebellar hypoplasia, is a more severe variant of PCH2 with prenatal onset even of clinical manifestations such as polyhydramnios, contractures, and babies usually do not live beyond the neonatal period [
10]. PCH1 B, C, and D share a common pathway involving the EXOSOME complex, while PCH2 A, B, C, F, PCH4, and PCH5 are related to mutations in the TSEN complex [
11].
Overall, MRI in PCH patients shows a variable degree of cerebellar hypoplasia with involvement of pons and cerebrum. On coronal imaging, the particular shape of the cerebellum, named “dragonfly” or “butterfly” appearance, was described by Barth et al. [
6]. The cerebellar hemispheres are particularly atrophic, with a substantial sparing of the vermis. Flattening of the caudate nuclear heads can be present [
5]. Additional inconstant findings are cerebellar cysts (PCH 1, 2, and 6), agenesis of the corpus callosum (PCH 9), simplified gyral pattern (PCH 6 and 10), and hypomyelination (PCH 1, 2, 6, and 9). In several PCH, pons could be relatively preserved, and in PCH10, the cerebellum is mostly normal 11. The presence of C-shaped inferior olive has been reported in PCH2, suggesting a prenatal onset 9 (
Figure 1).
A dragonfly-like pattern has been described in PCH 1, 2 (
Figure 1), 4, 6 (
Figure 2), and 9.
1.2. Infantile Neuroaxonal Dystrophy
Infantile neuroaxonal dystrophy (PLAN, PLA2G6-Associated Neurodegeneration) belongs to the neurodegeneration with brain iron accumulation (NBIA) with onset in the first or second year of life. It is a rare neurodegenerative disorder due to mutations in the gene PLA2G6. It is characterized by psychomotor regression, profound hypotonia, gait disturbance, cerebellar ataxia, pyramidal signs, and early optic atrophy with loss of visual acuity and ultimate blindness. A progressive peripheral neuropathy leads to hyporeflexia and dementia [
12,
13].
MRI shows global atrophy of the cerebellum, mainly involving the inferior part of the vermis. Associated features are hyperintensity of the cerebellar cortex and dentati nuclei, thinning of the optic chiasm, clava hypertrophy, and, inconstantly, iron deposition in the globus pallidus and the substantia nigra [
14,
15] (
Figure 3).
1.3. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS)
The autosomal recessive spastic ataxia of Charlevoix- Saguenay (ARSACS) is an early-onset neurodegenerative disease characterized by cerebellar ataxia, pyramidal tract involvement, and peripheral neuropathy 4. In late-onset cases [
12], patients missing pyramidal tract involvement [
13] and occasionally present with hearing loss [
14]. Retinal fibers hypermyelination observed at funduscopic examination is considered a pathognomonic hallmark [
15].
The responsible gene (
SACS) is located on chromosome 13q. It encodes for the Sacsin protein, which is widely expressed throughout the brain with a predilection for pyramidal neurons, Purkinje cells, thalamic and pontine nuclei, and reticular formation [
16].
MRI studies show characteristic features of the disease: atrophy of the superior cerebellar vermis and cervical spinal cord with bulky pons and increased thickness of the middle cerebellar peduncles. On T2w-FLAIR images, the pons shows a subtle hyperintensity (mainly in the raphe and lateral aspects) and exhibit hypointense stripes, acknowledged as the corticospinal tract (CST). Similar hypointensity is also observed in the medial lemnisci [
4]. Thinning of the corpus callosum and a rim of T2-hyperintensity around the thalami are also frequent findings (
Figure 4).
Analyses of diffusion tensor tractography (DTI) [
4,
17] revealed an over-representation of transverse pontine fibers (TPF), causing a bulky shape of the pons and thick middle cerebellar peduncles (MCP) and probably preventing the reconstruction of the CST at this level. Significant changes consistent with microstructural alterations were found in the supratentorial white matter, forceps, cingulum, and superior longitudinal fasciculus 4. Pathological correlations are still lacking.
1.4. CDG
Congenital disorders of glycosylation (CDG), which is the addition process of complex sugar chains to lipids and proteins, comprise most of the genetic disorders due by abnormal synthesis of glycoconjugates. These disorders involve different organ systems, including the nervous system [
18].
Congenital disorder of glycosylation type 1A (CDG1A) is the most common
N-linked CDG and is responsible for around 80% of all cases. This disorder is caused by mutations in
PMM2 gene. The CNS and peripheral nervous system (PNS) are the primary targets [
19].
As a laboratory finding, the isoelectric focusing of serum transferrin showed a consistent increase of isotransferrins with higher isoelectric points than normal [
20].
CDG1A is an important cause of cerebellar ataxia. In a study of posterior fossa imaging of 158 children with ataxia, 84 presented global cerebellar atrophy; 21 of them had PMM2 mutation, while 18 presented defects of the respiratory chain [
21].
Most children with CDG1A have small pontine protuberance, shrunken appearance of the superior vermis and flattened inferior vermis, and hyperintense hemispheric cerebellar cortex at birth. In milder cases, the initial radiological findings could be normal with a subsequent atrophic evolution (
Figure 5).
The cerebellar white matter is also slightly hyperintense compared with supratentorial white matter, leading to the typical “bright cerebellum” appearance. 1H-MRS shows a markedly reduced NAA peak [
22]. Pathological findings suggest very early-onset hypoplasia followed by progressive atrophy [
22,
23].
1.5. Cockayne Syndrome
Cockayne Syndrome (CS) is a rare autosomal recessive multisystem disorder caused by mutations in
CSA and
CSB genes, where proteins are involved in the nucleotide excision repair pathway. Clinical characteristics include neurodevelopmental delay, cutaneous photosensitivity, pigmentary retinopathy, neurosensory hearing loss. The disease encompasses a wide spectrum of clinical severity, divided into four overlapping clinical subgroups of decreasing severity: (1) COFS is the most severe form, starting in utero or the neonatal period with a rapidly fatal outcome; (2) CSII, begins in the first year of life and leads to death in infancy; (3) CSI is the classic form that manifests during infancy; death occurs in the first decade of life. (4) CSIII shows milder neurological signs, and affected patients may reach adulthood [
24].
Brain CT and MRI findings include diffuse cerebral and cerebellar atrophy, calcifications in the basal ganglia, and lack of myelination of the white matter [
25]. These cardinal neuroimaging features are not all simultaneously present: brain atrophy is probably the earliest detectable sign. Brain calcifications are typically bilateral and symmetric, ranging from punctate to large, and are most frequently seen in the putamen. Calcifications, which are progressive, may be seen in the white matter-cortex junction, dentate nuclei, pallida, and more rarely in white matter and thalami. Diffuse brain atrophy with white matter loss and ventricular dilatation is observed in the very early stages of the disease and always progresses; the brainstem and the cerebellum usually show moderate atrophy with enlarged cisterna magna. White matter abnormalities attributed to hypomyelination, or demyelination involve the corpus callosum, the posterior limb of the internal capsule and the external capsule, whereas the cerebellum and brainstem always show normal myelination. After several years of the disease, the signal intensity of the hemispheric white matter is very high on FLAIR images, suggesting progressive astrogliosis [
25].
1.6. Lysosomal Storage Disorders: GM2-Gangliosidosis and Neuronal Ceroid Lipofuscinoses
In lysosomal storage disorders (LSDs), neurodegeneration can involve multiple brain structures such as the thalami, cortex, hippocampus, and cerebellum. The selective vulnerability of Purkinje cells is responsible for the important damage of the cerebellar cortex. [
26,
27].
Late-onset GM2-gangliosidosis is the prototype of LSD characterized by isolated cerebellar atrophy. The disease is caused by beta-hexosaminidase A deficiency or combined beta-hexosaminidase A and B. In late-onset phenotype, neurological symptoms include cerebellar ataxia, psychiatric disorders, dystonia, and anterior motor neuron disease. The most common MRI findings show severe, isolated cerebello-vermian atrophy. In a minority of cases, imaging shows important cerebral atrophy and white matter abnormal signal intensity secondary to axonal degeneration [
27].
The neuronal ceroid lipofuscinoses (NCLs) are a group of neurodegenerative diseases characterized by the intracellular accumulation of autofluorescent ceroid lipopigments due to different gene mutations. Currently, 14 different genetic forms have been described [
28], divided in clinical groups based on the age of onset.
There are three main childhood NCL phenotypes: infantile, late infantile, and juvenile. The most common genetic form of infantile NCL is
CLN1 due to a mutation in the gene that encodes the enzyme palmitoyl protein thioesterase 1 (PPT1). Late infantile NCL is often caused by mutations in the
CLN2 gene, causing tripeptidyl-peptidase 1 deficiency (TPP1). The disease begins around the third year of life in previously healthy children, with epilepsy of sudden onset or unexpected delay of psychomotor development. Mutations cause juvenile NCL (JNCL) in
CLN3. The main clinical sign of this type of NCL (JNCL,
CLN3, juvenile-onset Batten’s disease, Spielmeyer–Vogt syndrome), starting around the age of 4 to 7 years, is a visual failure that leads to blindness usually within a few years [
29]. Overall, the most frequent forms of NCL are the juvenile form (Batten disease, CLN3) and late infantile form (Jansky–Bielschowsky disease, CLN2).
The neuroradiological findings in the different forms of NCL are quite similar: thalamic involvement with T2 hypointensity fading away with increasing age of onset; infra and supratentorial atrophy with distinct thinning of the cerebral cortex, and white matter T2 hyperintensity, usually not marked, with appearance only in adolescence and juvenile forms [
30] (
Figure 6).
1.7. Progressive Encephalopathy, Edema, Hypsarrhythmia, and Optic Atrophy (PEHO) Syndrome
Progressive encephalopathy, edema, hypsarrhythmia, and optic atrophy (PEHO) syndrome is a rare progressive neurodegenerative disease characterized by severe progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy [
31,
32]. The onset of the disease is in early infancy; dysmorphic facial features, mental retardation, edema of the face and limbs, and infantile spasms with hypsarrhythmia are commonly observed. MRI may show severe, progressive atrophy of the cerebellum and brainstem. Less frequently, mild cerebral atrophy and abnormal signal intensity in the cerebral white matter are noted [
33].
