
Lipid Vesicles and Other Polymolecular Aggregates—From Basic Studies of Polar Lipids to Innovative Applications


Abstract
:1. Introduction
2. Comparison of Aqueous Dispersions of a Few Selected Biological Polar Lipids
2.1. Polar Lipid Classification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polar Lipid 1 | Properties in Bulk Aqueous Solution | Properties at the Water–Air Interface |
|---|---|---|
| Class I | Practically insoluble, the molecules do not swell | Molecules spread to form stable monolayers |
| Palmitic acid | ||
| Oleic acid | ||
| Diolein | ||
| Class II | Practically insoluble, the molecules swell to form lyotropic liquid crystalline phases 3 | Molecules spread to form stable monolayers |
| DOPC | ||
| POPC | ||
| egg PC | ||
| DPPC | ||
| DOPA | ||
| DOPE | ||
| Monoolein | ||
| Class IIIA 2 | Soluble, formation of micelles at high water content above the CMC 4; formation of liquid crystalline phases 3 at low water content | Molecules spread but form unstable monolayers due to the solubility in water |
| Sodium oleate | ||
| Potassium oleate | ||
| Oleoyl-lyso-PC |
2.2. Oleic Acid and Palmitic Acid
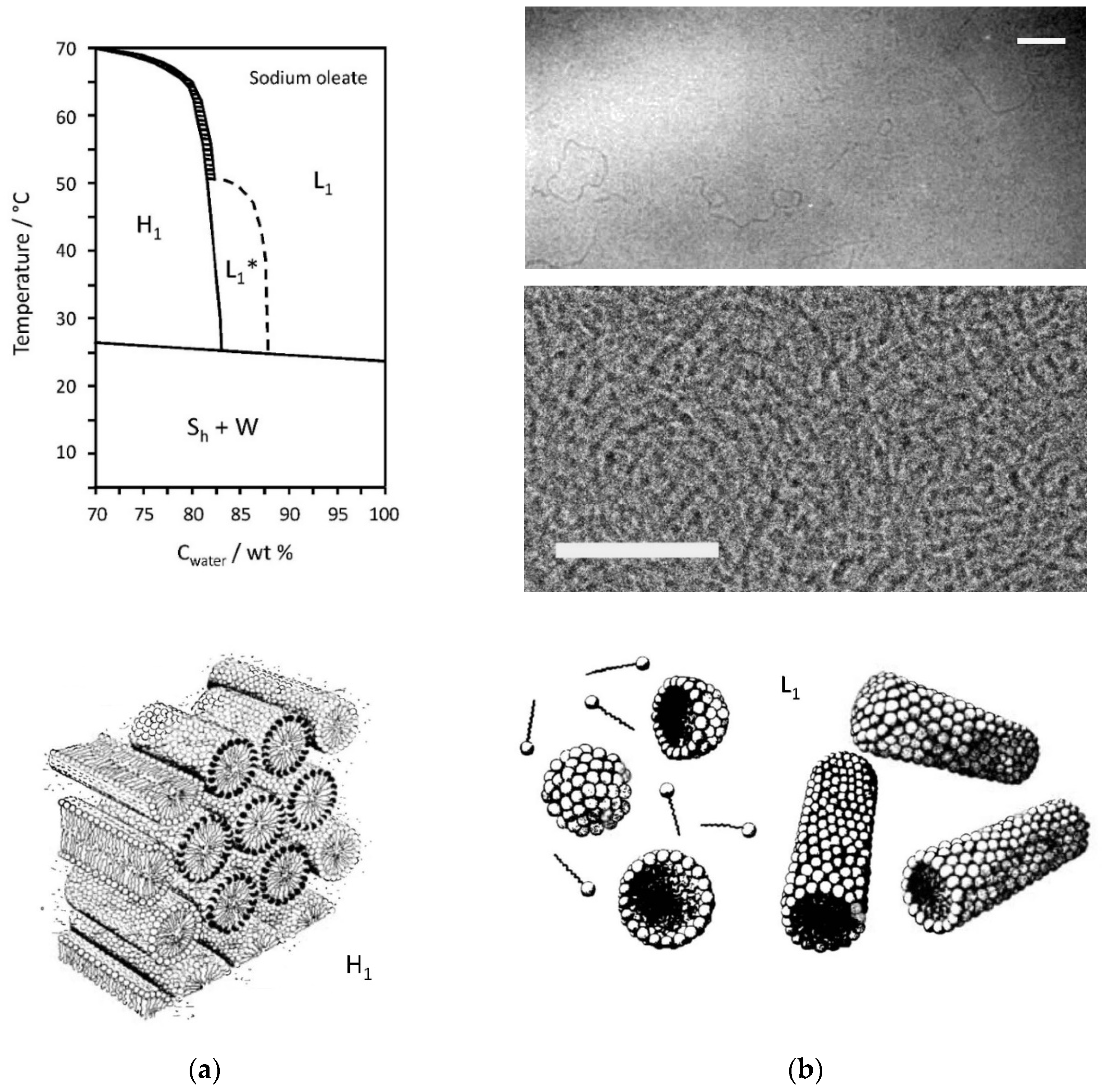
2.3. Sodium Oleate/Water Mixtures
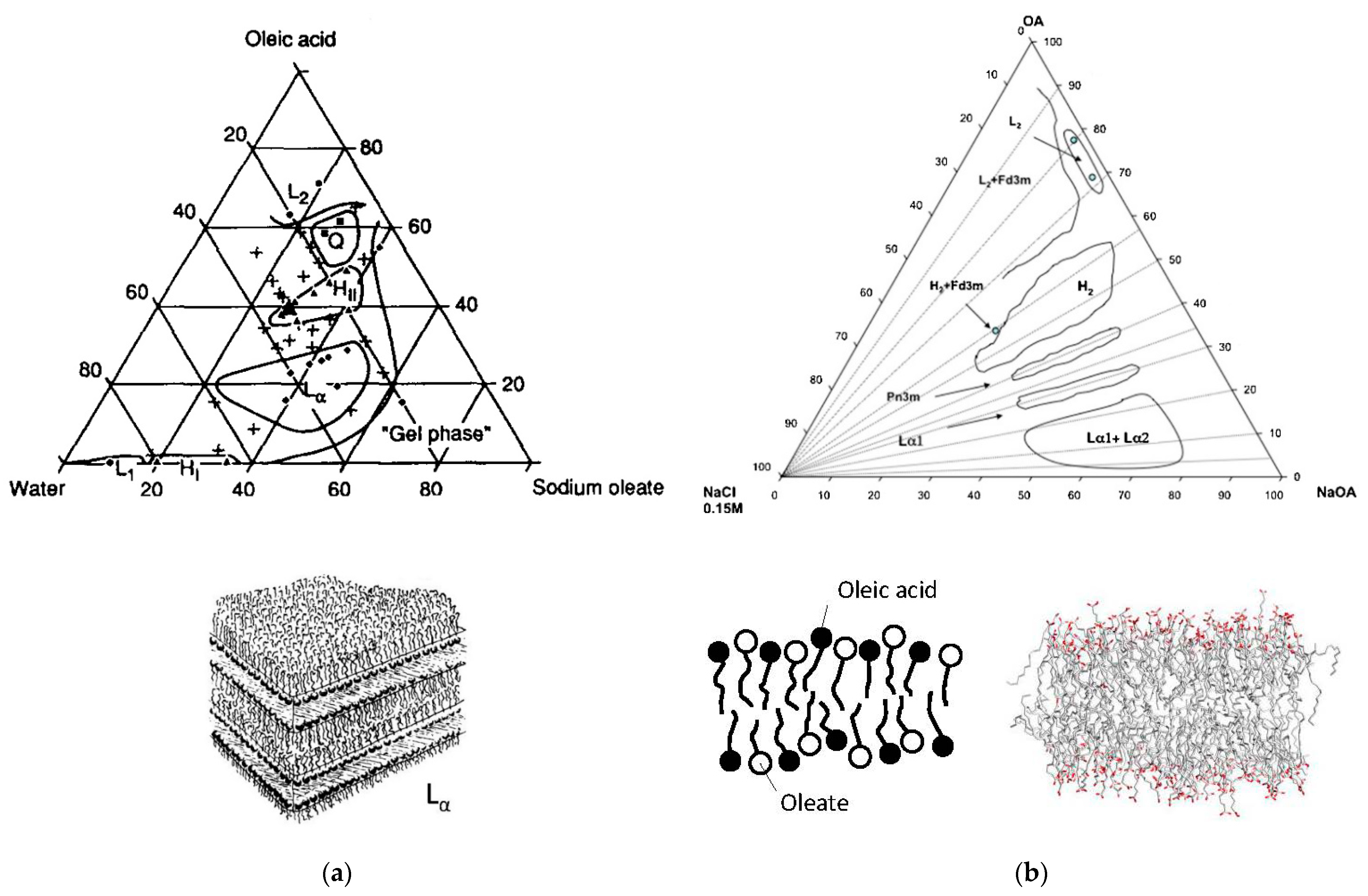
2.4. Sodium or Potassium Oleate/Oleic Acid/Water Mixtures
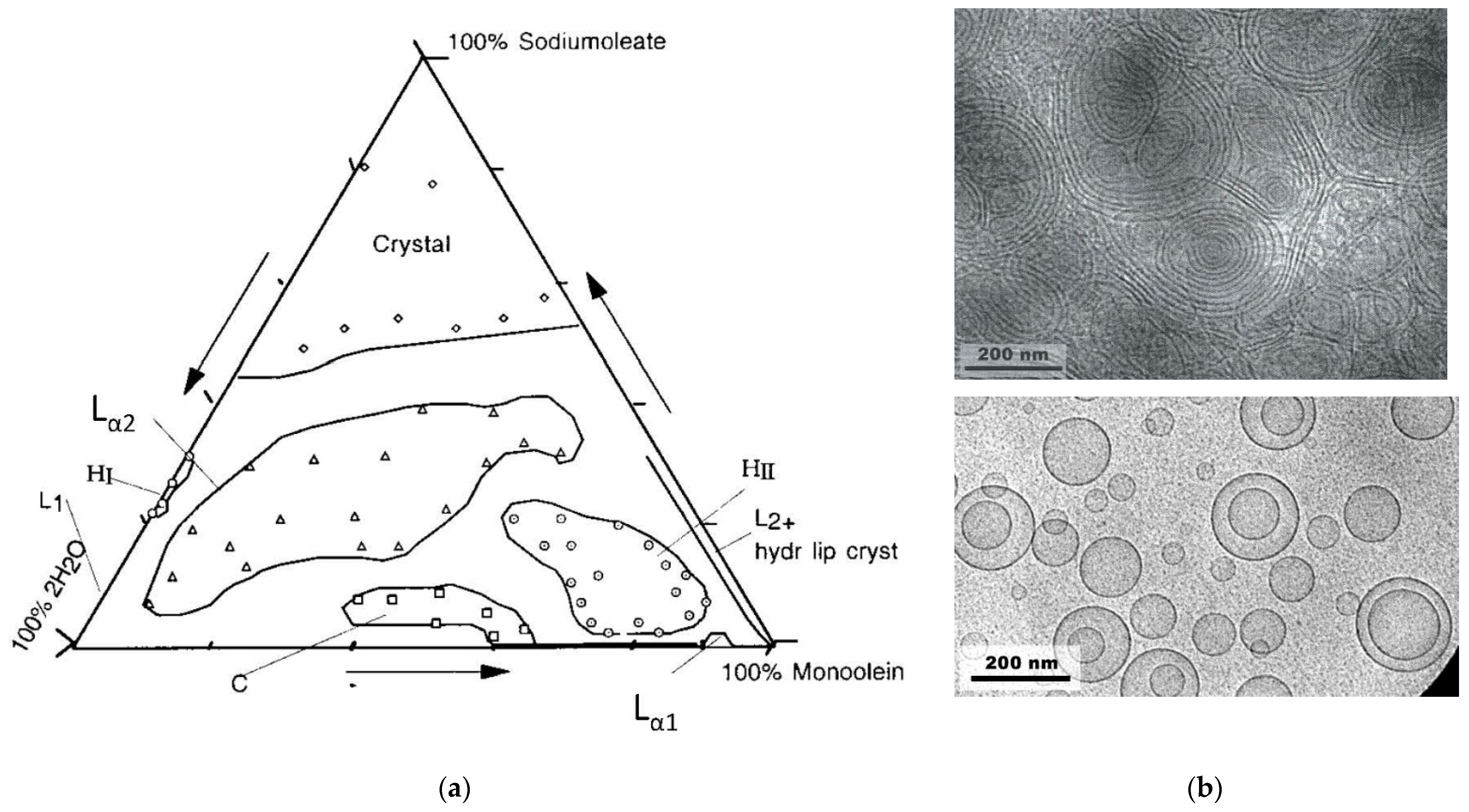
2.5. Monoolein/Water Mixtures
2.6. Monoolein/Sodium Oleate/Water Mixtures
2.7. Monoolein/Oleic Acid/Water and Monoolein/Oleic Acid/Sodium Oleate/Water Mixtures
2.8. Diolein
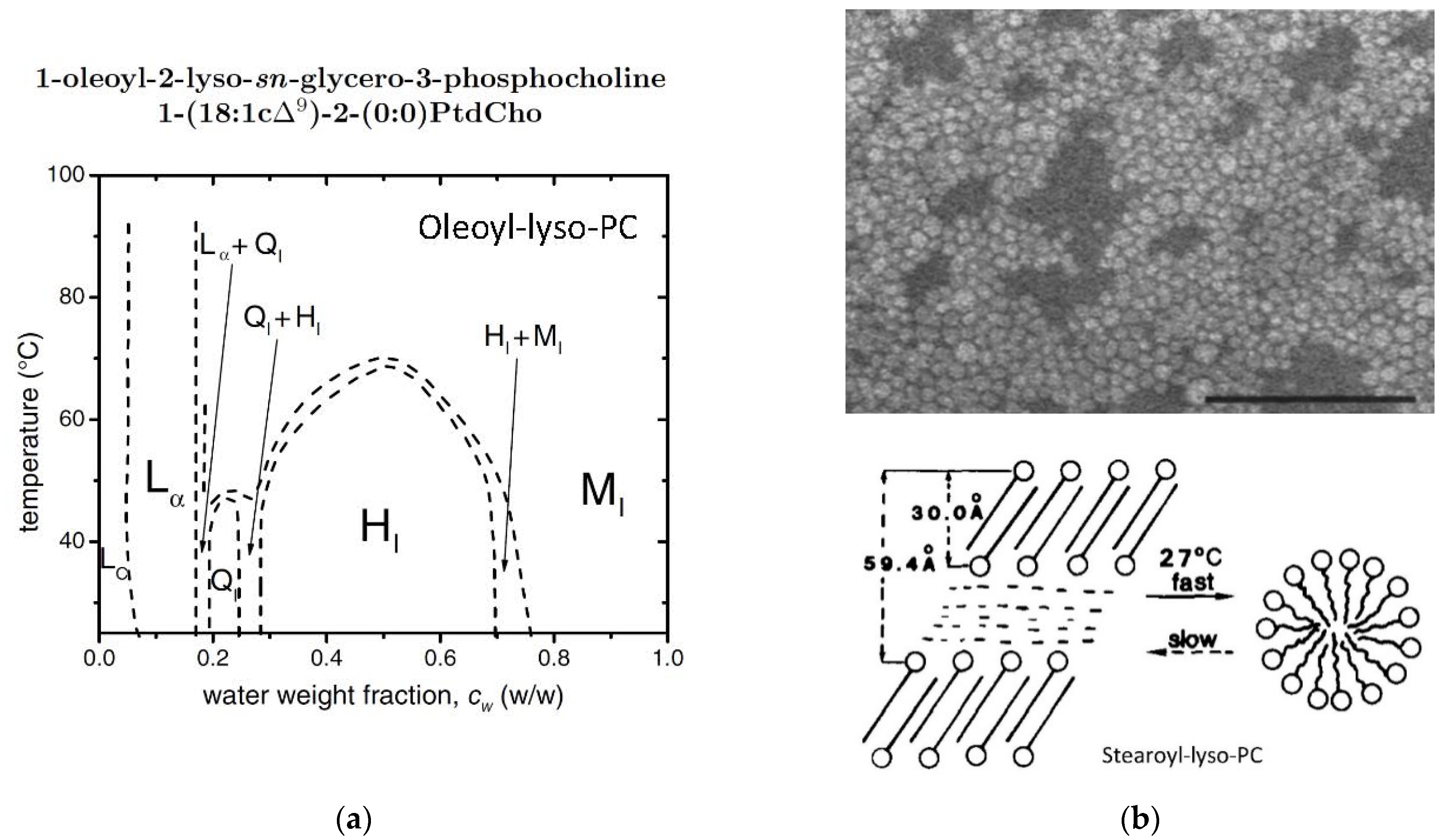
2.9. Oleoyl-Lyso-PC/Water Mixtures
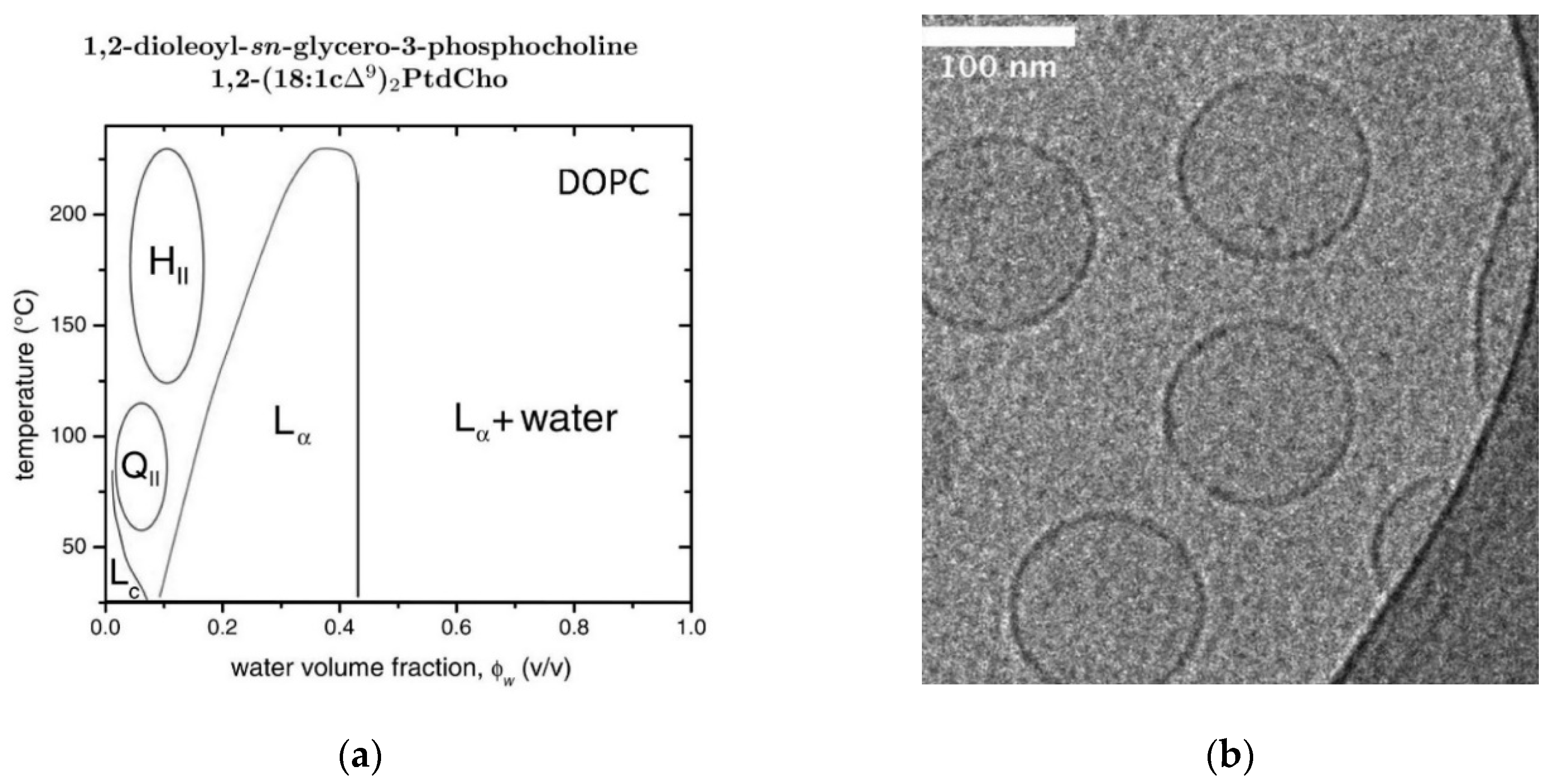
2.10. DOPC/Water Mixtures
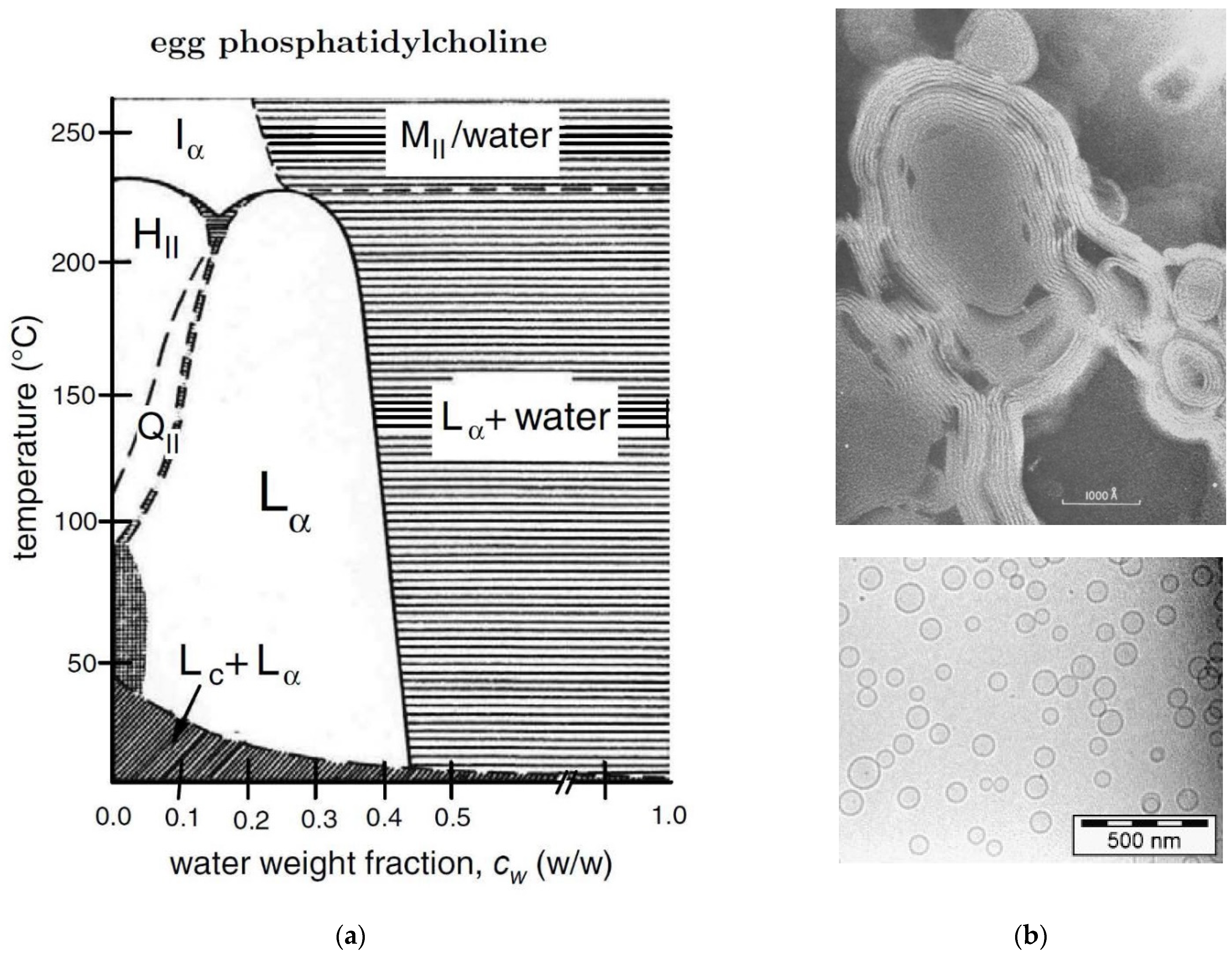
2.11. Egg PC/Water or POPC/Water Mixtures
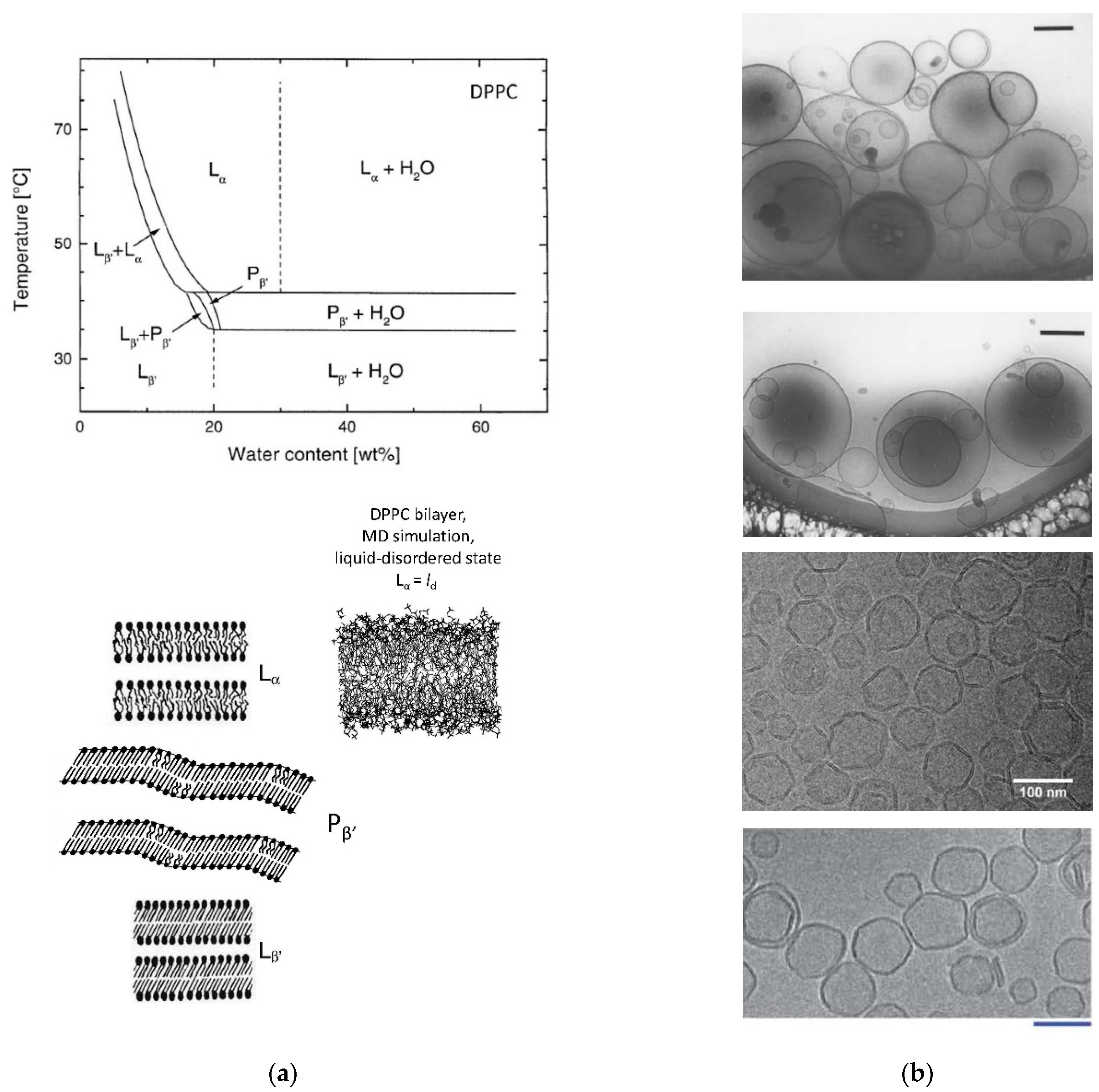
2.12. DPPC/Water Mixtures
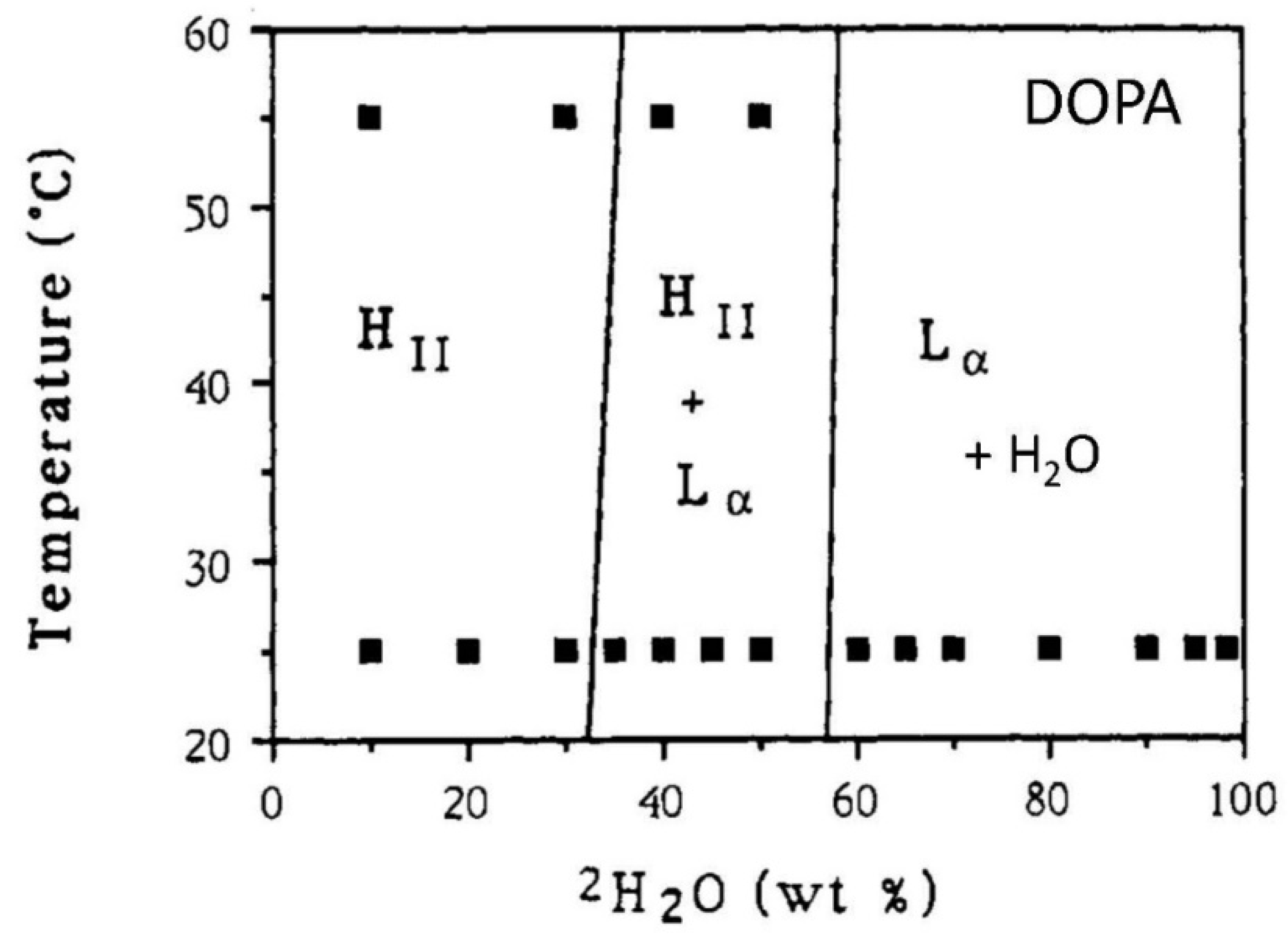
2.13. DOPA/Water Mixtures
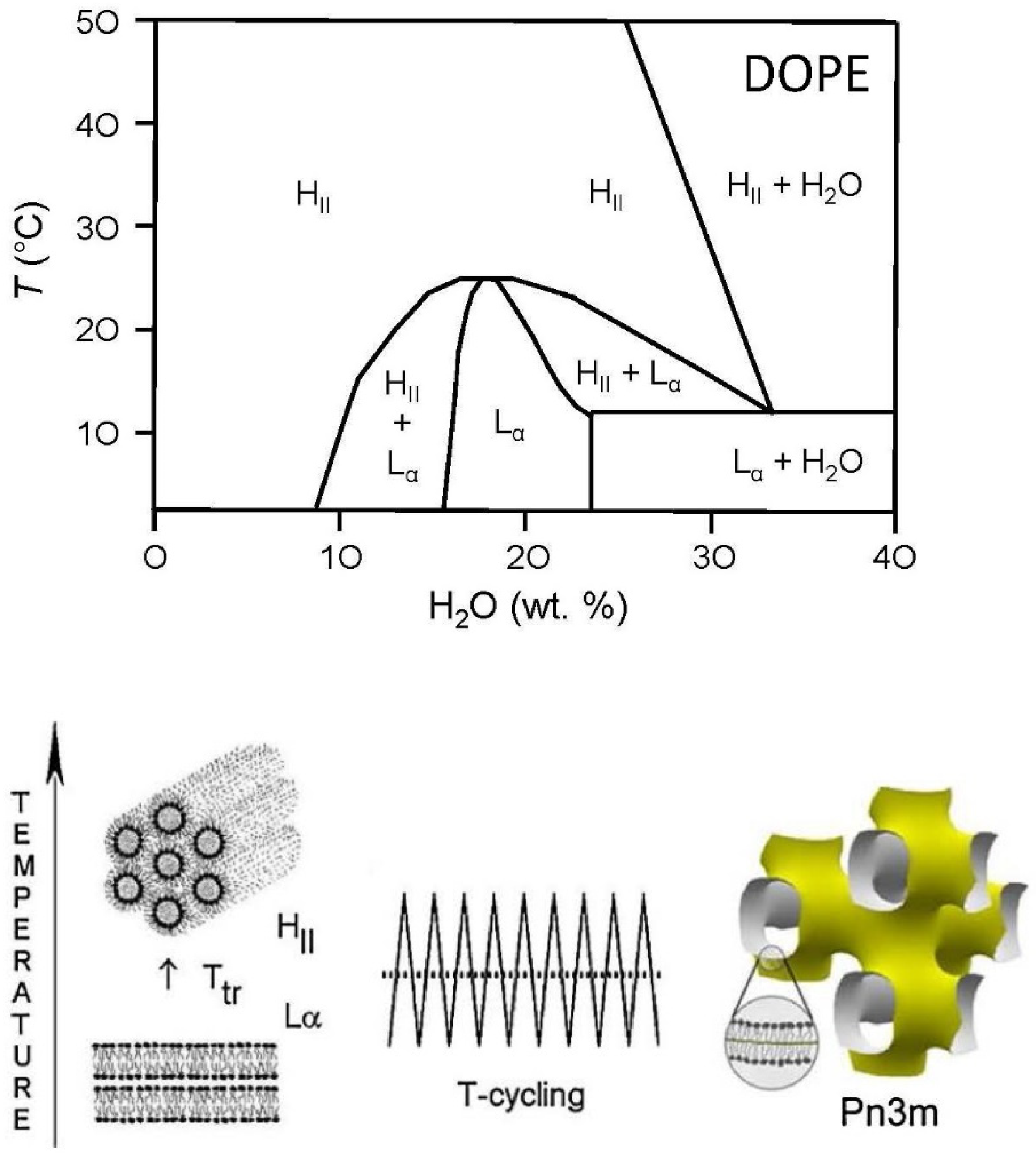
2.14. DOPE/Water Mixtures
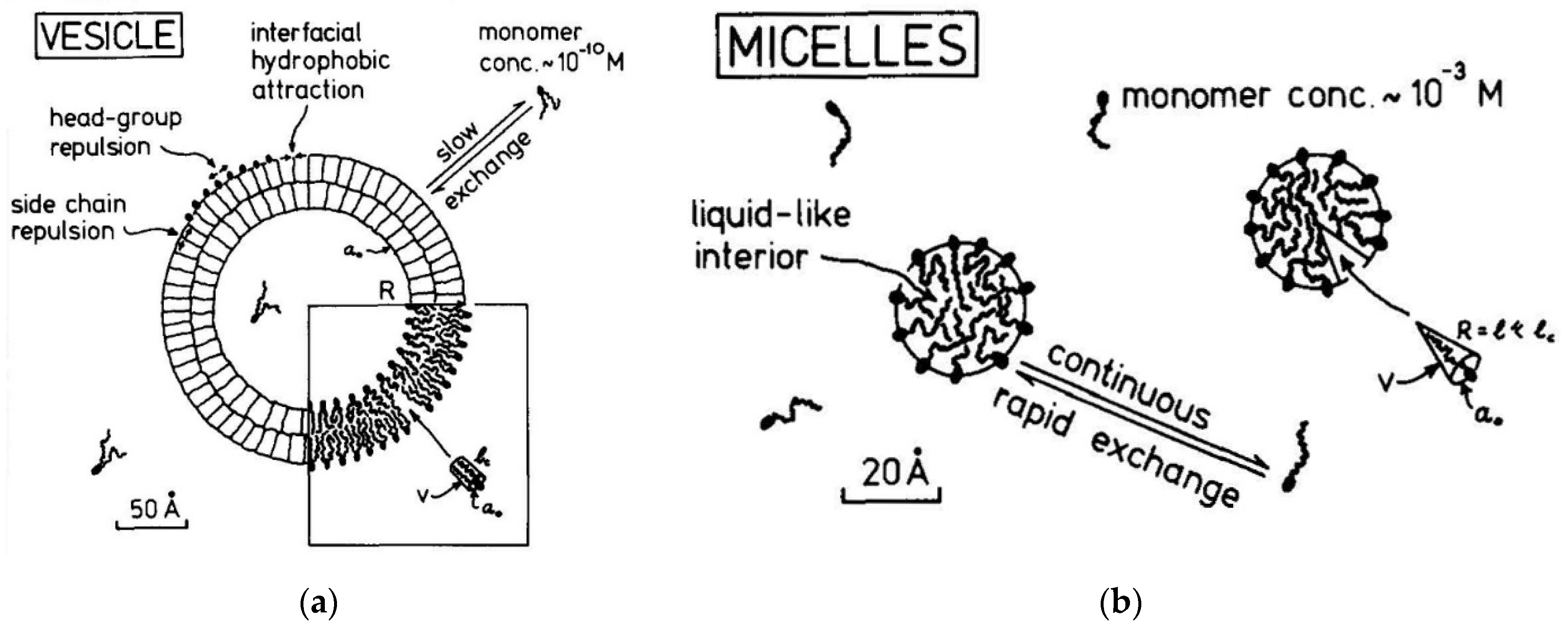
2.15. A comparison of Lipid Vesicles and Micelles
2.16. Summary
3. Increasing the Stability of Aqueous Dispersions of Lipid Vesicles and Cubosomes
4. Lipid Vesicle Dispersions Obtained by Guided Assembly
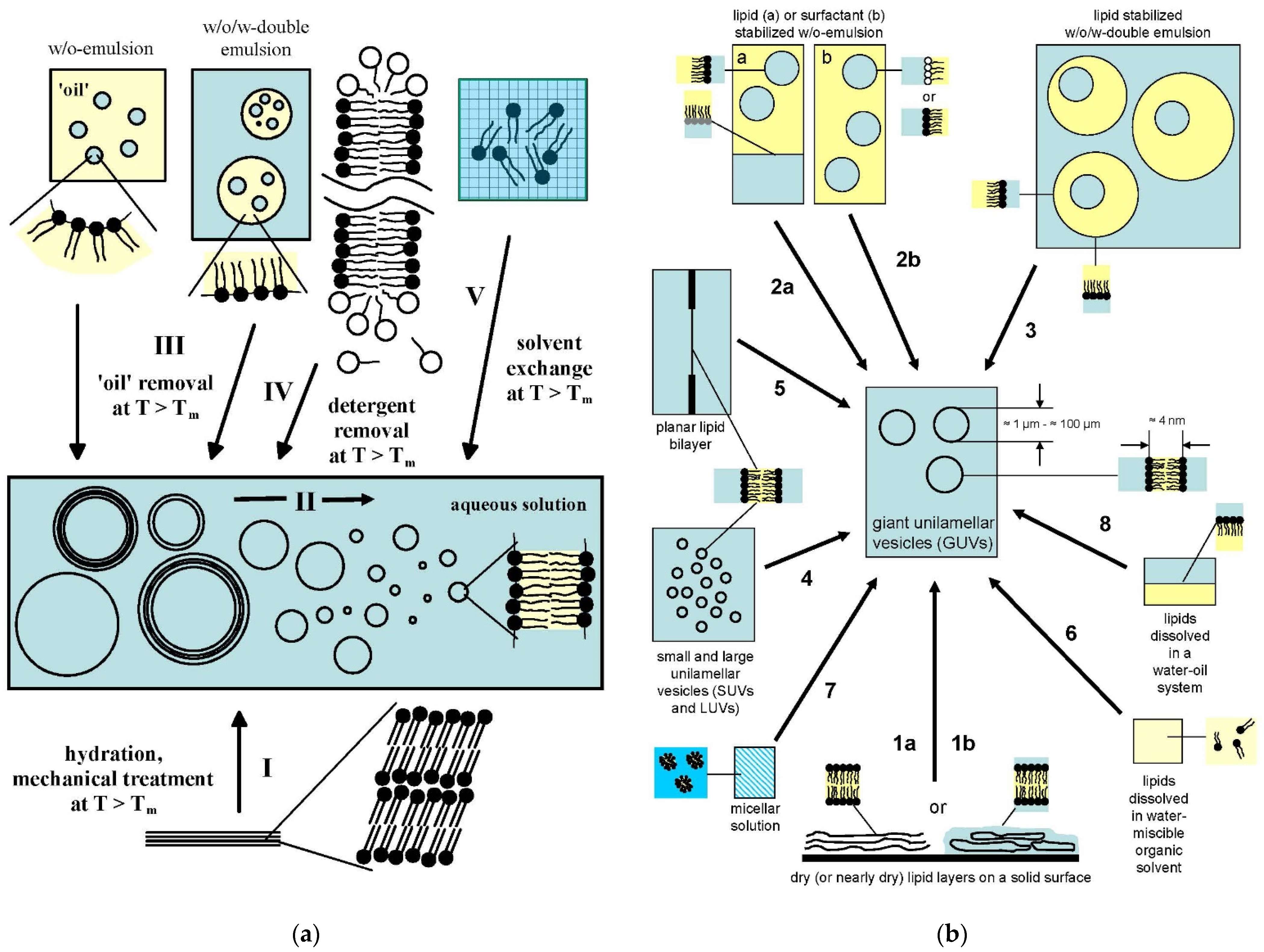
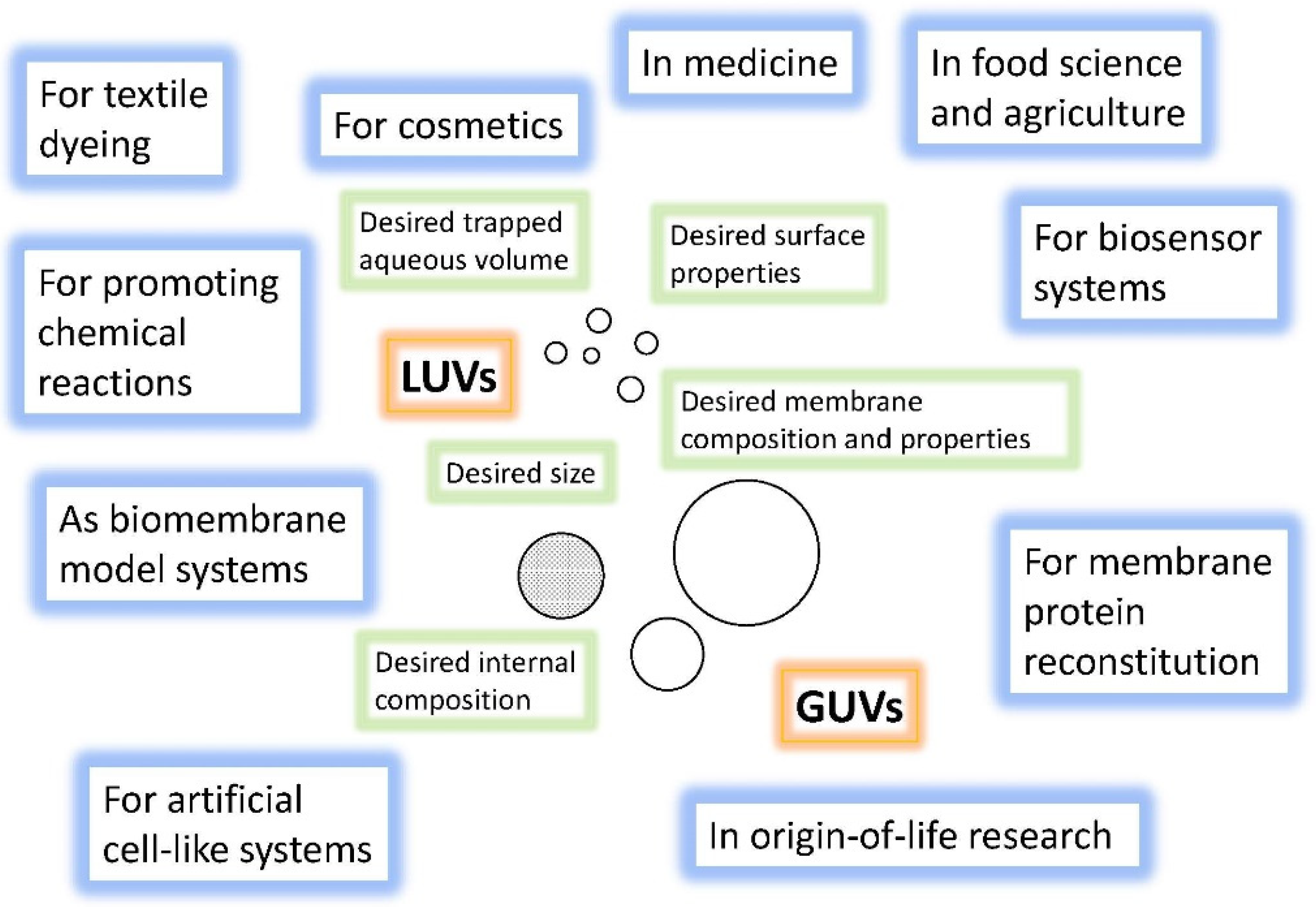
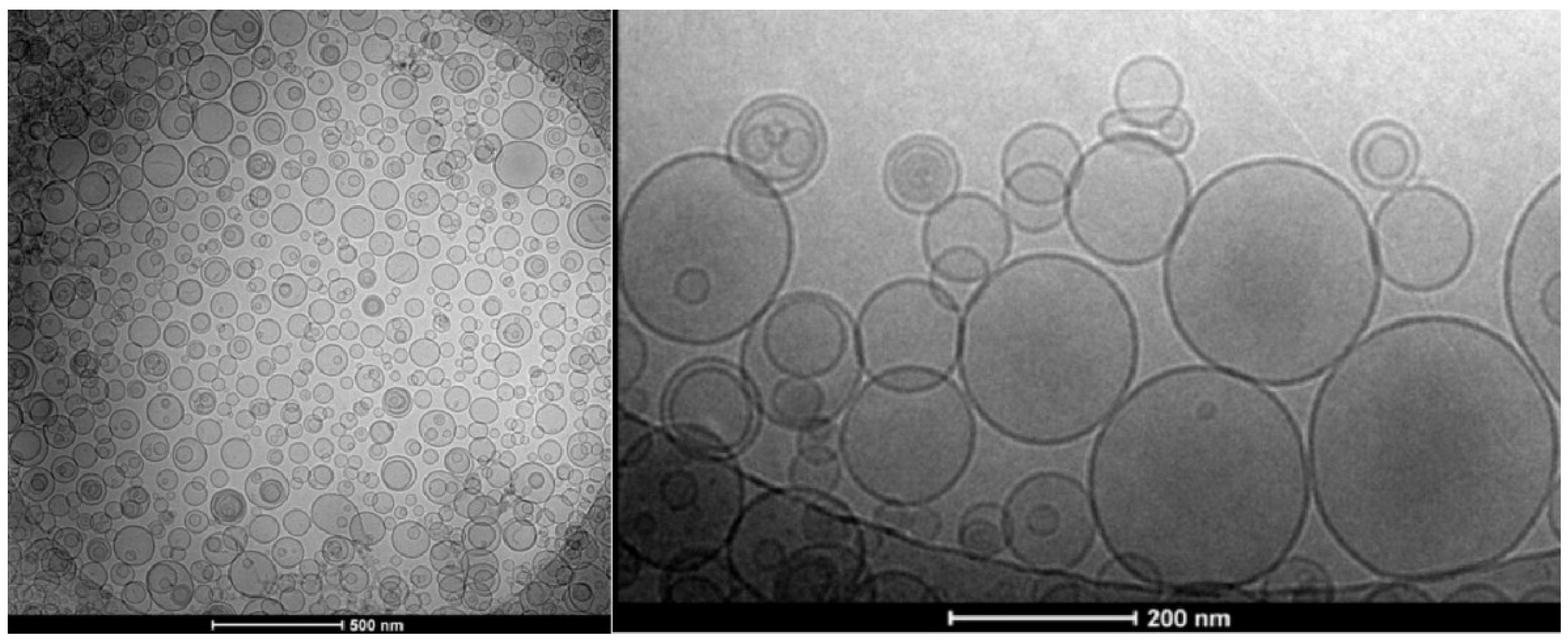
4.1. Overview of the Concepts for the Formation of Large or Giant Unilamellar Vesicles
4.2. Reproducible Large Scale Formation of LUV Dispersions with the “Ethanol Injecton Method”
4.3. Sophisticated Microfluidic Methods for the Formation of GUVs