1.8. Ataxia Telangiectasia
Ataxia-telangiectasia (AT) is the most common recessive ataxia in children under 5 years of age. It is characterized by prominent progressive ataxia and abnormalities of posture and tone, movement disorders for extrapyramidal dysfunction, and oculocutaneous telangiectasia appearing between the age of 2 and 8 years. The clinical spectrum of AT is characterized by recurrent infections due to immunodeficiency, skin and endocrine abnormalities, predisposition for malignancies, and radiation sensitivity [
34]. AT is caused by mutations in the ataxia-telangiectasia gene (ATM) involved in DNA repair [
35]. Neuropathological studies show cerebellar atrophy, mainly cortical, degeneration of spinal posterior columns, especially of the gracile fascicle, and axonal degeneration in the peripheral nerves; deep cerebellar nuclei and inferior olives abnormalities are inconstantly reported [
36].
Radiological imaging is limited to MRI since CT and studies with x-rays can increase the risk of genetic mutation in these patients highly sensitive to DNA damage by ionizing radiation [
37,
38]. MR images reveal diffuse cerebellar atrophy, with initial involvement of the superior vermis and lateral cerebellar hemispheres and unusual decreased cortical thickness along cerebellar hemispheres. Supratentorial brain shows no sign of volume loss. At the same time, confluent hyperintensities in T2/FLAIR and multiple focal hypointensities (thought to be deposits of haemosiderin related to capillary telangiectasia) are seen in deep white matter, mainly in adult patients [
39]. Hypoplasia of the inferior vermis and a large cisterna magna were also frequent signs [
40] (
Figure 7).
1.9. Ataxias with Oculomotor Apraxia (AOAs)
The AOAs include AOA1 (early-onset cerebellar ataxia with oculomotor apraxia type 1, ocular motor apraxia, peripheral neuropathy, and hypoalbuminemia), which is most frequent in Portugal and Japan, and AOA2 (ataxia with ocular apraxia type 2, ataxia with ocular apraxia and increased alpha-fetoprotein levels) [
41]. The onset of AOAs is mostly in childhood and early teens. AOA1 and AOA2 are associated with mutations in the aprataxin gene and the senataxin gene, respectively. In AOA1, cerebellar ataxia with atrophy of the cerebellum on MRI and sensorimotor axonal neuropathy is present in all patients, whereas oculomotor apraxia occurs in about 85% of cases [
42]. A typically choreic movement disorder, which is evident in the early stages and declines over time, is present in 50% of cases.
2. Mitochondrial Disorders
In MRI studies of several mitochondrial disorders, the cerebellum appears involved. In many instances, however, cerebellar ataxia is not the most characterizing feature. Patients with mitochondrial disease often present a combination of abnormalities: signal abnormalities in the basal ganglia, or the brainstem, or cerebellum; the pattern of distribution, however, is fairly characteristic and may suggest a specific disorder [
43].
Isolated or predominant cerebellar volume loss due to cerebellar hypoplasia or, more frequently, progressive cerebellar atrophy are common neuroradiological features in children with various mitochondrial disorders: complex I, complex II, complex III and complex IV deficiency, isolated or combined, multiple respiratory chain defects (RCDs) and partial coenzyme Q10 deficiency [
44] (
Figure 8).
Coenzyme Q10 transfers electrons along the respiratory chain; among other clinical manifestations, Coenzyme Q10 deficiency has been associated with cerebellar ataxia [
45]; 13 genes encode proteins involved in its biosynthesis, but only mutations in 10 genes have been associated with diseases. One of the most well-documented is mutations in the COQ8A gene (formerly ADCK3). In these patients, cerebellar atrophy is an almost universal finding [
46].
In POLG related disorders and myoclonic epilepsy with ragged red fibers (MERRF), white matter abnormalities and changes in the dentate nucleus can be observed. The latter has also been reported in Kearns–Sayre syndrome (KSS) [
43].
In other mitochondrial disorders, the cerebellum is the main target of the disease, sometimes in combination with brainstem or supratentorial abnormalities. In children, the most common mitochondrial disorder is Leigh’s syndrome, characterized by cerebellar atrophy with lesions in the dentate nuclei and lesions in the brainstem (periaqueductal area, pontine) tegmentum and medulla oblongata), lesions in the basal ganglia, particularly the posterior part of the putamen. In mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), cerebral atrophy and cerebellar volume loss may be associated with leukoencephalopathy. Lesions in MELAS are extensive cortical and subcortical hyperintensities in T2-w.i. in supra and infratentorial compartments compatible with stroke-like lesions but not confined to vascular territories. They do not evolve as ischemic infarcts and may initially completely regress. Progressive cerebellar atrophy is a characterizing feature [
44]. In an unsuspected adolescent with a first stroke-like episode, observation at MRI of cerebellar atrophy should suggest the possibility of MELAS.
2.1. Spinocerebellar Ataxia
Hereditary spinocerebellar ataxias define a genetically heterogeneous group of autosomal dominantly inherited progressive disorders characterized by loss of balance and coordination. Although the onset of ataxia usually occurs in the third or fourth decade of life [
47], several genetic SCAs have been described in children. Usually, imaging is poor, showing mainly cerebellar atrophy in all these forms.
CACNA1G calcium channel is known to cause adult-onset spinocerebellar ataxia [
42]; recent works report cerebellar ataxia associate with mutations in this gene [
48,
49]. MRI shows cerebellar atrophy [
49] (
Figure 9).
CACNA1A can cause a wide range of disorders, including episodic ataxia type 2 (EA2), familial hemiplegic migraine (FHM1), and progressive spinocerebellar ataxia (SCA6). Mild progressive cerebella atrophy has been described in mutations of this gene [
50].
In SCA 5, due to mutations, the SPTBN2 gene has been reported early ataxia associated with cerebellar atrophy [
51].
2.2. H-ABC (Hypomyelination with Atrophy of Basal Ganglia and Cerebellum)
H-ABC is an autosomal dominant hypomyelination leukodystrophy due to TUBB4A mutations [
52]. The characteristic clinical features are progressive dystonia with early onset associated with cerebellar and pyramidal signs and cognitive deficiency. Neuroradiological findings combine diffuse cerebellar atrophy, hypomyelination, and typical basal ganglia abnormalities characterized by progressive neostriatal atrophy, with a normal size of the pallidum and thalamus, until almost complete disappearance of the putamina and caudate [
53] (
Figure 10).
Hypomyelination is recognizable by the complete or almost complete absence of the normal T1 and T2 signal on MRI in all the stages of the disease. Cerebellar atrophy and/or neostriatal atrophy may be absent; mild hypomyelination or normal MRI have also been reported [
54].
2.3. H Leukodystrophy
RNA polymerase III (POLR3) is made of 17 subunits, with two major subunits, RPC1 and RPC2, encoded by the
POLR3A and
3B genes, respectively. POLR3-related leukodystrophy is a term that now encompasses all the hypomyelination diseases caused by mutations of these genes [
26,
55]. Biallelic variants in POLR3A cause 4H leukodystrophy, an autosomal recessive disorder characterized by hypomyelination, hypogonadotropic hypogonadism, and hypodontia, associated with early cerebellar ataxia. Pyramidal signs, cognitive decline, endocrine features, and myopia are usually present, and tooth agenesis, including hypodontia or oligodontia, is a characteristic manifestation.
Neuroimaging demonstrates cerebello-vermian atrophy with normal basal ganglia and T2 hyperintensity of middle cerebellar peduncles. Cases with POLR3A demonstrate less cerebellar atrophy, while cases with POLR3B show subtler degrees of hypomyelination 27 (
Figure 11).
3. Conclusions
MRI, supported by CT to demonstrate calcifications, offers all the imaging features to evaluate and suggest a specific diagnosis to the clinician. In recent years, advanced MR techniques such as 1H-MRS, fMRI, DTI, and volumetric studies have offered new insights into the pathogenesis of a few diseases with positive MRI findings. They have occasionally helped discover abnormalities in disorders in which conventional MRI fails to demonstrate any morphological abnormalities. Their diagnostic value, however, has not yet been systematically assessed and remains a matter of research.
Conventional MRI, however, remains valuable support in a diagnostic flowchart to address the proper diagnosis.