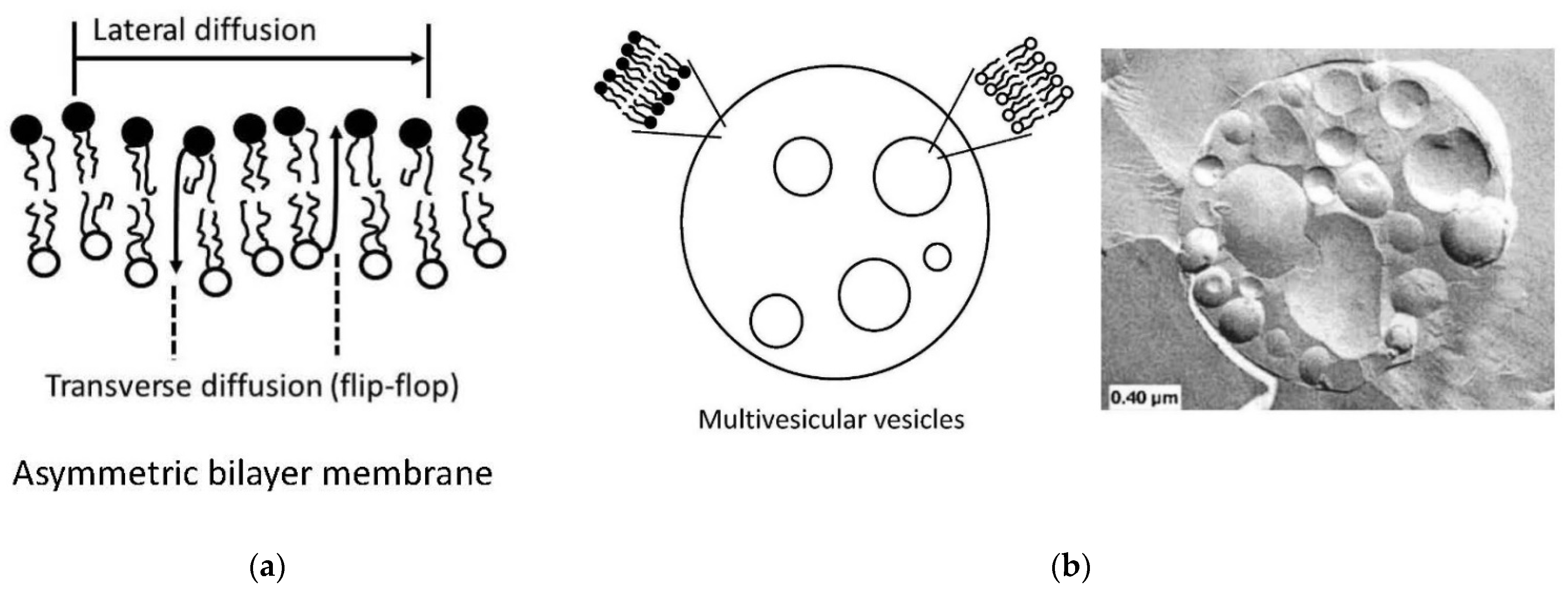
4.4. Preparation of Vesicles with Asymmetric Membranes and/or with Internal Vesicles
5. Functionalization of Lipid Vesicles and Possible Bicelle Formation
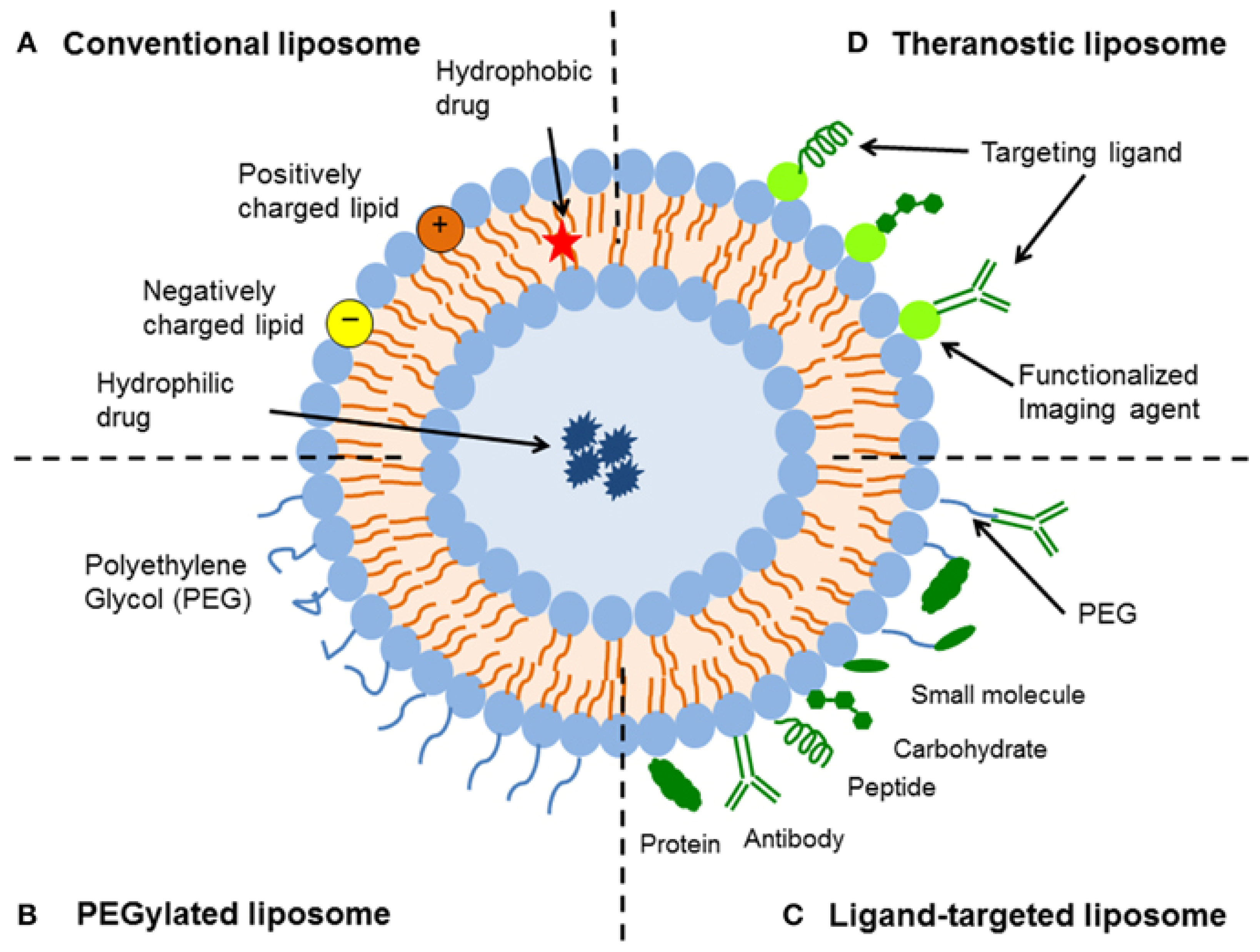
5.1. Opportunities for the Functionalization of Lipid Vesicles
- First of all, hydrophilic molecules can be entrapped in the interior aqueous volume of the vesicles, for example, low molar mass pharmaceutically active compounds or therapeutic enzymes. Such entrapment can be achieved during the vesicle preparation, followed by separation of non-entrapped compounds (see Chapter 4). In special cases, loading of the vesicles is also possible after vesicles formation (so-called “remote loading”); see Section 5.4. Independent from the way desired water-soluble molecules are entrapped inside vesicles of desired size, lamellarity, and membrane composition, the entrapped molecules are separated from the bulk solution by a lipid bilayer, which acts as protective permeability barrier for the entrapped molecules.
- Second, hydrophobic compounds can be embedded within the vesicle membrane. This can again be achieved either during vesicle preparation or after vesicle formation. Depending on the type of membrane-embedded compound, it may alter the physico-chemical properties of the membrane in a desired way. The embedding of cholesterol into a fluid phospholipid bilayer, for example, can result in a significant bilayer rigidification, or it can lead to domain formation within the membrane due to a non-homogeneous distribution of the cholesterol and lipid molecules; see Section 5.3.
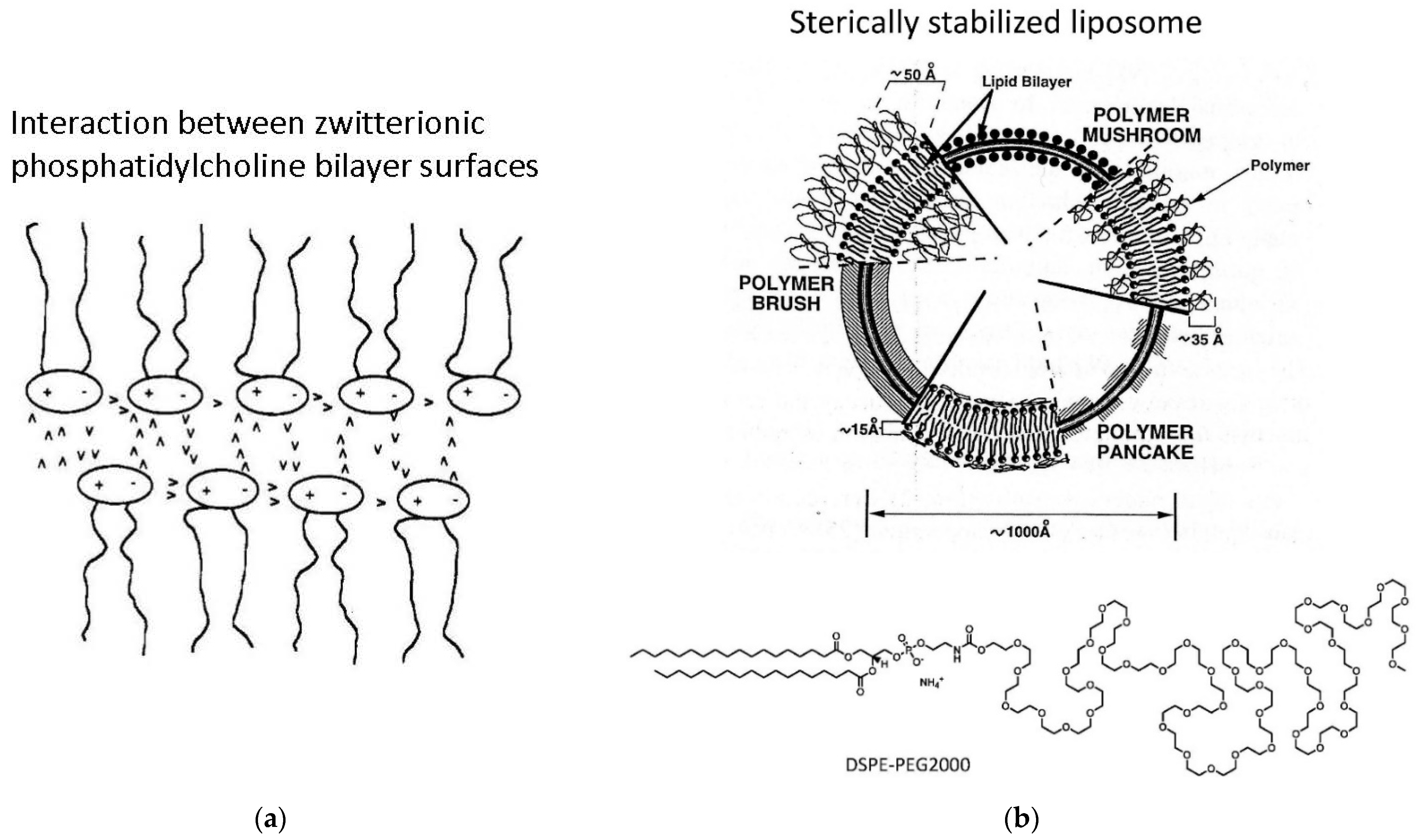
- Third, the external vesicle surface can be functionalized. Such surface functionalization can be achieved either during vesicle preparation by using amphiphiles with a desired functionalized head group (for example PEGylated lipids, as mentioned in Chapter 3, Figure 16b) or by chemically modifying the outer vesicle surface after vesicle formation.
5.2. Vesicle Functionalization May Lead to the Formation of Bicelles
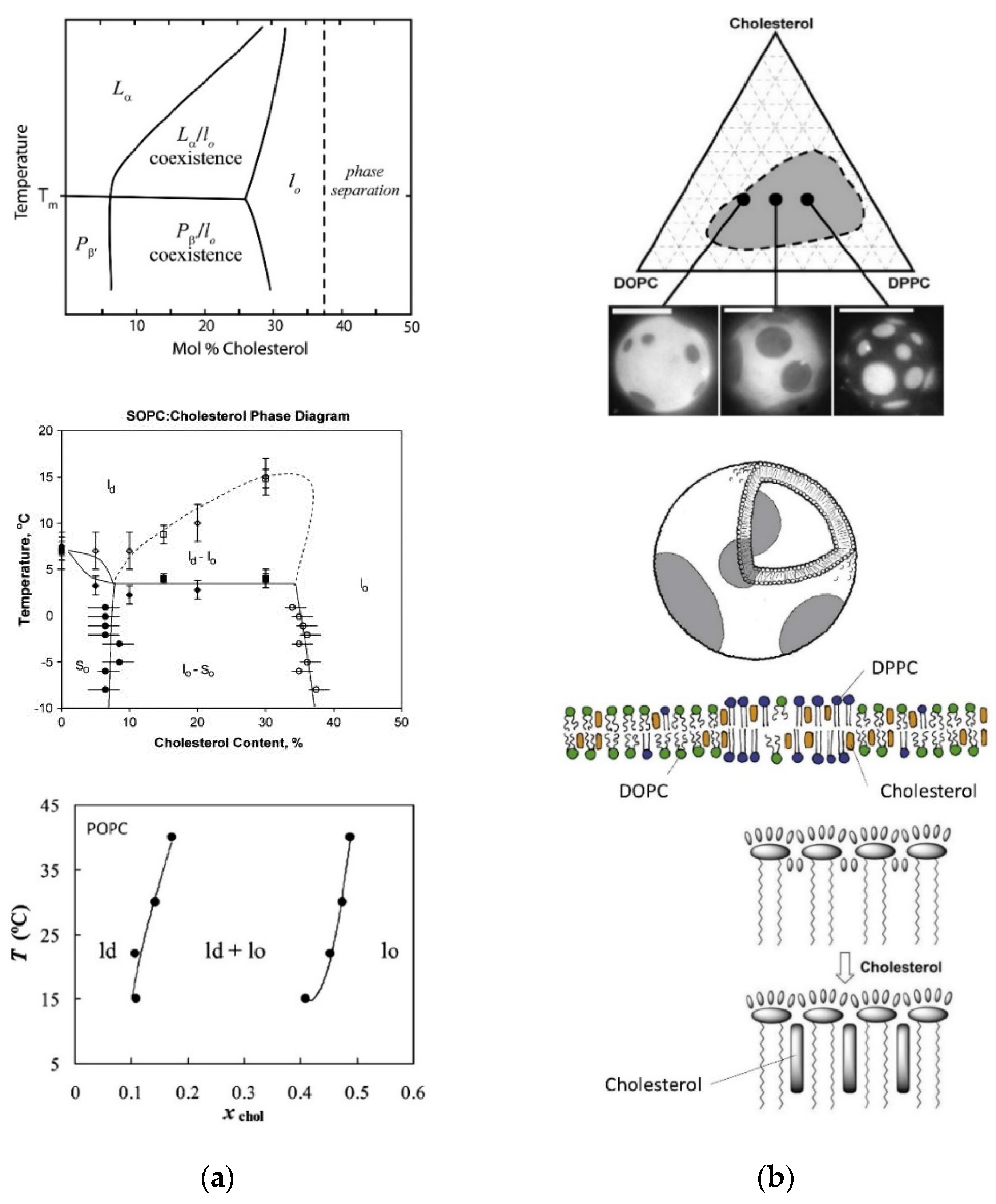
5.3. Domain formation within Vesicle Membranes May Occur
5.4. The “Remote Loading” of Lipid Vesicles with Certain Water-Soluble Compounds
6. Selected Examples of Applications of Lipid Vesicles and Related Lipid Aggregates
6.1. Overview
- The good level of understanding of the correlation between chemical structure of an amphiphile and types of aggregates that can be formed in an aqueous medium and of the aggregates’ thermodynamic and kinetic properties.