Author Contributions
Conceptualization, L.C. and M.M.; methodology, L.C. and M.M.; investigation, L.C. and M.M.; resources, L.C. and M.M.; writing—original draft preparation, L.C. and M.M.; writing—review and editing, L.C. and M.M. Both authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Ethical review and approval were waived for this study because this study involves only anonymizated images.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mascalchi, M.; Cosottini, M.; Lolli, F.; Salvi, F.; Tessa, C.; Macucci, M.; Tosetti, M.; Plasmati, R.; Ferlini, A.; Tassinari, C.A.; et al. Proton MR spectroscopy of the cerebellum and pons in patients with degenerative ataxia. Radiology 2002, 223, 371–378. [Google Scholar] [CrossRef]
- Guerrini, L.; Belli, G.; Mazzoni, L.; Foresti, S.; Ginestroni, A.; Della Nave, R.; Diciotti, S.; Mascalchi, M. Impact of cerebrospinal fluid contamination on brain metabolites evaluation with 1H-MR spectroscopy: A single voxel study of the cerebellar vermis in patients with degenerative ataxias. J. Magn. Reson. Imaging 2009, 30, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Taoka, T.; Kin, T.; Nakagawa, H.; Sakamoto, M.; Wada, T.; Takayama, K.; Wuttikul, C.; Iwasaki, S.; Ueno, S.; Kichikawa, K. Diffusivity and diffusion anisotropy of cerebellar peduncles in cases of spinocerebellar degenerative disease. Neuroimage 2007, 37, 387–393. [Google Scholar] [CrossRef]
- Prodi, E.; Grisoli, M.; Panzeri, M.; Minati, L.; Fattori, F.; Erbetta, A.; Uziel, G.; D’Arrigo, S.; Tessa, A.; Ciano, C.; et al. Supratentorial and pontine MRI abnormalities characterize recessive spastic ataxia of Charlevoix-Saguenay. A comprehensive study of an Italian series. Eur. J. Neurol. 2013, 20, 138–146. [Google Scholar] [CrossRef]
- Namavar, Y.; Barth, P.G.; Kasher, P.R.; Van Ruissen, F.; Brockmann, K.; Bernert, G.; Writzl, K.; Ventura, K.; Cheng, E.Y.; Ferriero, D.M.; et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 2011, 134, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Barth, P.G. Pontocerebellar hypoplasia—How many types? Eur. J. Paediatr. Neurol. 2000, 4, 161–162. [Google Scholar] [CrossRef]
- Bacrot, S.; Mechler, C.; Talhi, N.; Martin-Coignard, D.; Roth, P.; Michot, C.; Ichkou, A.; Alibeu, O.; Nitschke, P.; Thomas, S.; et al. Whole exome sequencing diagnoses the first fetal case of Bainbridge-Ropers syndrome presenting as pontocerebellar hypoplasia type 1. Birth Defects Res. 2018, 110, 538–542. [Google Scholar] [CrossRef]
- Barth, P.G.; Blennow, G.; Lenard, H.G.; Begeer, J.H.; Van Der Kley JMHanefeld, F.; Peters, A.; Valk, J. The syndrome of autosomal recessive pontocerebellar hypoplasia, microcephaly, and extrapyramidal dyskinesia (Pontocerebellar hypoplasia type 2): Compiled data from 10 pedigrees. Neurology 1995, 45, 311–317. [Google Scholar] [CrossRef]
- Barth, P.G.; Aronica, E.; Vries, L.; Nikkels, P.G.J.; Scheper, W.; Hoozemans, J.J.; Poll-The, B.T.; Troost, D. Pontocerebellar hypoplasia type 2: A neuropathological update. Acta Neuropathol. 2007, 114, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Cassandrini, D.; Biancheri, R.; Tessa, A.; Di Rocco, M.; Di Capua, M.; Bruno, C.; Denora, P.S.; Sartori, S.; Rossi, A.; Nozza, P.; et al. Pontocerebellar hypoplasia: Clinical, pathologic, and genetic studies. Neurology 2010, 75, 1459–1464. [Google Scholar] [CrossRef]
- Rüsch, C.T.; Bölsterli, B.K.; Kottke, R.; Steinfeld, R.; Boltshauser, E. Pontocerebellar hypoplasia: A pattern recognition approach. Cerebellum 2020, 19, 569–582. [Google Scholar] [CrossRef]
- Baets, J.; Deconinck, T.; Smets, K.; Goossens, D.; Van Den Bergh, P.; Dahan, K.; Schmedding, E.; Santens, P.; Rasic, V.M.; Van Damme, P.; et al. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology 2010, 75, 1181–1188. [Google Scholar] [CrossRef]
- Shimazaki, H.; Takiyama, Y.; Sakoe, K.; Ando, Y.; Nakano, I. A phenotype without spasticity in sacsin-related ataxia. Neurology 2005, 64, 2129–2131. [Google Scholar] [CrossRef]
- Breckpot, J.; Takiyama, Y.; Thienpont, B.; Van Vooren, S.; Vermeesch, J.R.; Ortibus, E.; Devriendt, K. A novel genomic disorder: A deletion of the SACS gene leading to Spastic Ataxia of Charlevoix-Saguenay. Eur. J. Hum. Genet. 2008, 16, 1050–1054. [Google Scholar] [CrossRef]
- Bouchard, J.P.; Richter, A.; Mathieu, J.; Brunet, D.; Hudson, T.J.; Morgan, K.; Melançon, S.B. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuromuscul. Disord. 1998, 8, 474–479. [Google Scholar] [CrossRef]
- Engert, J.C.; Bérubé, P.; Mercier, J.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 2000, 24, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Gazulla, J.; Benavente, I.; Vela, A.C.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. New findings in the ataxia of charlevoix-saguenay. J. Neurol. 2012, 259, 869–878. [Google Scholar] [CrossRef]
- Freeze, H.H.; Eklund, E.A.; Ng, B.G.; Patterson, M.C. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012, 11, 453–466. [Google Scholar] [CrossRef] [Green Version]
- Haeuptle, M.A.; Hennet, T. Congenital disorders of glycosylation: An update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum. Mutat. 2009, 30, 1628–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stibler, H.; Jaeken, J. Carbohydrate deficient serum transferrin in a new systemic hereditary syndrome. Arch. Dis. Child. 1990, 65, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Boddaert, N.; Desguerre, I.; Bahi-Buisson, N.; Romano, S.; Valayannopoulos, V.; Saillour, Y.; Seidenwurm, D.; Grevent, D.; Berteloot, L.; Lebre, A.S.; et al. Apport de l’IRM dans les syndromes cérébelleux chez l’enfant: À propos de 158 cas. J. Neuroradiol. 2010, 37, 220–230. [Google Scholar] [CrossRef]
- Feraco, P.; Mirabelli-Badenier, M.; Severino, M.; Alpigiani, M.G.; Di Rocco, M.; Biancheri, R.; Rossi, A. The shrunken, bright cerebellum: A characteristic MRI finding in congenital disorders of glycosylation type 1a. Am. J. Neuroradiol. 2012, 33, 2062–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoun, H.; Villeneuve, N.; Gelot, A.; Panisset, S.; Adamsbaum, C. Cerebellar atrophy: An important feature of carbohydrate deficient glycoprotein syndrome type 1. Pediatr. Radiol. 1999, 29, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Nouspikel, T. Nucleotide excision repair and neurological diseases. DNA Repair 2008, 7, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Koob, M.; Laugel, V.; Durand, M.; Fothergill, H.; Dalloz, C.; Sauvanaud, F.; Dollfus, H.; Namer, I.J.; Dietemann, J.L. Neuroimaging in Cockayne syndrome. Am. J. Neuroradiol. 2010, 31, 1623–1630. [Google Scholar] [CrossRef] [Green Version]
- Alves, C.A.P.F.; Fragoso, D.C.; Gonçalves, F.G.; Marussi, V.H.; do Amaral, L.L.F. Cerebellar ataxia in children: A clinical and MRI approach to the differential diagnosis. Top. Magn. Reson. Imaging 2018, 27, 275–302. [Google Scholar] [CrossRef] [PubMed]
- Blaser, S.I.; Steinlin, M.; Al-Maawali, A.; Yoon, G. The pediatric cerebellum in inherited neurodegenerative disorders: A pattern-recognition approach. Neuroimaging Clin. 2016, 26, 373–416. [Google Scholar] [CrossRef]
- Mole, S.E.; Cotman, S.L. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2237–2241. [Google Scholar] [CrossRef] [Green Version]
- Marshall, F.J.; De Blieck, E.A.; Mink, J.W.; Dure, L.; Adams, H.; Messing, S.; Rothberg, P.G.; Levy, E.; McDonough, T.; DeYoung, J.; et al. A clinical rating scale for Batten disease: Reliable and relevant for clinical trials. Neurology 2005, 65, 275–279. [Google Scholar] [CrossRef]
- Vanhanen, S.-L.; Puranen, J.; Autti, T.; Raininko, R.; Liewendahl, K.; Nikkinen, P.; Santavuori, P.; Suominen, P.; Vuori, K.; Häkkinen, A.M. Neuroradiological findings (MRS, MRI, SPECT) in infantile neuronal ceroid-lipofuscinosis (infantile CLN1) at different stages of the disease. Neuropediatrics 2004, 35, 27–35. [Google Scholar] [CrossRef]
- Salonen, R.; Somer, M.; Haltia, M.; Lorentz, M.; Norio, R. Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO syndrome). Clin. Genet. 1991, 39, 287–293. [Google Scholar] [CrossRef]
- Riikonen, R. The PEHO syndrome. In Brain and Development; Elsevier: Amsterdam, The Netherlands, 2001; Volume 23, pp. 765–769. [Google Scholar]
- Huisman, T.A.G.M.; Klein, A.; Werner, B.; Straube, T.; Boltshauser, E. Serial MR imaging, diffusion tensor imaging, and MR spectroscopic findings in a child with progressive encephalopathy, edema, hypsarrhythmia, and optic atrophy (PEHO) syndrome. Am. J. Neuroradiol. 2006, 27, 1555–1558. [Google Scholar] [PubMed]
- Crawford, T.O. Ataxia telangiectasia. Semin. Pediatr. Neurol. 1998, 5, 287–294. [Google Scholar] [CrossRef]
- Smirnov, D.A.; Cheung, V.G. ATM gene mutations result in both recessive and dominant expression phenotypes of genes and microRNAs. Am. J. Hum. Genet. 2008, 83, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhagen, M.M.M.; Martin, J.-J.; van Deuren, M.; Ceuterick-de Groote, C.; Weemaes, C.M.R.; Kremer, B.H.P.H.; Taylor, M.A.R.; Willemsen, M.A.A.P.; Lammens, M. Neuropathology in classical and variant ataxia-telangiectasia. Neuropathology 2012, 32, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Sahama, I.; Sinclair, K.; Pannek, K.; Lavin, M.; Rose, S. Radiological imaging in ataxia telangiectasia: A review. Cerebellum 2014, 13, 521–530. [Google Scholar] [CrossRef]
- Vijapura, C.; Saad Aldin, E.; Capizzano, A.A.; Policeni, B.; Sato, Y.; Moritani, T. Genetic syndromes associated with central nervous system tumors. RadioGraphics 2017, 37, 258–280. [Google Scholar] [CrossRef]
- Lin, D.D.M.; Barker, P.B.; Lederman, H.M.; Crawford, T.O. Cerebral abnormalities in adults with ataxia-telangiectasia. Am. J. Neuroradiol. 2014, 35, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Farina, L.; Uggetti, C.; Ottolini, A.; Martelli, A.; Bergamaschi, R.; Sibilla, L.; Zappoli, F.; Egitto, M.G.; Lanzi, G. Ataxia-telangiectasia: MR and CT findings. J. Comput. Assist. Tomogr. 1994, 18, 724–727. [Google Scholar] [CrossRef]