- The remarkable size range within which lipid vesicles can be prepared, from ~30 nm to 100 μm (and more), and the knowledge about the concepts and methods that have been elaborated for obtaining stable vesicles with a relatively defined diameter (preparation methods).
- The knowledge about the concepts that can be used for loading vesicles with desired water- and/or membrane-soluble ingredients.
- The possibilities of tuning lipid vesicles, basically at will, by incorporating functionalized, partially synthetic amphiphiles in the membrane, or by modifying some of the vesicle-forming amphiphiles present in the membrane after vesicle formation.
- The expected costs for the development of a product might be unacceptably high due to the molecular complexity of specifically engineered vesicles and the method of vesicle preparation and purification, as compared to the expected quantity that one would need to produce and the price consumers would be willing to pay for a specific product.
- The loading of the vesicles with desired active compounds might not be as efficient as required.
6.2. Liposome (and “Lipid Nanoparticle”) Applications in Medicine
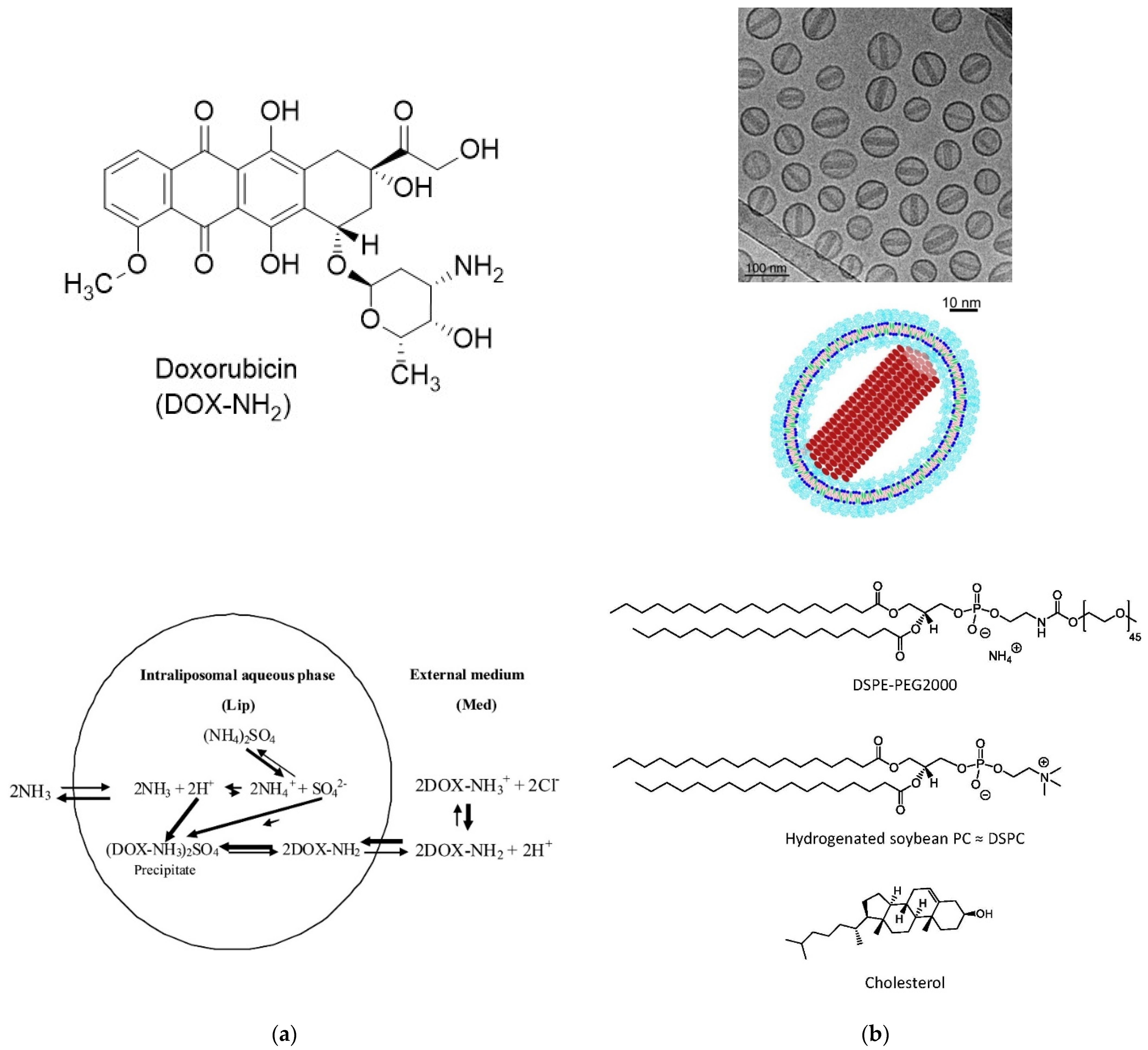
6.2.1. Doxil®, AmBisome®, Visudyne®, Exparel®, and Inflexal® V
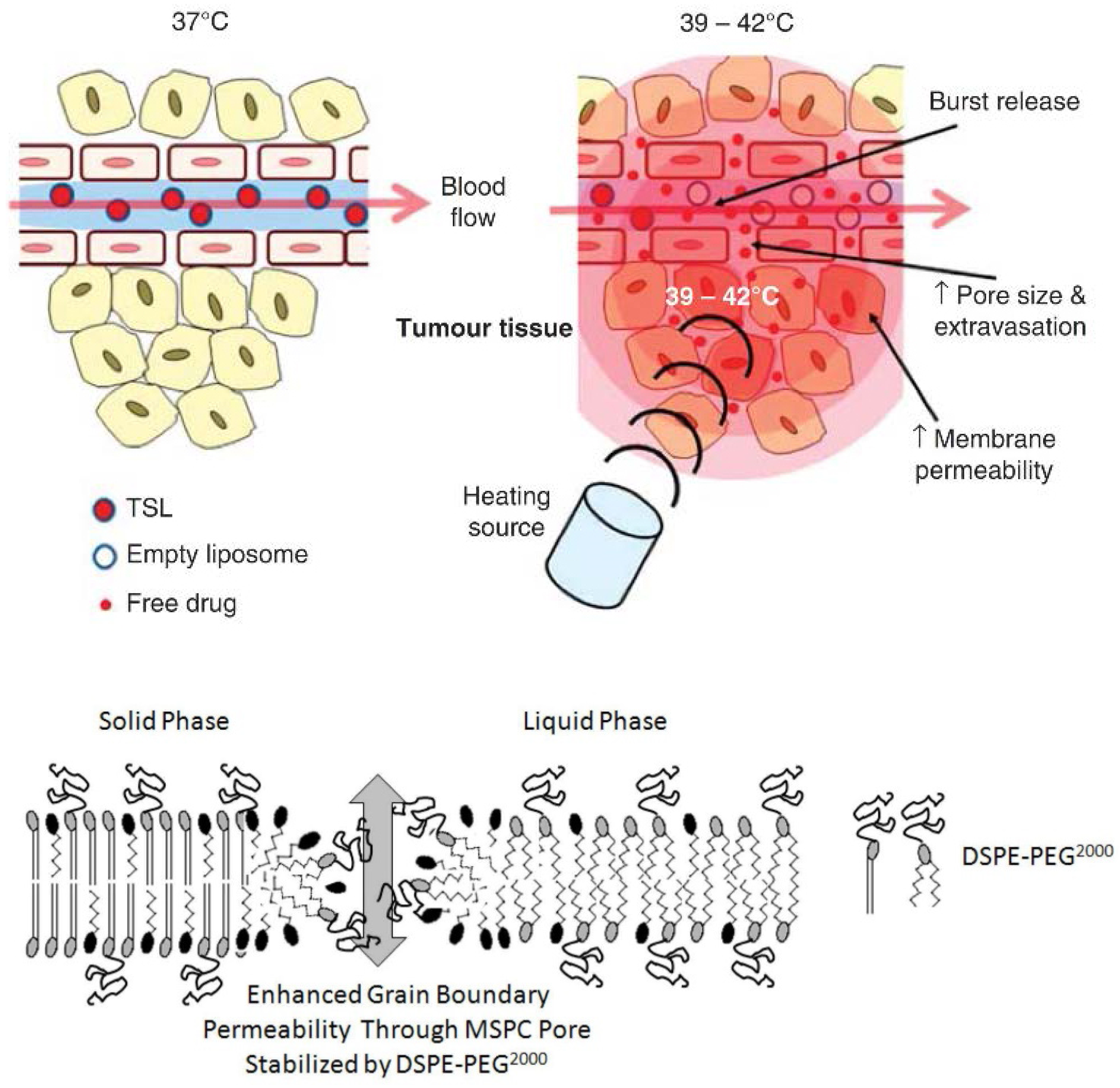
6.2.2. ThermoDox®
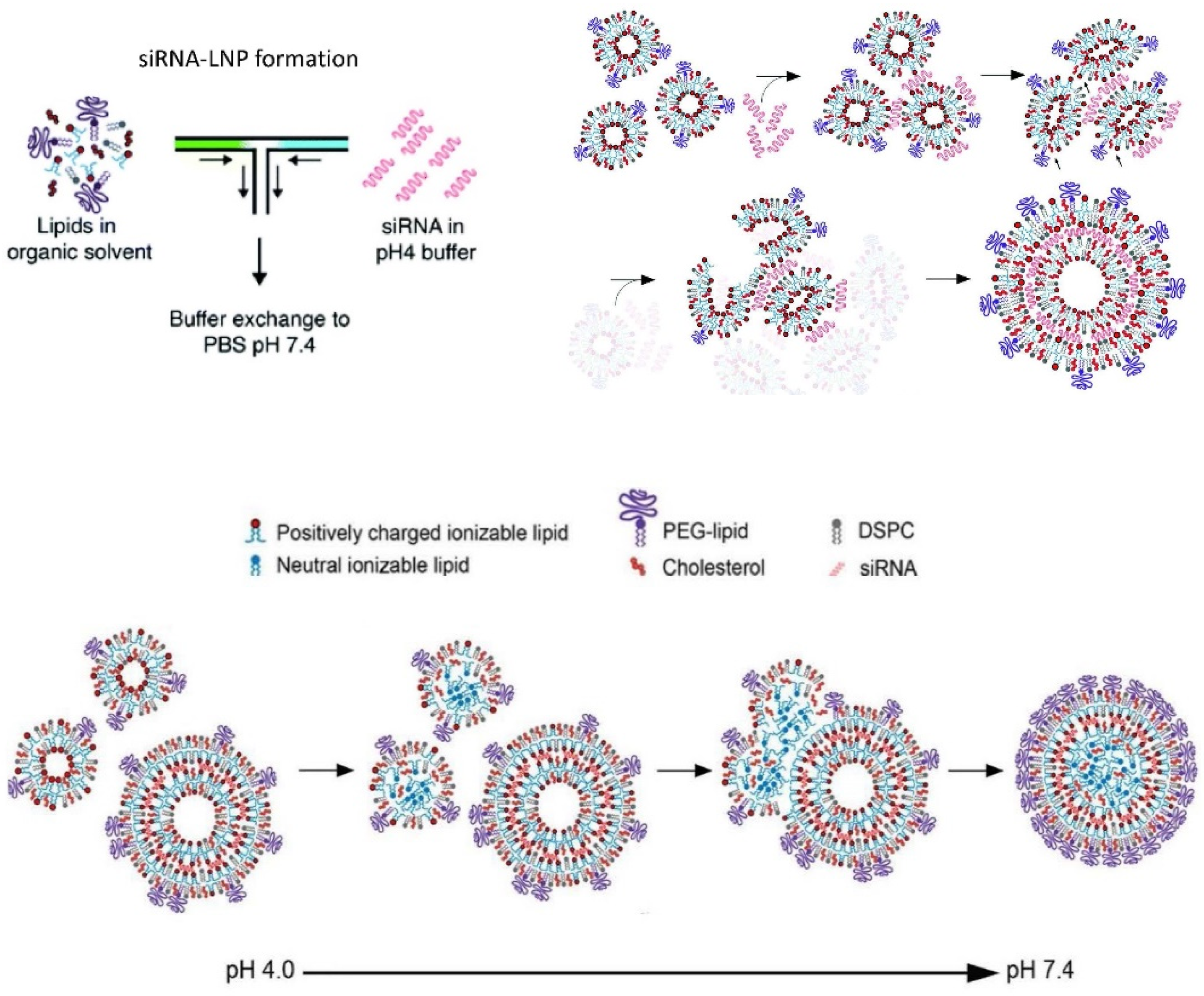
6.2.3. Onpattro®
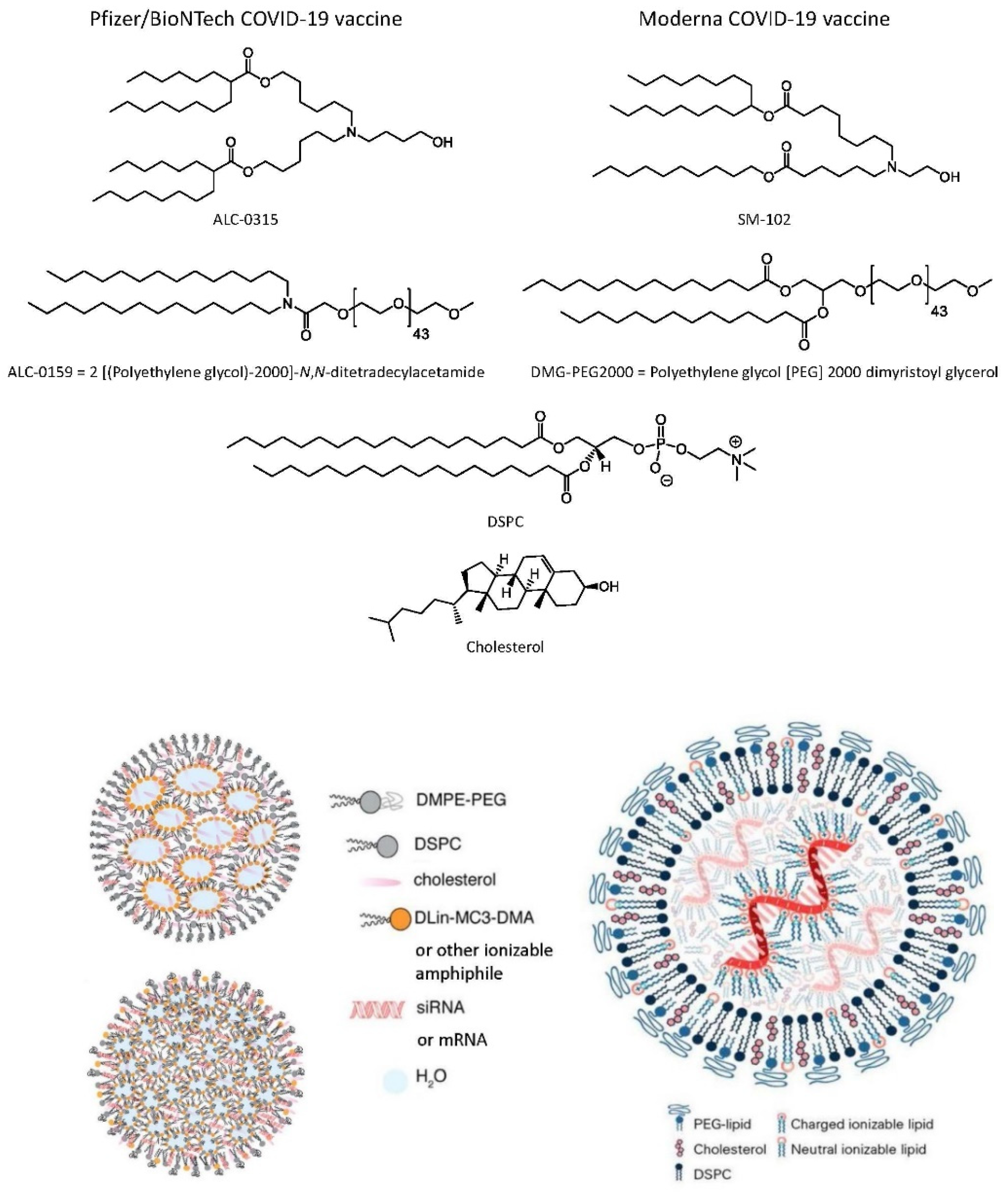
6.2.4. Lipid Nanoparticle (LNP) mRNA COVID-19 Vaccines
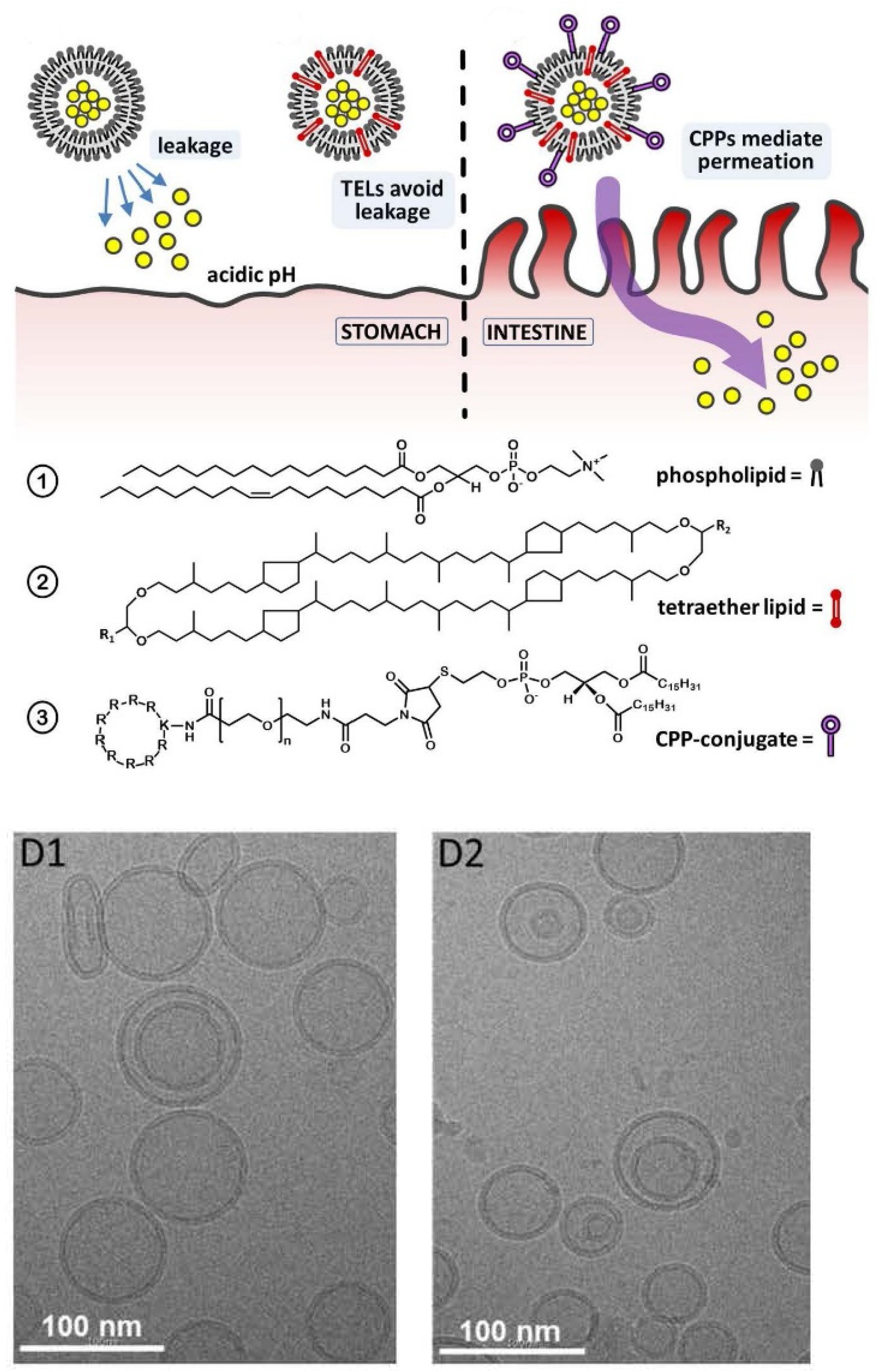
6.2.5. Lipid Vesicles for Transdermal or Oral Drug Delivery Applications
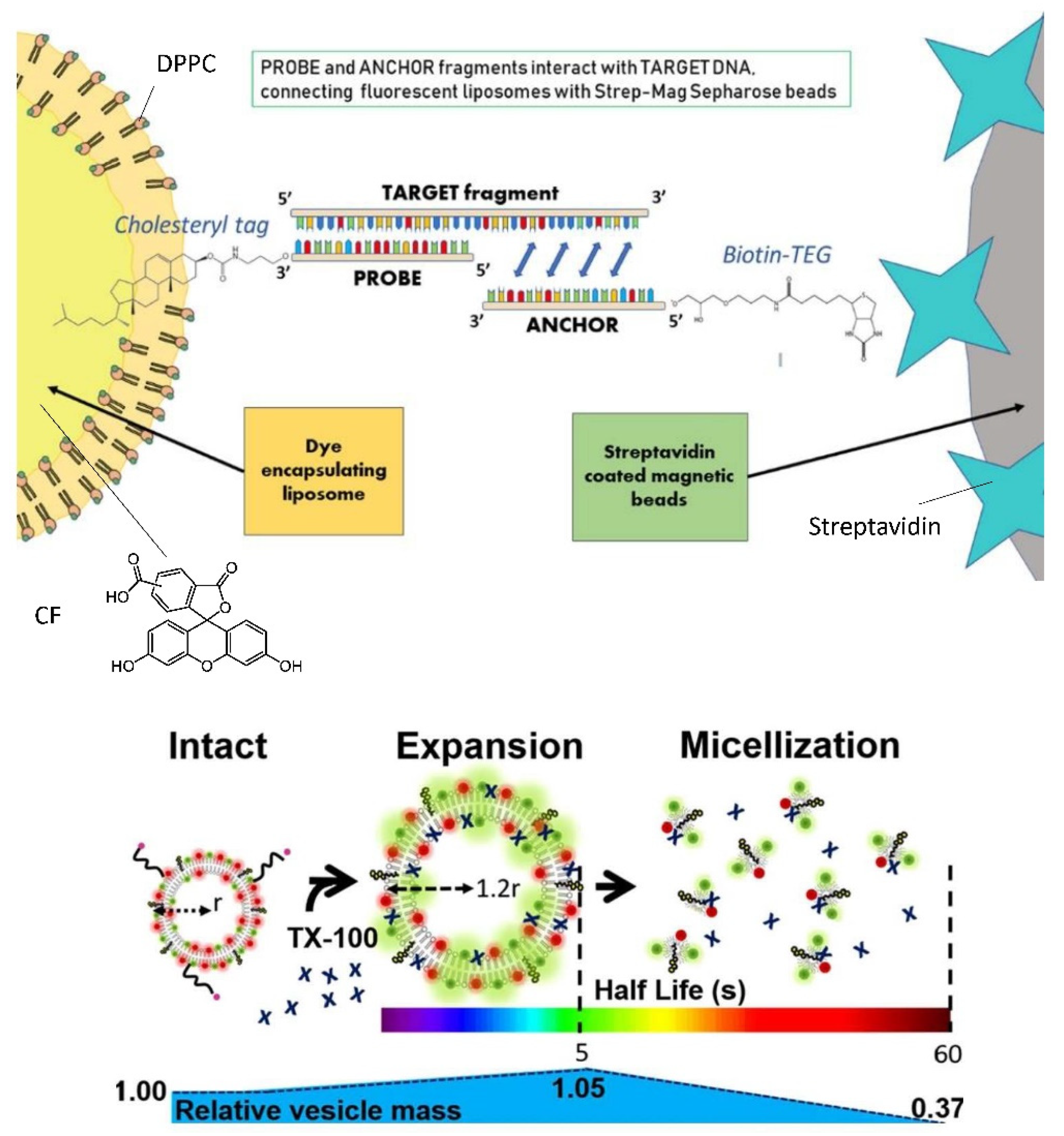
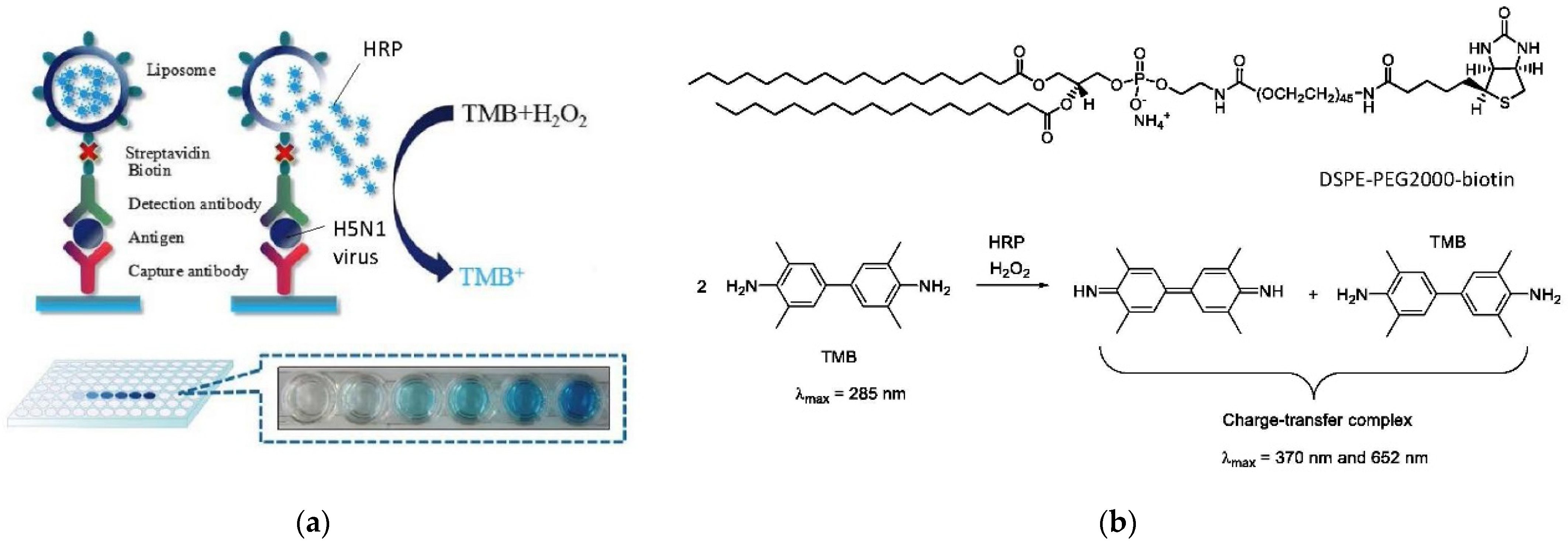
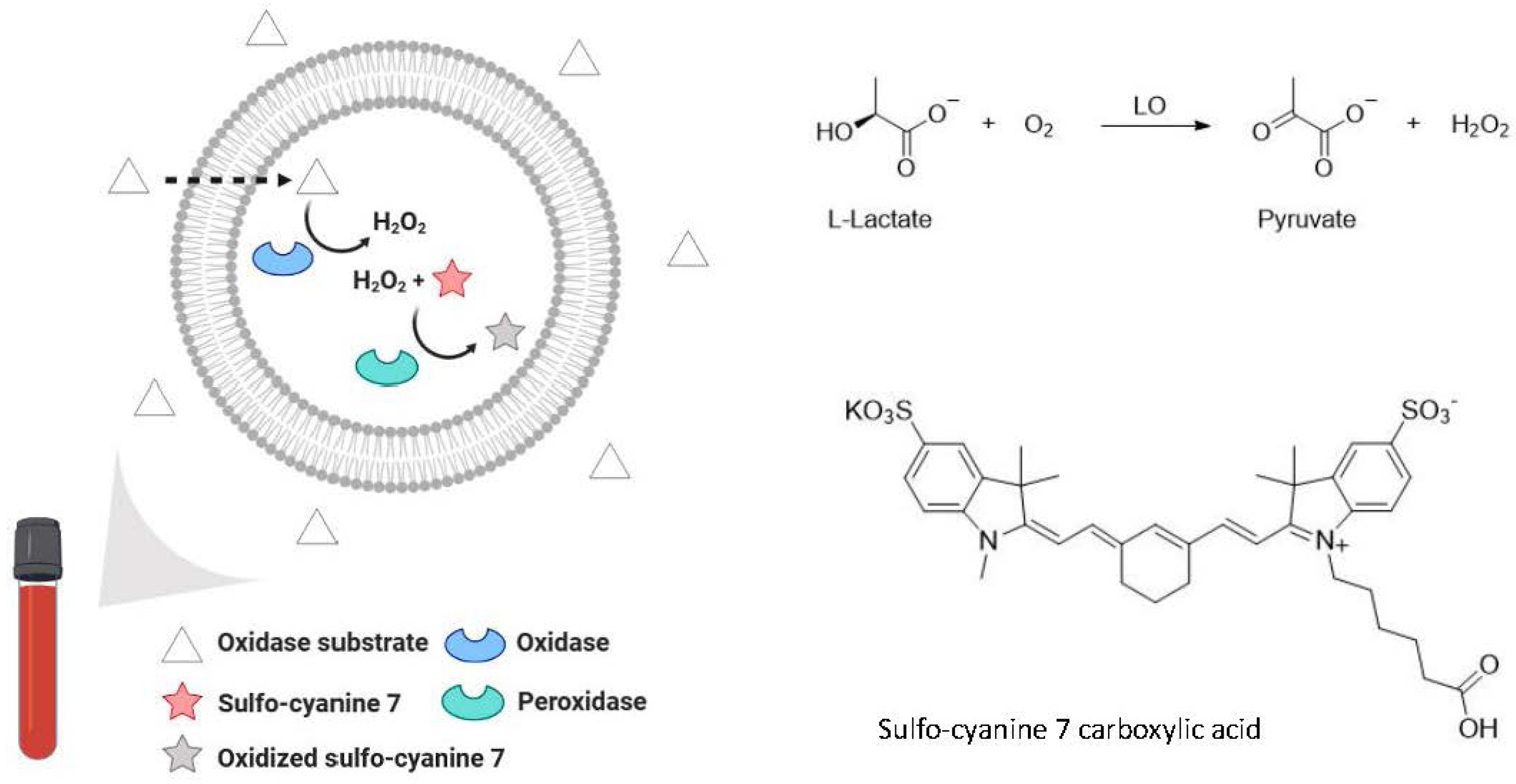
6.3. Bioanalytical Applications of Lipid Vesicles (Bioassays and Biosensors)

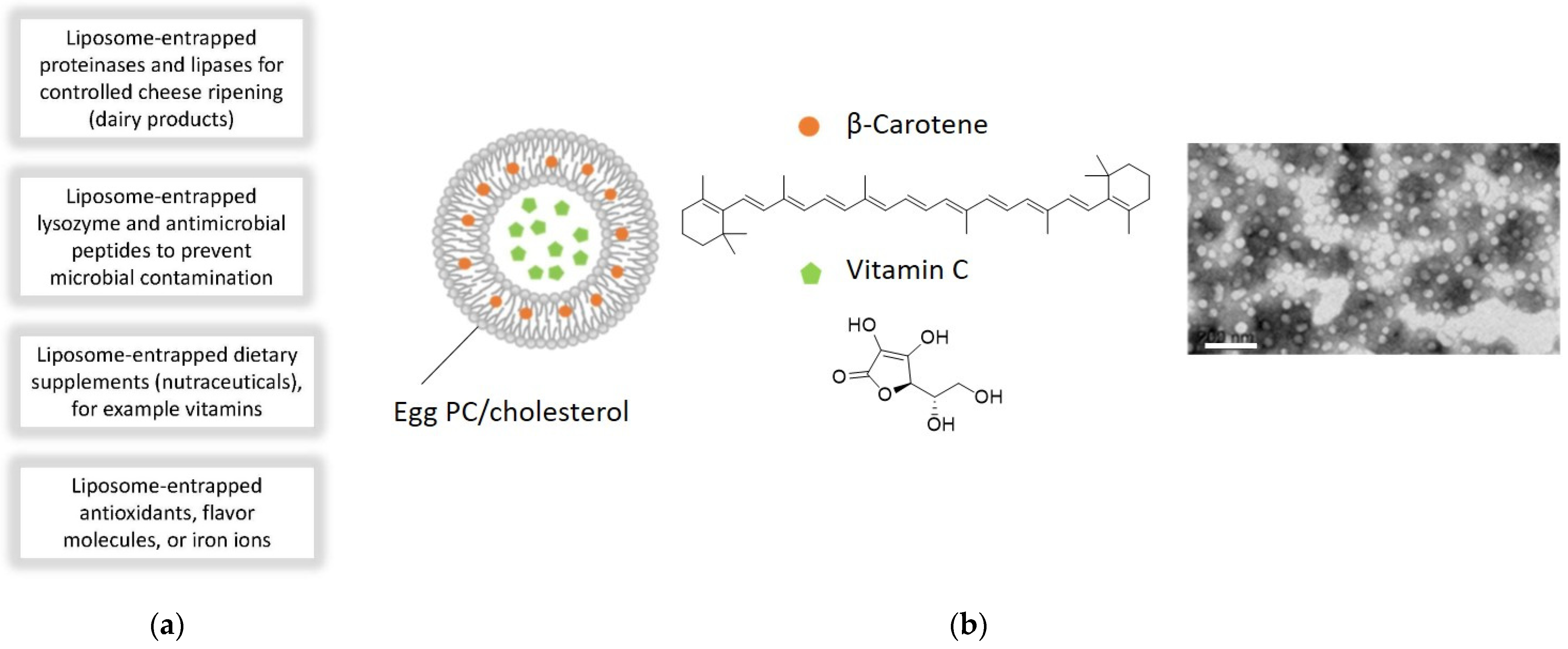
6.4. Application of Lipid Vesicles in Food Processing and Nutrition
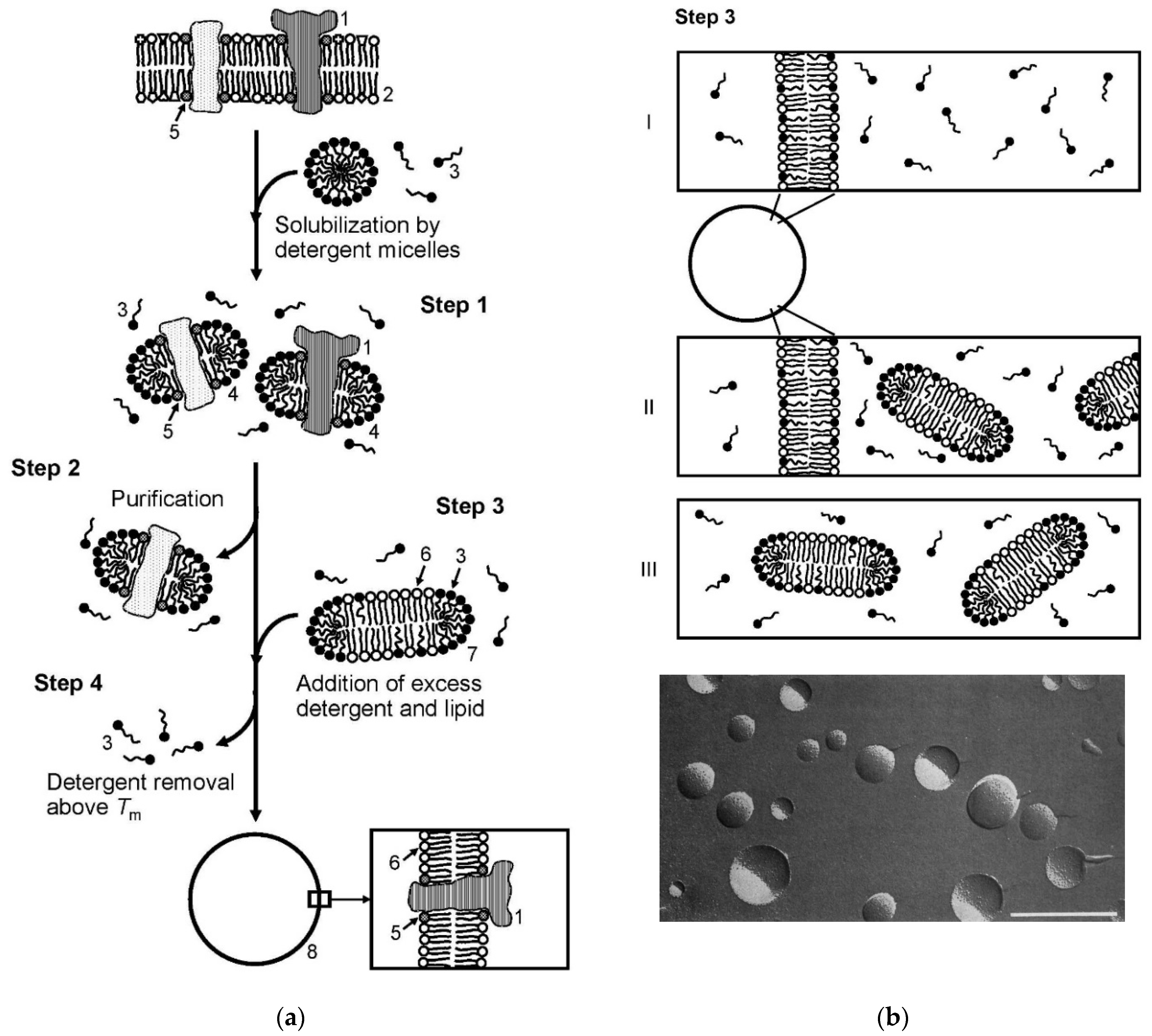
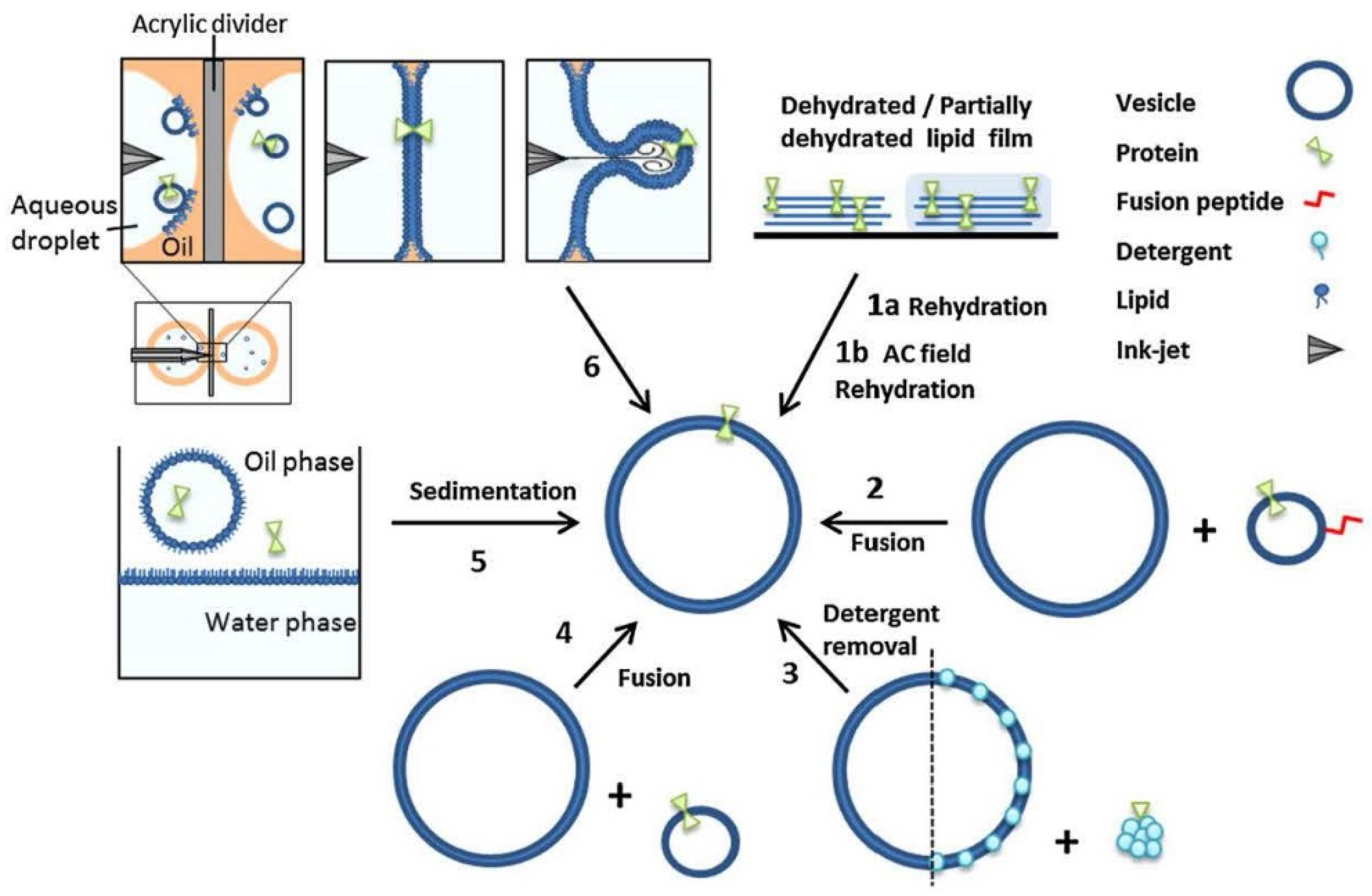
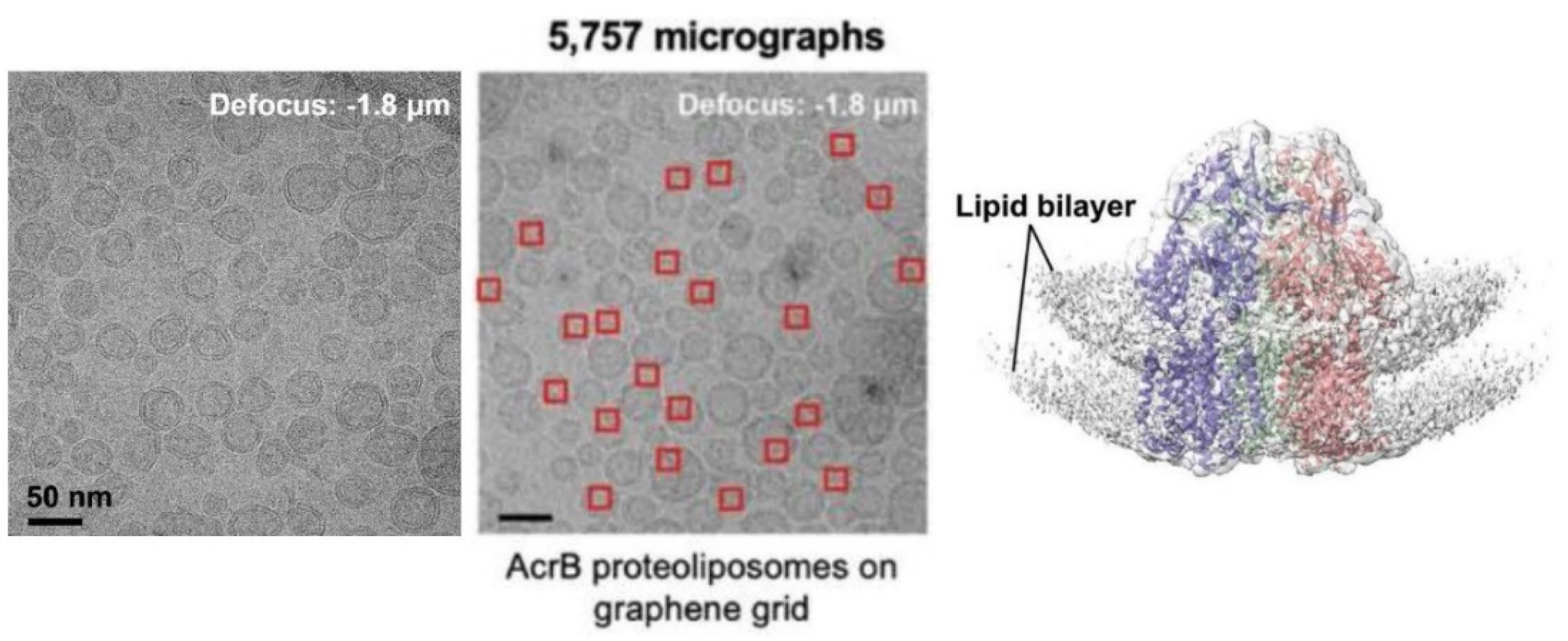
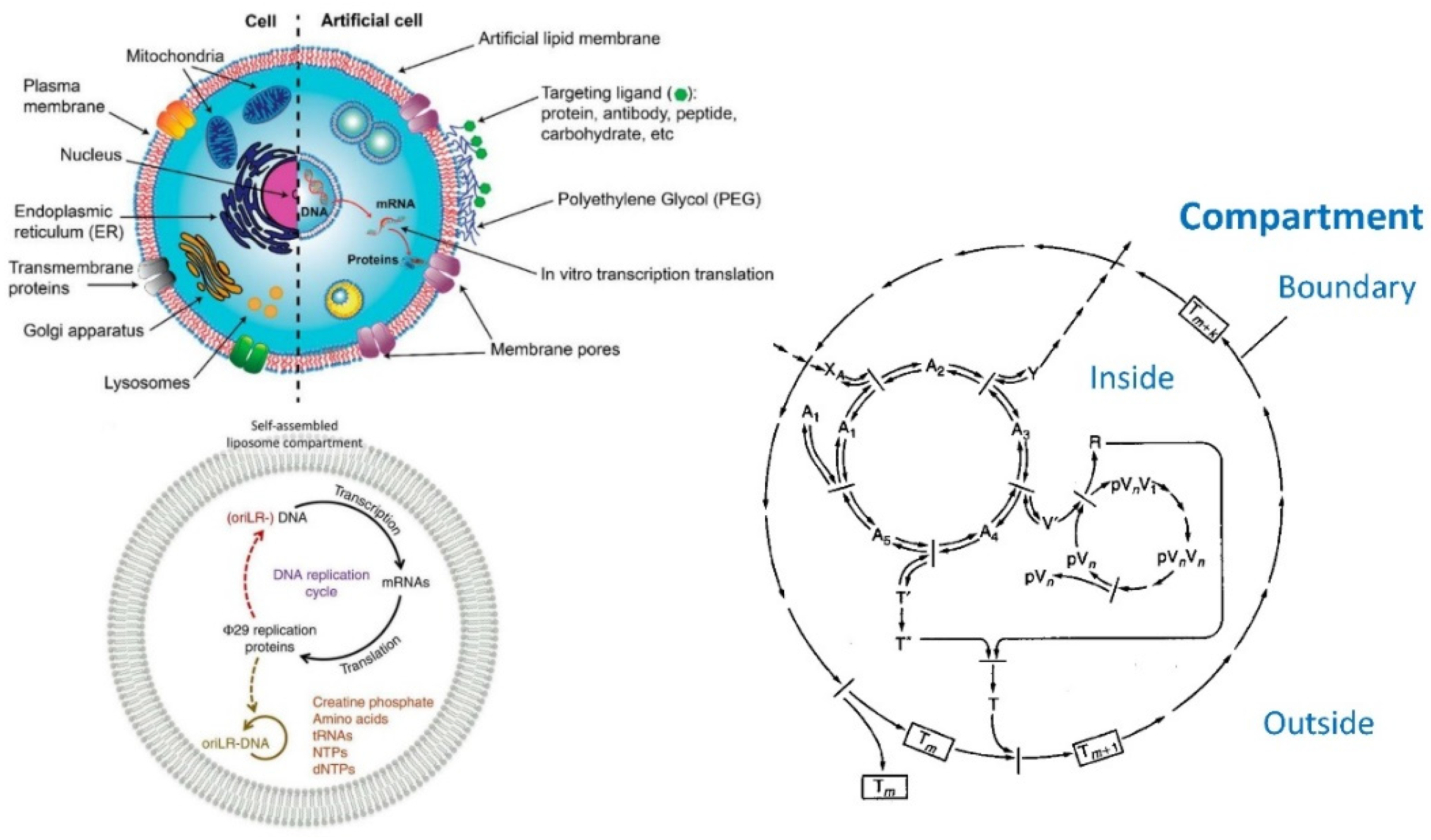
6.5. Application of Lipid Vesicles for Membrane Protein Reconstitution, for the Preparation of Synthetic Cell-like Compartments (“Artificial Cells”), and as Models of “Protocells”

7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, M.D.; Carrasco, M.J.; Alishetty, S.; Paige, M.; Alameh, M.G.; Weissman, D. Nanomaterial Delivery Systems for mRNA Vaccines. Vaccines 2021, 9, 65. [Google Scholar] [CrossRef]
- Park, K.S.; Sun, X.; Aikins, M.E.; Moon, J.J. Non-viral COVID-19 vaccine delivery systems. Adv. Drug Deliv. Rev. 2021, 169, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef] [PubMed]
- Mannino, R.J.; Allebach, E.S.; Strohl, W.A. Encapsulation of High Molecular Weight DNA in Large Unilamellar Phospholipid Vesicles. FEBS Lett. 1979, 101, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Wilson, T.; Papahadjopoulos, D.; Taber, R. The Introduction of Poliovirus RNA into Cells via Lipid Vesicles (Liposomes). Cell 1979, 17, 77–84. [Google Scholar] [CrossRef]
- Dimitriadis, G.J. Entrapment of plasmid DNA in liposomes. Nucleic Acids Res. 1979, 6, 2697–2705. [Google Scholar] [CrossRef] [Green Version]
- Straubinger, R.M.; Papahadjopoulos, D. Liposomes as Carriers for Intracellular Delivery of Nucleic Acids. Methods Enzymol. 1983, 101, 512–527. [Google Scholar] [PubMed]
- Gershon, H.; Ghirlando, R.; Guttman, S.B.; Minsky, A. Mode of Formation and Structural Features of DNA-Cationic Liposome Complexes Used for Transfection. Biochemistry 1993, 32, 7143–7151. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, B.; Sorgi, F.L.; Huang, L. New structures in complex formation between DNA and cationic liposomes visualized by freeze-fracture electron microscopy. FEBS Lett. 1994, 356, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, J.; Arvidson, G.; Karlsson, G.; Almgren, M. Complexes between cationic liposomes and DNA visualized by cryo-TEM. Biochim. Biophys. Acta 1995, 1235, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Gregoriadis, G.; Saffie, R.; Hart, S.L. High Yield Incorporation of Plasmid DNA within Liposomes: Effect on DNA Integrity and Transfection Efficiency. J. Drug. Target. 1996, 3, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Monnard, P.-A.; Oberholzer, T.; Luisi, P.L. Entrapment of nucleic acids in liposomes. Biochim. Biophys. Acta 1997, 1329, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Perrie, Y.; Gregoriadis, G. Liposome-entrapped plasmid DNA: Characterisation studies. Biochim. Biophys. Acta 2000, 1475, 125–132. [Google Scholar] [CrossRef]