- Le Ber, I.; Bouslam, N.; Rivaud-Péchoux, S.; Guimarães, J.; Benomar, A.; Chamayou, C.; Goizet, C.; Moreira, M.C.; Klur, S.; Yahyaoui, M.; et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: A clinical and genetic study in 18 patients. Brain 2004, 127, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arrigo, S.; Riva, D.; Bulgheroni, S.; Chiapparini, L.; Castellotti, B.; Gellera, C.; Pantaleoni, C. Ataxia with oculomotor apraxia type 1 (AOA1): Clinical and neuropsychological features in 2 new patients and differential diagnosis. J. Child Neurol. 2008, 23, 895–900. [Google Scholar] [CrossRef]
- Vernon, H.J.; Bindoff, L.A. Mitochondrial ataxias. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 155, pp. 129–141. [Google Scholar]
- Scaglia, F.; Wong, L.-J.C.; Vladutiu, G.D.; Hunter, J.V. Predominant cerebellar volume loss as a neuroradiologic feature of pediatric respiratory chain defects. Am. J. Neuroradiol. 2005, 26, 1675–1680. [Google Scholar] [PubMed]
- Lamperti, C.; Naini, A.; Hirano, M.; De Vivo, D.C.; Bertini, E.; Servidei, S.; Valeriani, M.; Lynch, D.; Banwell, B.; Berg, M.; et al. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology 2003, 60, 1206–1208. [Google Scholar] [CrossRef] [PubMed]
- Traschütz, A.; Schirinzi, T.; Laugwitz, L.; Murray, N.H.; Bingman, C.A.; Reich, S.; Kern, J.; Heinzmann, A.; Vasco, G.; Bertini, E.; et al. Clinico-genetic, imaging and molecular delineation of COQ8A-ataxia: A multicenter study of 59 patients. Ann. Neurol. 2020, 88, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Prim. 2019, 5, 1–21. [Google Scholar] [CrossRef]
- Li, X.; Zhou, C.; Cui, L.; Zhu, L.; Du, H.; Liu, J.; Wang, C.; Fang, S. A case of a novel CACNA1G mutation from a Chinese family with SCA42: A case report and literature review. Medicine 2018, 97, e12148. [Google Scholar] [CrossRef]
- Barresi, S.; Dentici, M.L.; Manzoni, F.; Bellacchio, E.; Agolini, E.; Pizzi, S.; Ciolfi, A.; Tarnopolsky, M.; Brady, L.; Garone, G.; et al. Infantile-onset syndromic cerebellar ataxia and CACNA1G mutations. Pediatr. Neurol. 2020, 104, 40–45. [Google Scholar] [CrossRef]
- Romaniello, R.; Zucca, C.; Tonelli, A.; Bonato, S.; Baschirotto, C.; Zanotta, N.; Epifanio, R.; Righini, A.; Bresolin, N.; Bassi, M.T.; et al. A wide spectrum of clinical, neurophysiological and neuroradiological abnormalities in a family with a novel CACNA1A mutation. J. Neurol. Neurosurg. Psychiatry 2010, 81, 840–843. [Google Scholar] [CrossRef]
- Zonta, A.; Brussino, A.; Dentelli, P.; Brusco, A. A novel case of congenital spinocerebellar ataxia 5: Further support for a specific phenotype associated with the p.(Arg480Trp) variant in SPTBN2. BMJ Case Rep. 2020, 13, e238108. [Google Scholar] [CrossRef]
- Simons, C.; Wolf, N.I.; McNeil, N.; Caldovic, L.; Devaney, J.M.; Takanohashi, A.; Crawford, J.; Ru, K.; Grimmond, S.M.; Miller, D.; et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am. J. Hum. Genet. 2013, 92, 767–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonduti, D.; Aiello, C.; Renaldo, F.; Dorboz, I.; Saaman, S.; Rodriguez, D.; Fettah, H.; Elmaleh, M.; Biancheri, R.; Barresi, S.; et al. TUBB4A-related hypomyelinating leukodystrophy: New insights from a series of 12 patients. Eur. J. Paediatr. Neurol. 2016, 20, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.M.; Polder, E.; Vanderver, A.; Naidu, S.; Schiffmann, R.; Fisher, K.; Raguž, A.B.; Blumkin, L.; Van Berkel, C.G.; Waisfisz, Q.; et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: Further delineation of the phenotype and genotype-phenotype correlation. Brain 2014, 137, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Osaka, H.; Sasaki, M.; Takanashi, J.I.; Hamada, K.; Yamashita, A.; Shibayama, H.; Shiina, M.; Kondo, Y.; Nishiyama, K.; et al. Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am. J. Hum. Genet. 2011, 89, 644–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
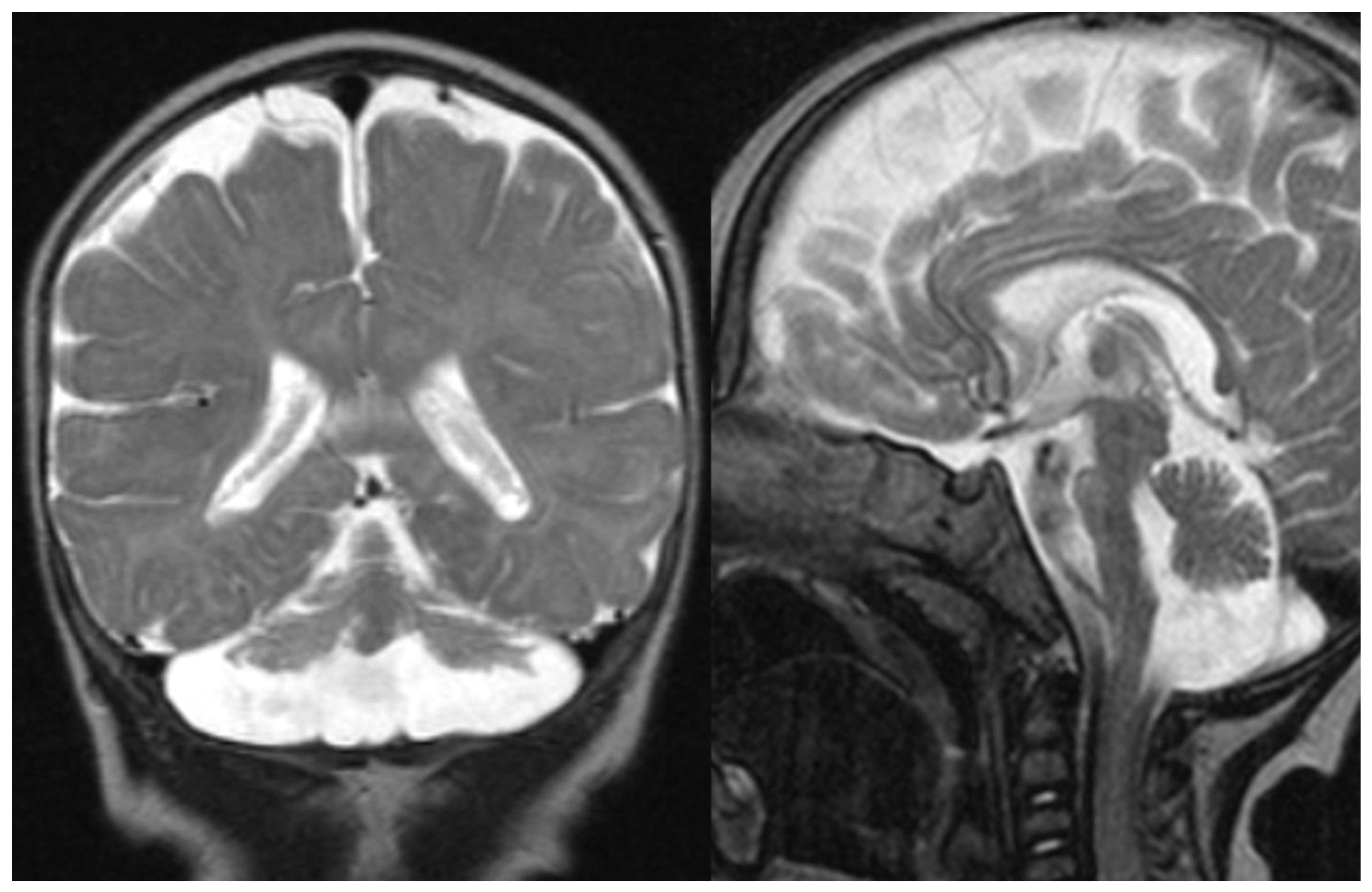
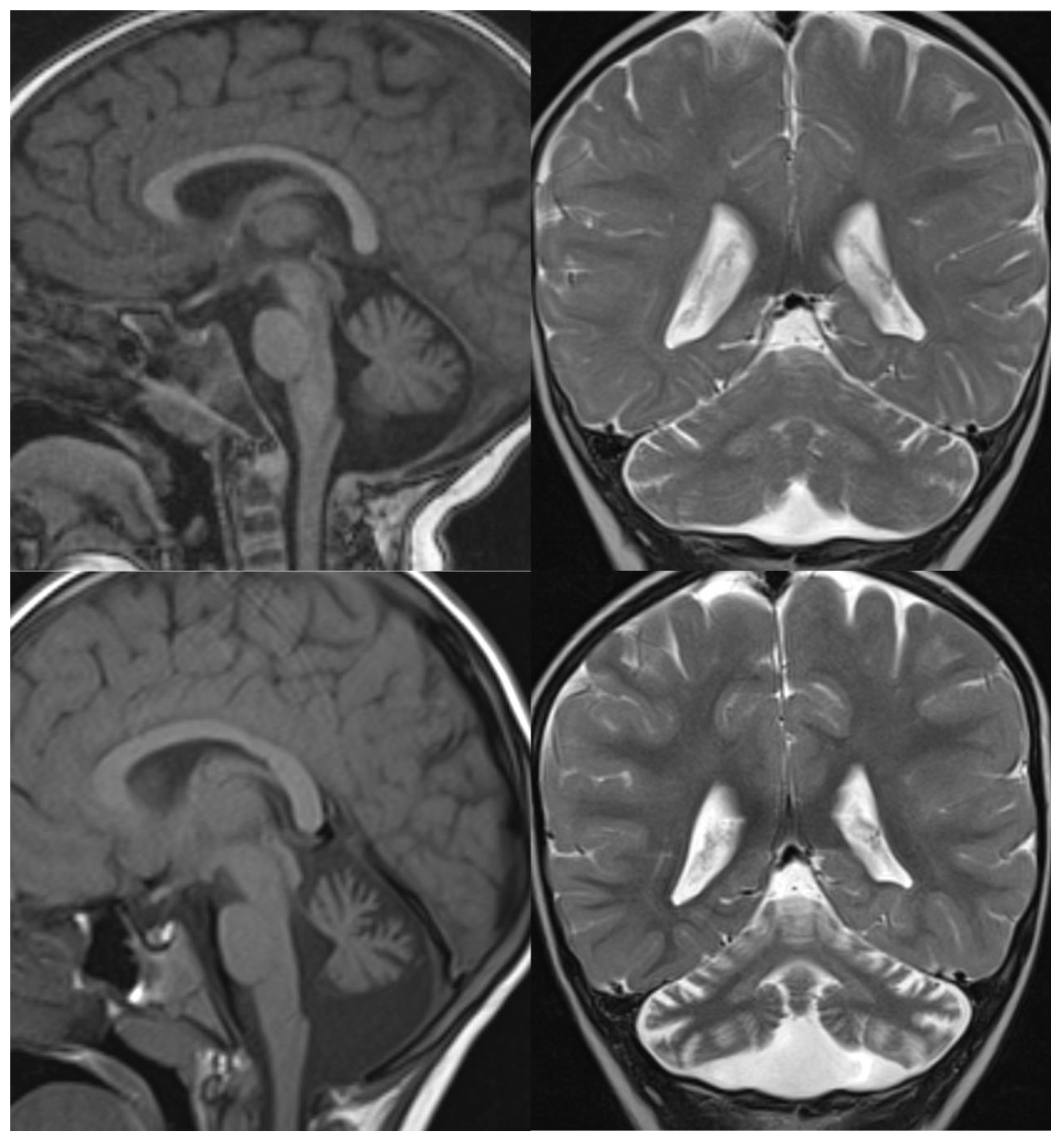
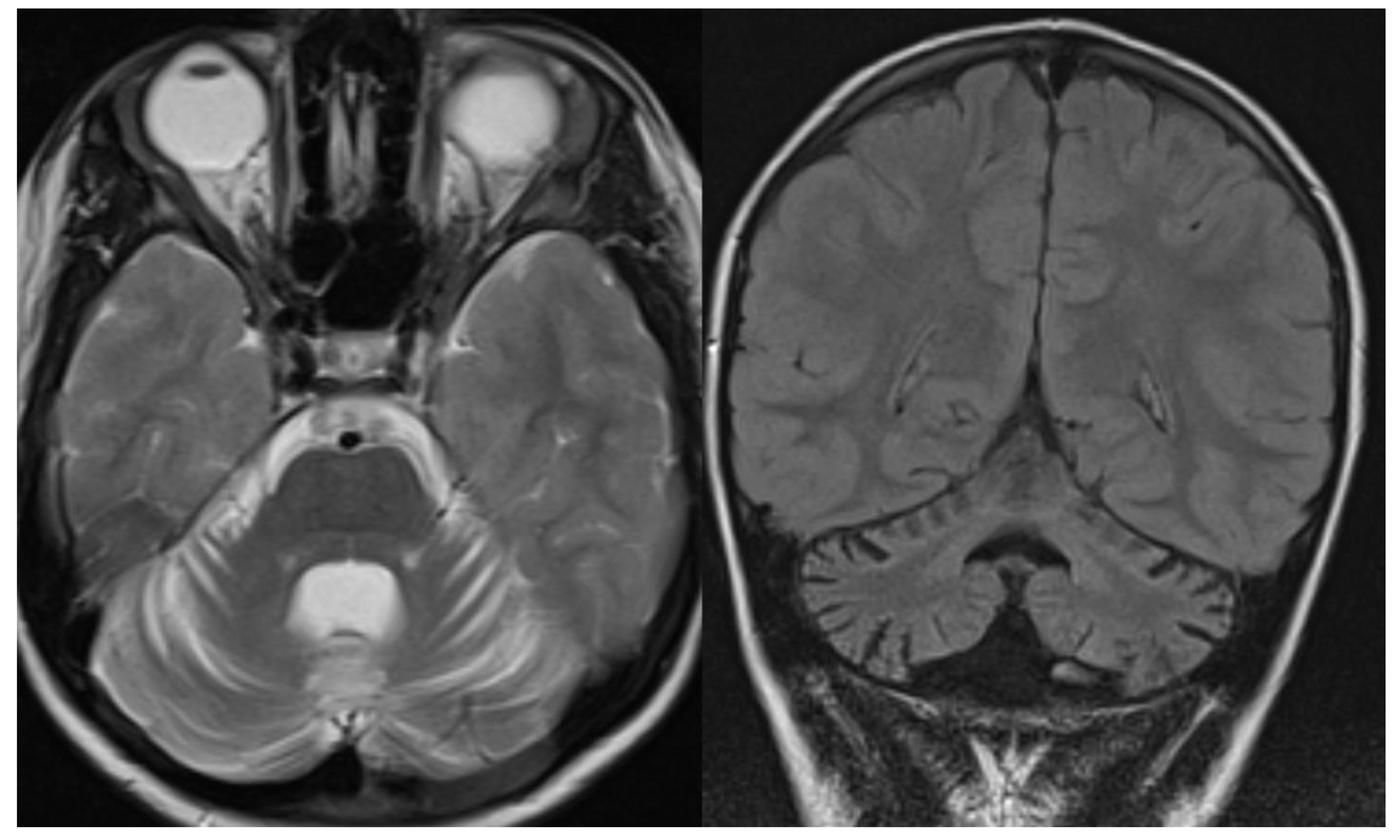
Figure 1.
PCH 2. Brain MRI scan of a 1-year-old girl with TSEN 54 mutation. MR images demonstrate the characteristic “dragonfly-like” cerebellar pattern: severe hypoplasia of the cerebellar hemispheres with a relative sparing of the vermis. Note the flat ventral surface of the pons and thin corpus callosum.
Figure 1.
PCH 2. Brain MRI scan of a 1-year-old girl with TSEN 54 mutation. MR images demonstrate the characteristic “dragonfly-like” cerebellar pattern: severe hypoplasia of the cerebellar hemispheres with a relative sparing of the vermis. Note the flat ventral surface of the pons and thin corpus callosum.
Figure 2.
PCH6. Brain MRI scan of a 5-year-old boy. The size of the pons is almost normal, the vermis grossly diminished with enlarged interfolial spaces. Vermis and hemispheres are equally hypoplastic.
Figure 2.
PCH6. Brain MRI scan of a 5-year-old boy. The size of the pons is almost normal, the vermis grossly diminished with enlarged interfolial spaces. Vermis and hemispheres are equally hypoplastic.
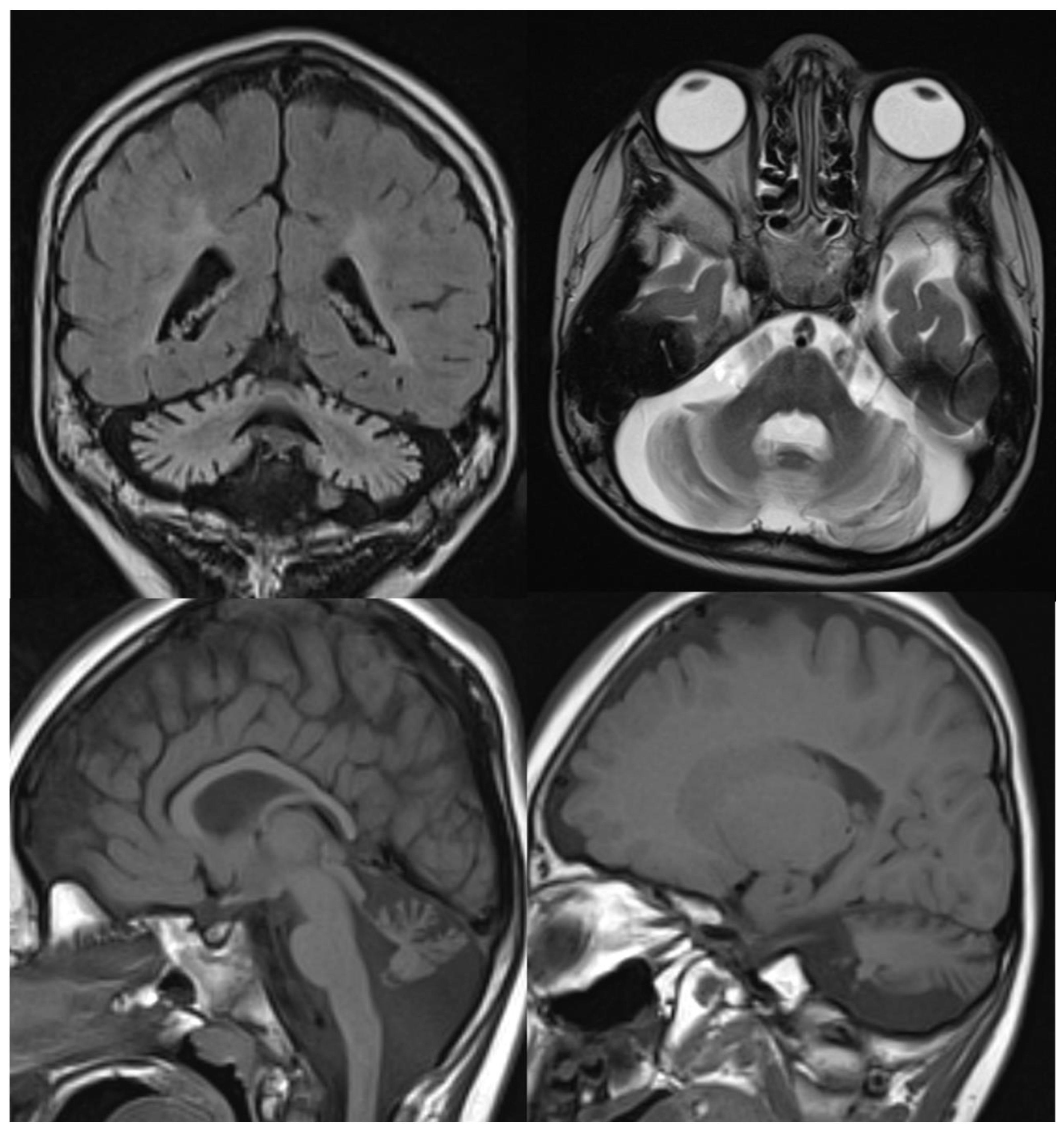
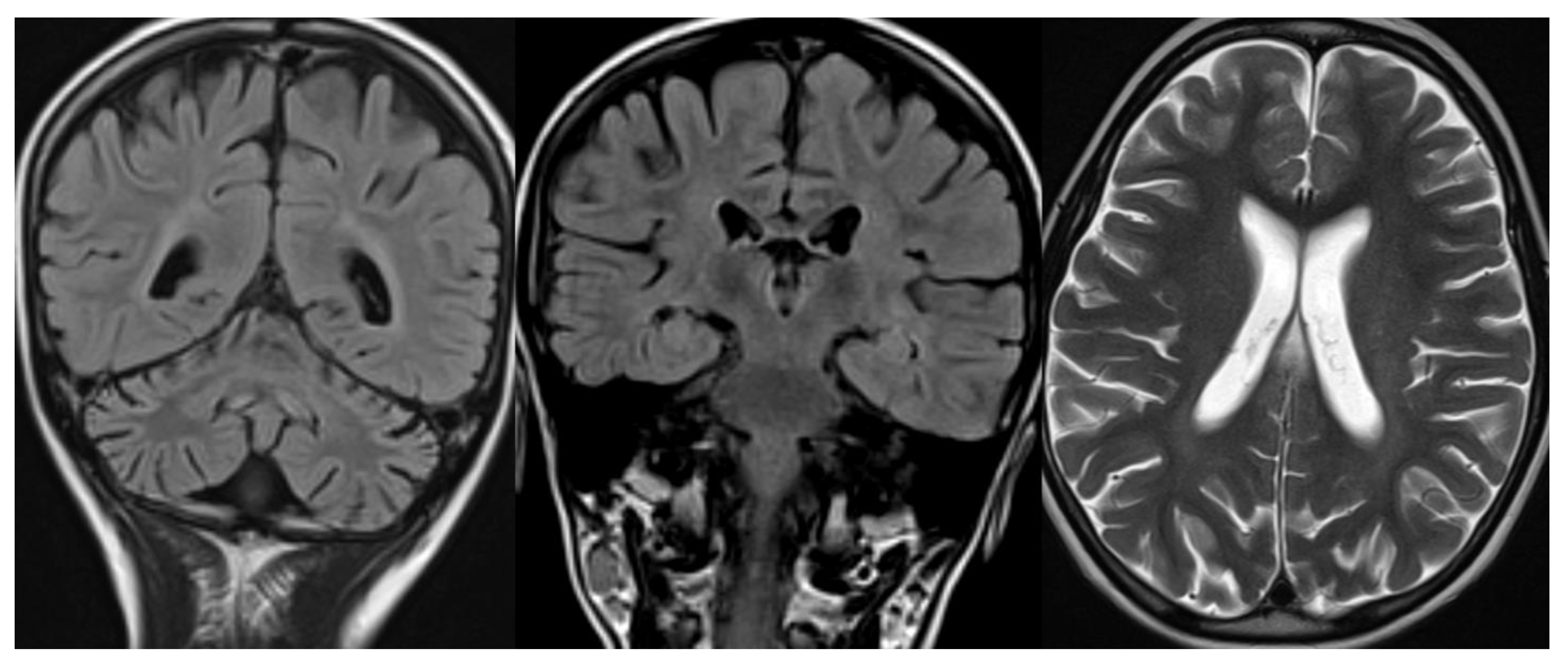
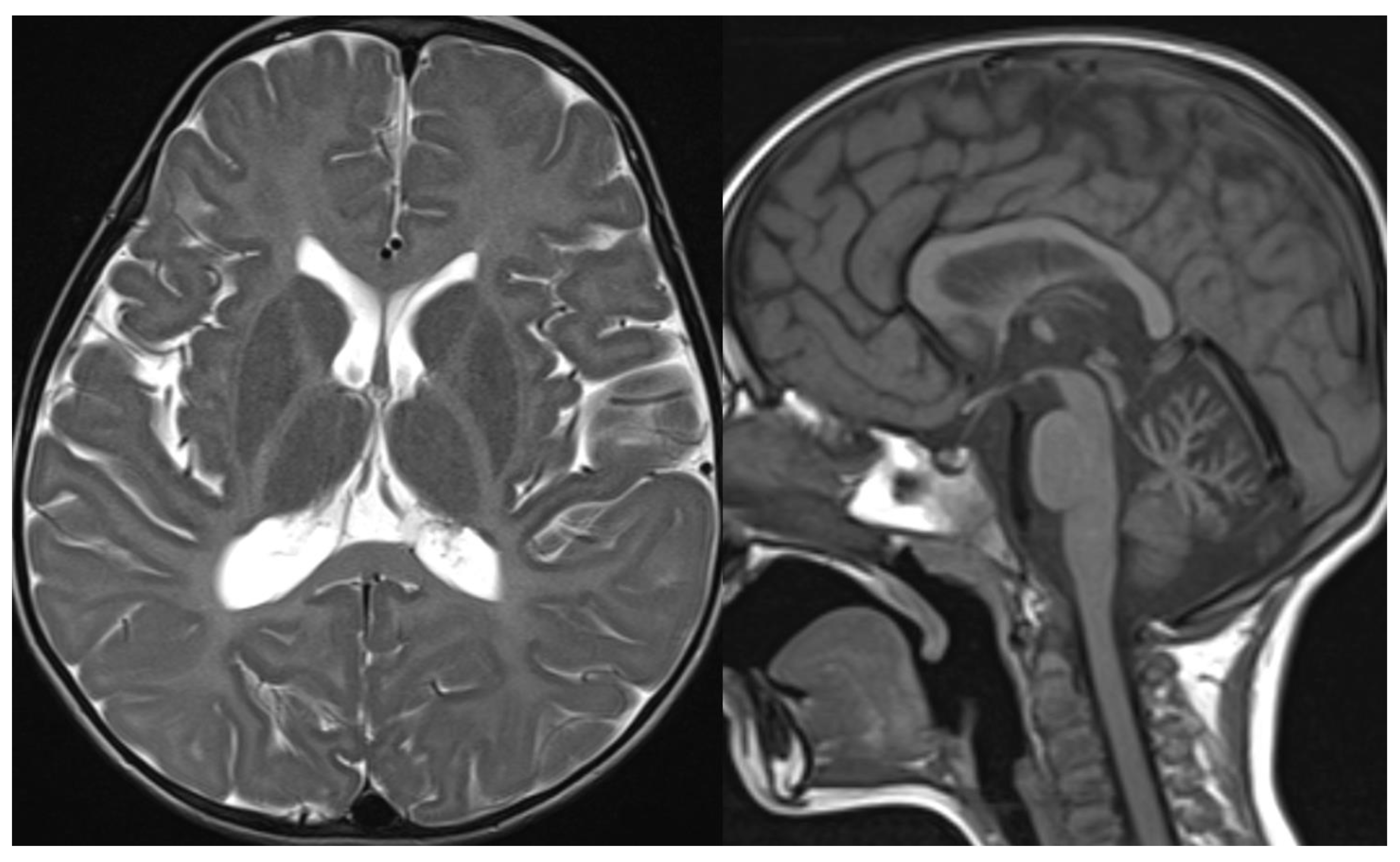
Figure 3.
INAD. Brain MRI scan of a 6-year-old girl. MR images demonstrate global cerebellar atrophy and cerebellar cortex hyperintensity, very thin optic chiasm, and “clava hypertrophy” due to enlargement of gracile and cuneate nuclei by axonal swellings and spheroids.
Figure 3.
INAD. Brain MRI scan of a 6-year-old girl. MR images demonstrate global cerebellar atrophy and cerebellar cortex hyperintensity, very thin optic chiasm, and “clava hypertrophy” due to enlargement of gracile and cuneate nuclei by axonal swellings and spheroids.