- Deamer, D.W. From “Banghasomes” to liposomes: A memoir of Alec Bangham, 1921–2010. FASEB J. 2010, 24, 1308–1310. [Google Scholar] [CrossRef]
- Heap, B.; Gregoriadis, G. Alec Douglas Bangham. Biogr. Mems Fell. R. Soc. 2011, 57, 25–43. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Kučerka, N.; Nieh, M.-P.; Katsaras, J. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochim. Biophys. Acta 2011, 1808, 2761–2771. [Google Scholar] [CrossRef]
- Blocher, M.; Walde, P.; Dunn, I.J. Modeling of Enzymatic Reactions in Vesicles: The Case of α-Chymotrypsin. Biotechnol. Bioeng. 1999, 62, 36–43. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent Advances with Liposomes as Pharmaceutical Carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregoriadis, G. Liposome research in drug delivery: The early days. J. Drug Target. 2008, 16, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Dawidczyk, C.M.; Kim, C.; Park, J.H.; Russell, L.M.; Lee, K.H.; Pomper, M.G.; Searson, P.C. State-of-the-art in design rules for drug delivery platforms: Lessons learned from FDA-approved nanomedicines. J. Control. Release 2014, 187, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouwstra, J.A.; Honeywell-Nguyen, P.L.; Gorris, G.S.; Ponec, M. Structure of the skin barrier and its modulation by vesicular formulations. Prog. Lipid Res. 2003, 42, 1–36. [Google Scholar] [CrossRef]
- Cevc, G. Lipid vesicles and other colloids as drug carriers on the skin. Adv. Drug Deliv. Rev. 2004, 56, 675–711. [Google Scholar] [CrossRef] [PubMed]
- El Maghraby, G.M.; Barry, B.W.; Williams, A.C. Liposomes and skin: From drug delivery to model membranes. Eur. J. Pharm. Sci. 2008, 34, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Cevc, G.; Vierl, U. Nanotechnology and the transdermal route. A state of the art review and critical appraisal. J. Control. Release 2010, 141, 277–299. [Google Scholar] [CrossRef]
- Kaul, S.; Gulkati, N.; Verma, D.; Mukherjee, S.; Nagaich, U. Role of Nanotechnology in Cosmeceuticals: A Review of Recent Advances. Hindawi J. Pharm. 2018, 3420204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, V.V.; Moon, J.-Y.; Lee, Y.-C. Liposomes for delivery of antioxidants in cosmeceuticals: Challenges and development strategies. J. Control. Release 2019, 300, 114–140. [Google Scholar] [CrossRef] [PubMed]
- Souto, E.B.; Fernandes, A.R.; Martins-Gomes, C.; Coutinho, T.E.; Durazzo, A.; Lucarini, M.; Souto, S.B.; Silva, A.M.; Santini, A. Nanomaterials for Skin Delivery of Cosmeceuticals and Pharmaceuticals. Appl. Sci. 2020, 10, 1594. [Google Scholar] [CrossRef] [Green Version]
- Noireaux, V.; Libchaber, A. A vesicle bioreactor as a step toward an artificial cell assembly. Proc. Natl. Acad. Sci. USA 2004, 101, 17669–17674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luisi, P.L.; Stano, P. Approaches to semi-synthetic minimal cells: A review. Naturwissenschaften 2006, 93, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Walde, P. Building artificial cells and protocell models: Experimental approaches with lipid vesicles. BioEssays 2010, 32, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Elani, Y. Construction of membrane-bound artificial cells using microfluidics: A new frontier in bottom-up synthetic biology. Biochem. Soc. Trans. 2016, 44, 723–730. [Google Scholar] [CrossRef] [PubMed]
- van Nies, P.; Westerlaken, I.; Blanken, D.; Salas, M.; Mencía, M.; Danelon, C. Self-replication of DNA by its encoded protens in liposome-based synthetic cells. Nat. Commun. 2018, 9, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göpfrich, K.; Platzman, I.; Spatz, J.P. Mastering Complexity: Towards Bottom-up Construction of Multifunctional Eukaryotic Synthetic Cells. Trends Biotechnol. 2018, 36, 938–951. [Google Scholar] [CrossRef] [Green Version]
- Stano, P. Gene Expression Inside Liposomes: From Early Studies to Current Protocols. Chem. Eur. J. 2019, 25, 7798–7814. [Google Scholar] [CrossRef]
- Dijkgraaf, R. Knowledge Is Infrastructure. Sci. Am. 2017, 316, 8. [Google Scholar] [CrossRef]
- Small, D.M. The Physical Chemistry of Lipids (Handbook of Lipid Research); Hanahan, D.J., Ed.; Plenum Press: New York, NY, USA, 1986; Volume 4. [Google Scholar]
- Jones, M.N.; Chapman, D. Micelles, Monolayers, and Biomembranes; WILEY-LISS: New York, NY, USA, 1995. [Google Scholar]
- Holmberg, K.; Jönsson, B.; Kronberg, B.; Lindman, B. Surfactants and Polymers in Aqueous Solution, 2nd ed.; John Wiley & Sons: Chichester, UK, 2003. [Google Scholar]
- Mahieu, N.; Canet, D.; Cases, J.M.; Boubel, J.C. Micellization of Sodium Oleate in D2O As Probed by Proton Longitudinal Magnetic Relaxation and Self-Diffusion Measurements. J. Phys. Chem. 1991, 95, 1844–1846. [Google Scholar] [CrossRef]
- Bergstrand, N.; Edwards, K. Aggregate Structure in Dilute Dispersions of Phospholipids, Fatty Acids, and Lysophospholipids. Langmuir 2001, 17, 3245–3253. [Google Scholar] [CrossRef]
- Vorum, H.; Brodersen, R.; Kragh-Hansen, U.; Pedersen, A.O. Solubility of long-chain fatty acids in phosphate buffer at pH 7.4. Biochim. Biophys. Acta 1992, 1126, 135–142. [Google Scholar] [CrossRef]
- Antunes, F.E.; Coppola, L.; Gaudio, D.; Nicotera, I.; Oliviero, C. Shear rheology and phase behavior of sodium oleate/water mixtures. Colloids Surf. A 2007, 297, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Vold, R.D. The Phase Rule Behavior of Concentrated Aqueous Systems of a Typical Colloidal Electrolyte: Sodium Oleate. J. Phys. Chem. 1939, 43, 1213–1231. [Google Scholar] [CrossRef]
- McBain, J.W.; Sierichs, W.C. The Solubility of Sodium and Potassium Soaps and the Phase Diagrams of Aqueous Potassium Soaps. I. Am. Chem. Oil Chem. Soc. 1948, 25, 221–225. [Google Scholar] [CrossRef]
- Suga, K.; Kondo, D.; Otsuka, Y.; Okamoto, Y.; Umakoshi, H. Characterization of Aqueous Oleic Acid/Oleate Dispersions by Fluorescent Probes and Raman Spectroscopy. Langmuir 2016, 32, 7606–7612. [Google Scholar] [CrossRef]
- Edwards, K.; Silvander, M.; Karlsson, G. Aggregate Structure in Dilute Aqueous Dispersions of Oleic Acid/Sodium Oleate and Oleic Acid/Sodium Oleate/Egg Phosphaidylcholine. Biochim. Biophys. Acta 1995, 1126, 135–142. [Google Scholar]
- Tatini, D.; Raudino, M.; Ambrosio, M.; Carretti, E.; Davidovich, I.; Talmon, Y.; Ninham, B.W.; LoNostro, P. Physicochemical characterization of green sodium oleate-based formulations. Part 1. structure and rheology. J. Colloid Interface Sci. 2021, 590, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Rosevear, F.B. Liquid Crystals: The Mesomorphic Phases of Surfactant Compositions. J. Soc. Cosmet. Chem. 1968, 19, 581–594. [Google Scholar]
- McBain, J.W.; Stewart, A. Conductivity in the Three-component System Oleic Acid–Potassium Oleate–Water. J. Chem. Soc. 1933, 928–932. [Google Scholar] [CrossRef]
- Small, D.M. A Classification of Biological Lipids Based upon Their Interaction in Aqueous Systems. J. Am. Oil Chem. Soc. 1968, 45, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Engblom, J.; Engström, S.; Fontell, K. The effect of the skin penetration enhancer Azone® on fatty acid-sodium soap-water mixtures. J. Control. Release 1995, 33, 299–305. [Google Scholar] [CrossRef]
- Mele, S.; Söderman, O.; Ljusberg-Wahrén, H.; Thuresson, K.; Monduzzi, M.; Nylander, T. Phase behavior in the biologically important oleic acid/sodium oleate/water system. Chem. Phys. Lipids 2018, 211, 30–36. [Google Scholar] [CrossRef]
- Koynova, R.; Tenchov, B. Phase Transitions and Phase Behavior of Lipids. In Encyclopedia of Biophysics; Roberts, G.C.K., Ed.; European Biophysical Societies Association (EBSA); Springer: Berlin/Heidelberg, Germany, 2013; pp. 1841–1854. [Google Scholar]
- Han, S. Molecular dynamics simulation of oleic acid/oleate bilayers: An atomistic model for a ufasome membrane. Chem. Phys. Lipids 2013, 175–176, 79–83. [Google Scholar] [CrossRef]