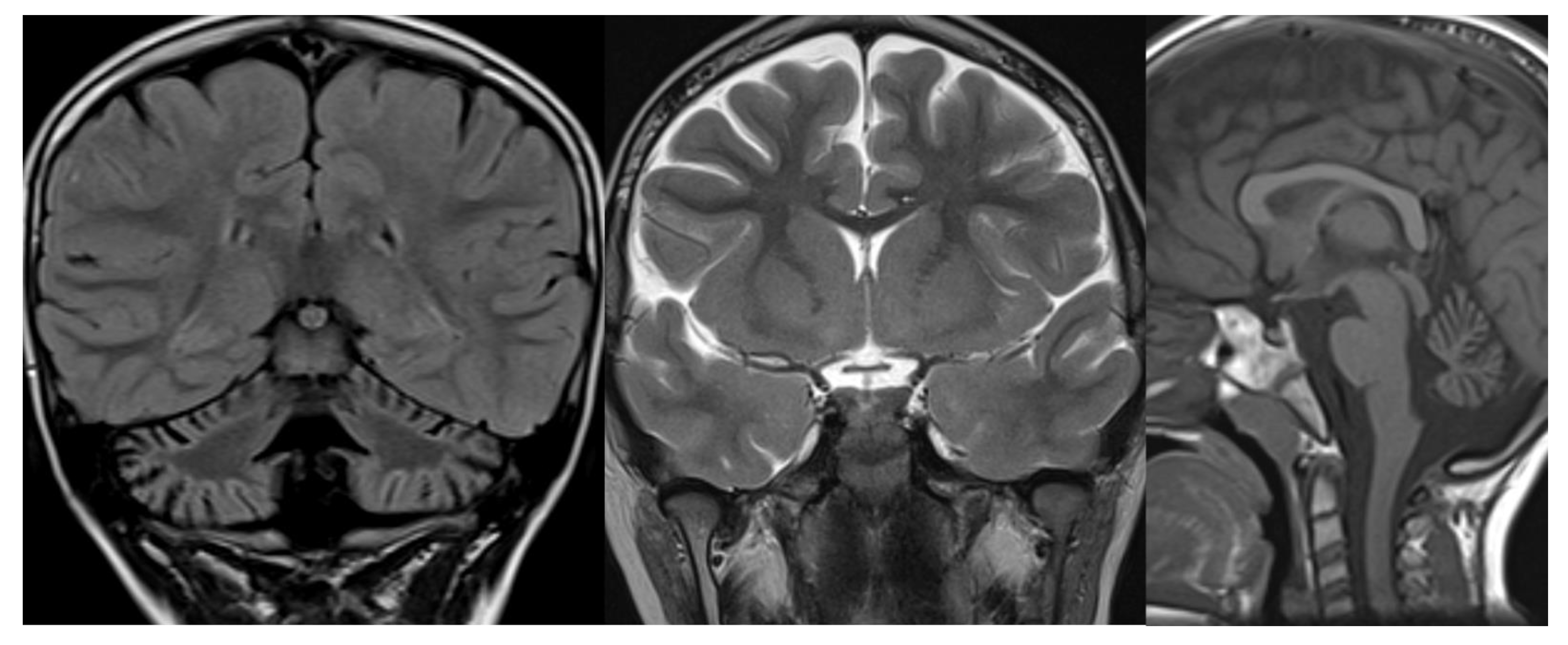
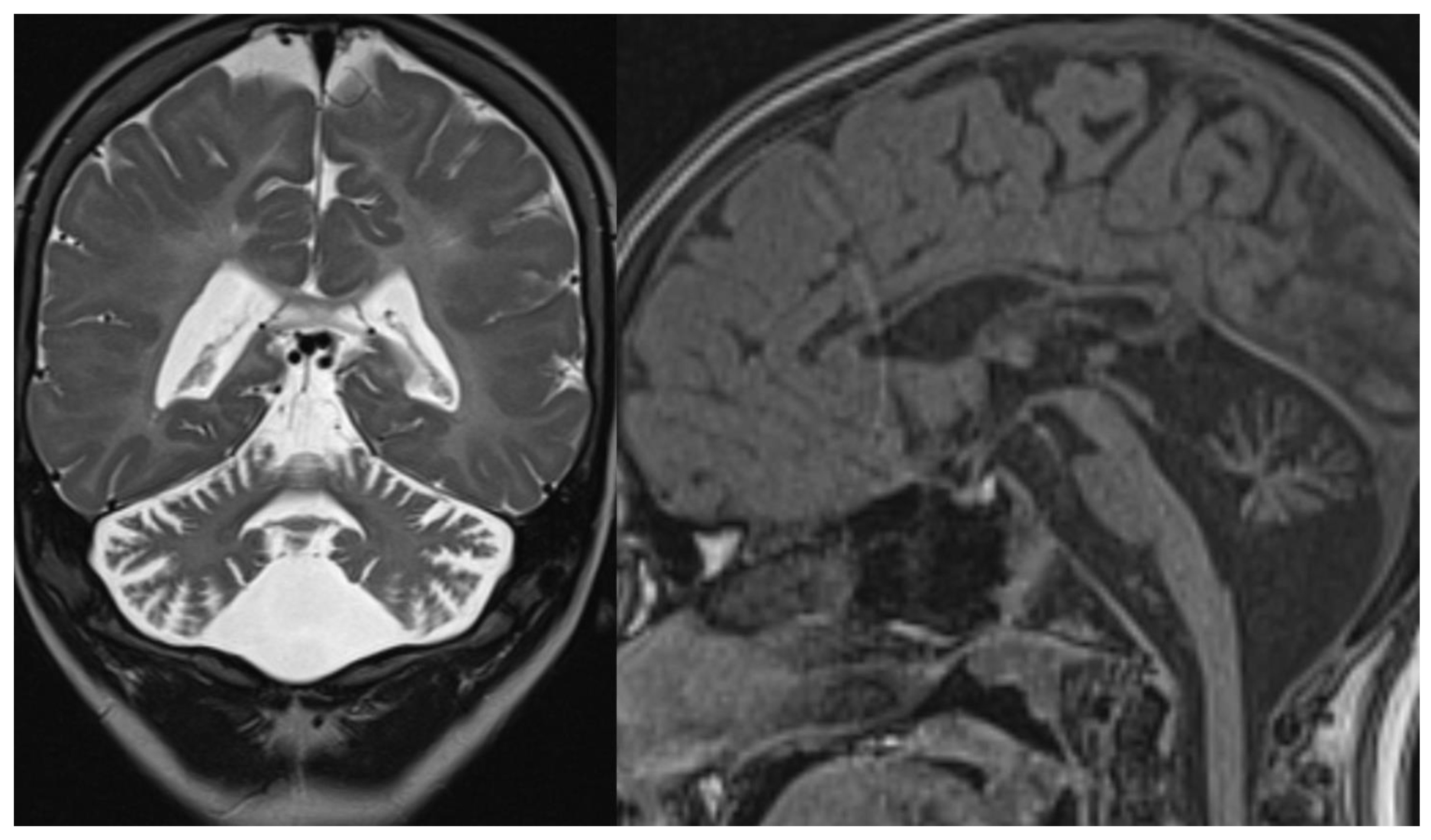
Figure 4.
ARSACS. Brain MRI scan of a 15-year-old boy. The images demonstrate the superior vermis and cervical spinal cord atrophy, bulky pons, and a thin corpus callosum. T2-wi at the level of the pons show diffuse, slight hyperintensity mostly in the lateral part of the pons and in the raphe. Corticospinal tracts are visible as hypointense parallel, thin stripes. The medial lemnisci also presented similar hypointensity. Note in the last image a rim of T2-hyperintensity around the thalami.
Figure 4.
ARSACS. Brain MRI scan of a 15-year-old boy. The images demonstrate the superior vermis and cervical spinal cord atrophy, bulky pons, and a thin corpus callosum. T2-wi at the level of the pons show diffuse, slight hyperintensity mostly in the lateral part of the pons and in the raphe. Corticospinal tracts are visible as hypointense parallel, thin stripes. The medial lemnisci also presented similar hypointensity. Note in the last image a rim of T2-hyperintensity around the thalami.
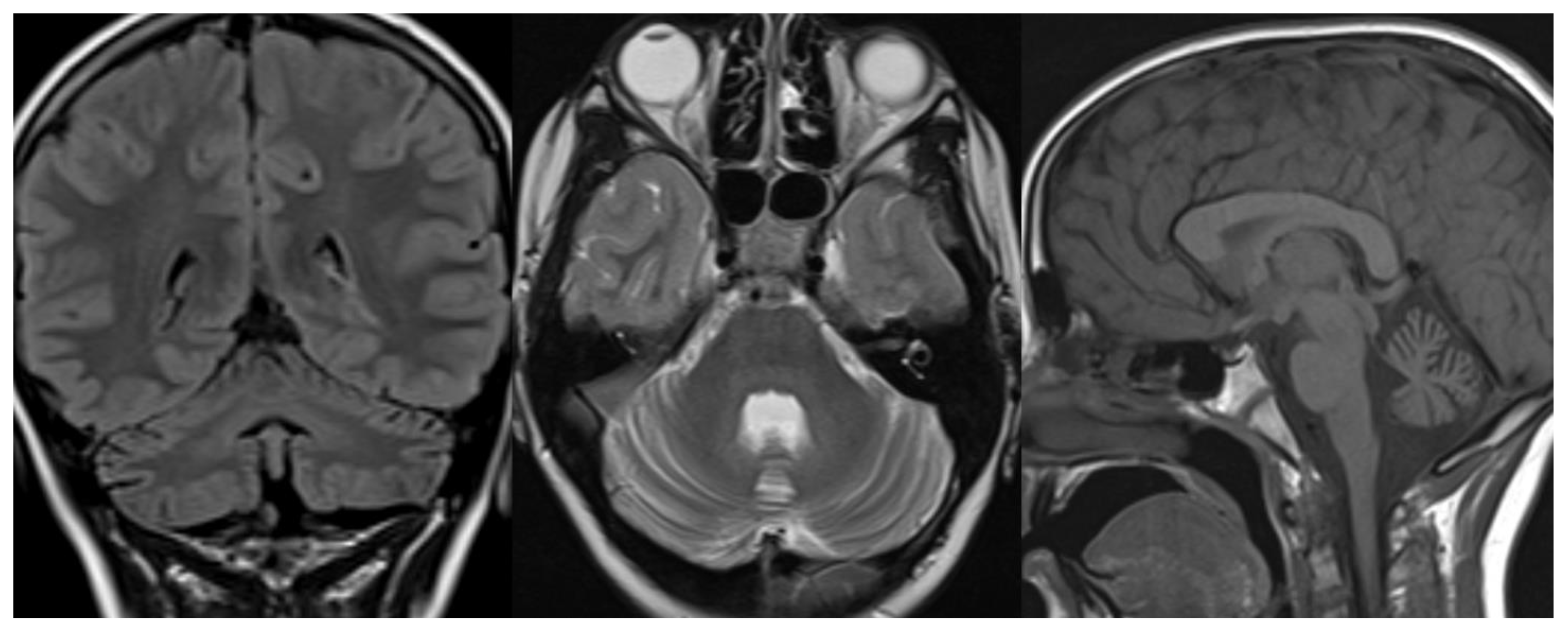
Figure 5.
GDC1a. Brain MRI scan of a boy. At 10 years (top), the exam demonstrates only slight cerebellar atrophy and hyperintensity in the dentate nuclei. The exam at 12 years (bottom) shows a severe worsening of cerebellar atrophy with the persistence of normal brainstem volume.
Figure 5.
GDC1a. Brain MRI scan of a boy. At 10 years (top), the exam demonstrates only slight cerebellar atrophy and hyperintensity in the dentate nuclei. The exam at 12 years (bottom) shows a severe worsening of cerebellar atrophy with the persistence of normal brainstem volume.
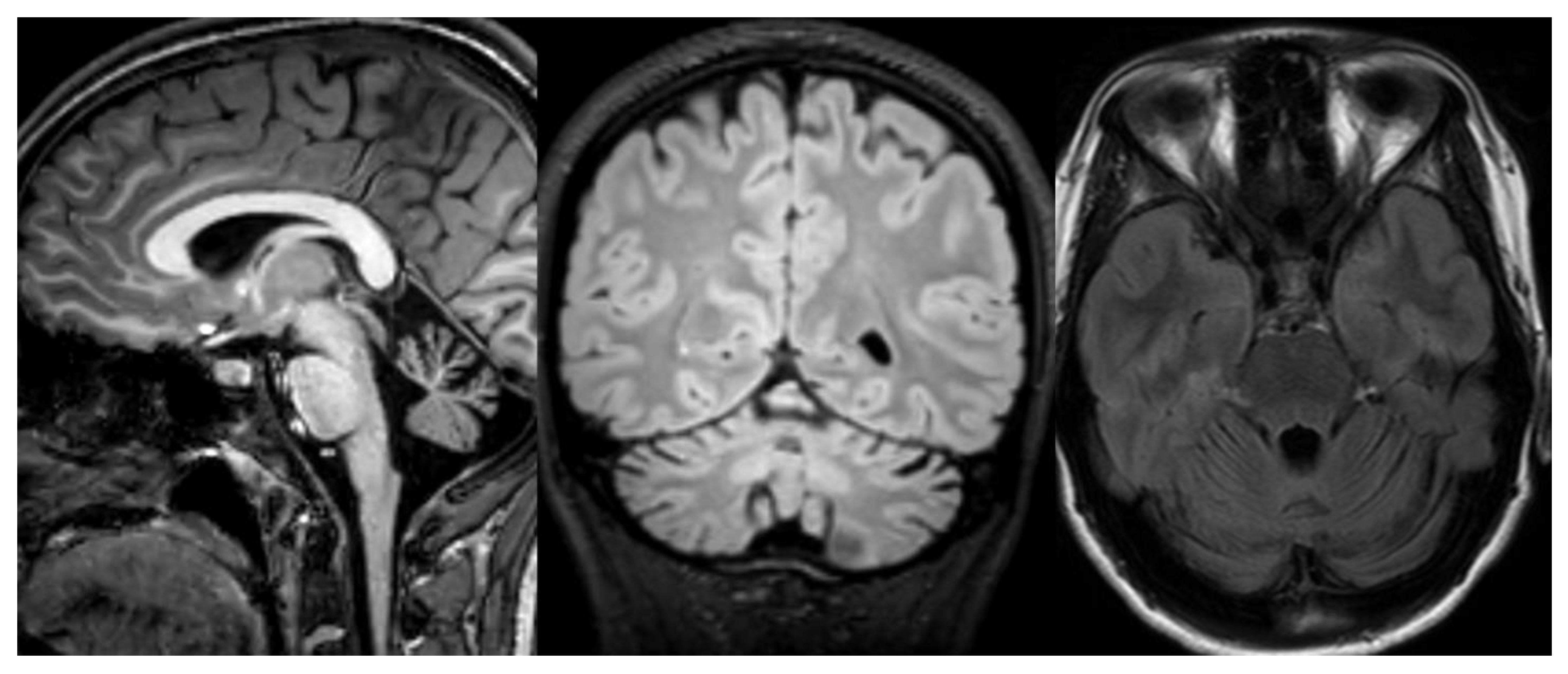
Figure 6.
Infantile NCL (CLN6). Brain MRI scan of a 6-year-old girl. MR images demonstrate diffuse cerebellar and cerebral atrophy associated with the markedly decreased signal intensity of the thalami. The last image shows hyperintensity in the deep and periventricular white matter.
Figure 6.
Infantile NCL (CLN6). Brain MRI scan of a 6-year-old girl. MR images demonstrate diffuse cerebellar and cerebral atrophy associated with the markedly decreased signal intensity of the thalami. The last image shows hyperintensity in the deep and periventricular white matter.
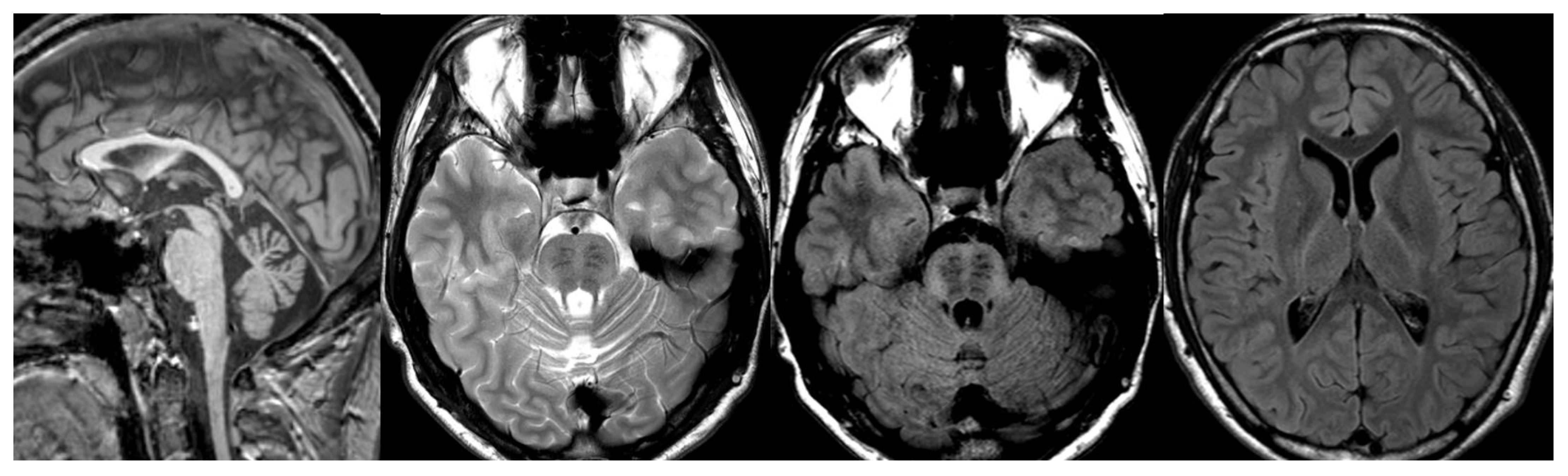
Figure 7.
Ataxia telangiectasia (AT). Brain MRI scan of a 10-year-old girl. MR images reveal severe atrophy of the cerebellar hemispheres, decreased thickness of the cortex of superior cerebellar hemispheres, and hypoplasia of inferior vermis.
Figure 7.
Ataxia telangiectasia (AT). Brain MRI scan of a 10-year-old girl. MR images reveal severe atrophy of the cerebellar hemispheres, decreased thickness of the cortex of superior cerebellar hemispheres, and hypoplasia of inferior vermis.
Figure 8.
Coenzyme Q10 deficiency. Brain MRI scan of a 15-year-old girl. MR images demonstrate not specific diffuse cerebellar atrophy and a normal volume of the brainstem.
Figure 8.
Coenzyme Q10 deficiency. Brain MRI scan of a 15-year-old girl. MR images demonstrate not specific diffuse cerebellar atrophy and a normal volume of the brainstem.
Figure 9.
CACNA1G mutation. Brain MRI scan of an 11-year-old girl. MR images demonstrate not specific diffuse cerebellar atrophy.
Figure 9.
CACNA1G mutation. Brain MRI scan of an 11-year-old girl. MR images demonstrate not specific diffuse cerebellar atrophy.
Figure 10.
HABC. Brain MRI scan of a 6-year-old girl. MR images show an almost complete lack of myelin, characterized by a strikingly hyperintense signal on a T2-weighted image. At this age, the putamen and caudate nucleus are normal-sized. Note the global cerebellar atrophy.
Figure 10.
HABC. Brain MRI scan of a 6-year-old girl. MR images show an almost complete lack of myelin, characterized by a strikingly hyperintense signal on a T2-weighted image. At this age, the putamen and caudate nucleus are normal-sized. Note the global cerebellar atrophy.
Figure 11.
POLR3B mutation. Brain MRI scan of a 10-year-old girl. MR images show marked global cerebellar atrophy with a thin corpus callosum and hypomyelination throughout the white matter, both cerebral and cerebellar.
Figure 11.
POLR3B mutation. Brain MRI scan of a 10-year-old girl. MR images show marked global cerebellar atrophy with a thin corpus callosum and hypomyelination throughout the white matter, both cerebral and cerebellar.
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