- Gebicki, J.M.; Hicks, M. Ufasomes are Stable Particles surrounded by Unsaturated Fatty Acid Membranes. Nature 1973, 243, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, W.R.; Deamer, D.W. Liposomes from Ionic, Single-Chain Amphiphiles. Biochemistry 1978, 17, 3759–3768. [Google Scholar] [CrossRef]
- Cistola, D.P.; Hamilton, J.A.; Jackson, D.; Small, D.M. Ionization and Phase Behavior of Fatty Acids in Water: Application of the Gibbs Phase Rule. Biochemistry 1988, 27, 1881–1888. [Google Scholar] [CrossRef]
- Walde, P.; Wick, R.; Fresta, M.; Mangone, A.; Luisi, P.L. Autopoietic Self-Reproduction of Fatty Acid Vesicles. J. Am. Chem. Soc. 1994, 116, 11649–11654. [Google Scholar] [CrossRef]
- Chen, I.A.; Szostak, J.W. A Kinetic Study of the Growth of Fatty Acid Vesicles. Biophys. J. 2004, 87, 988–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walde, P.; Namani, T.; Morigaki, K.; Hauser, H. Formation and Properties of Fatty Acid Vesicles (Liposomes). In Liposome Technology, 3rd ed.; Gregoriadis, G., Ed.; Informa Healthcare: New York, NY, USA, 2006; Volume I, pp. 1–19. [Google Scholar]
- Morigaki, K.; Walde, P. Fatty Acid Vesicles. Curr. Opin. Colloid Interface Sci. 2007, 12, 75–80. [Google Scholar] [CrossRef]
- Mansy, S.S.; Szostak, J.W. Thermostability of model protocell membranes. Proc. Natl. Acad. Sci. USA 2008, 105, 13351–13355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salentinig, S.; Sagalowicz, L.; Glatter, O. Self-Assembled Structures and pKa Value of Oleic Acid in Systems of Biological Relevance. Langmuir 2010, 26, 11670–11679. [Google Scholar] [CrossRef]
- Ferreira, D.A.; Bentley, M.V.L.B.; Karlsson, G.; Edwards, K. Cryo-TEM investigation of phase behaviour and aggregate structure in dilute dispersions of monoolein and oleic acid. Int. J. Pharm. 2006, 310, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Monnard, P.-A.; Deamer, D.W. Preparation of Vesicles from Nonphospholipid Amphiphiles. Methods Enzymol. 2003, 372, 133–151. [Google Scholar]
- Ganem-Quintanar, A.; Quintanar-Guerrero, D.; Buri, P. Monoolein: A Review of the Pharmaceutical Applications. Drug Dev. Ind. Pharm. 2000, 26, 809–820. [Google Scholar] [CrossRef]
- Kulkarni, C.V.; Wachter, W.; Iglesias-Salto, G.; Engelskirchen, S.; Ahualli, S. Monoolein: A magic lipid? Phys. Chem. Chem. Phys. 2011, 13, 3004–3021. [Google Scholar] [CrossRef]
- Garti, N.; Libster, D.; Aserin, A. Lipid polymorphism in lyotropic liquid crystals for triggered release of bioactives. Food Funct. 2012, 3, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Milak, S.; Zimmer, A. Glycerol monooleate liquid crystalline phases used in drug delivery systems. Int. J. Pharm. 2015, 478, 569–587. [Google Scholar] [CrossRef] [PubMed]
- Hyde, S.T.; Andersson, S.; Ericsson, B.; Larsson, K. A cubic structure consisting of a lipid bilayer forming an infinite periodic minimum surface of the gyroid type in the glycerolmonooleat-water system. Z. Kristallogr. 1985, 168, 213–219. [Google Scholar]
- Briggs, J.; Chung, H.; Caffrey, M. The Temperature-Composition Phase Diagram and Mesophase Structure Characterization of the Monoolein/Water System. J. Phys. II 1996, 6, 723–751. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Caffrey, M. The phase diagram of the monoolein/water system: Metastability and equilibrium aspects. Biomaterials 2000, 21, 223–234. [Google Scholar] [CrossRef]
- Tenchov, B.; Koynova, R. Cubic phases in phosphatidylethanolamine dispersions: Formation, stability and phase transitions. Chem. Phys. Lipids 2017, 208, 65–74. [Google Scholar] [CrossRef]
- Borné, J.; Nylander, T.; Khan, A. Vesicle formation and other structures in aqueous dispersions of monoolein and sodium oleate. J. Colloid Interface Sci. 2003, 257, 310–320. [Google Scholar] [CrossRef]
- Cong, J.Y.T.; Mulet, X.; Boyd, B.J.; Drummond, C.J. Steric Stabilizers for Cubic Phase Lyotropic Liquid Crystal Nanodispersions (Cubosomes). Adv. Planar Lipid Bilayers Liposomes 2015, 21, 131–187. [Google Scholar]
- Borné, J.; Nylander, T.; Khan, A. Phase Behavior and Aggregate Formation for the Aqueous Monoolein System Mixed with Sodium Oleate and Oleic Acid. Langmuir 2001, 17, 7742–7751. [Google Scholar] [CrossRef]
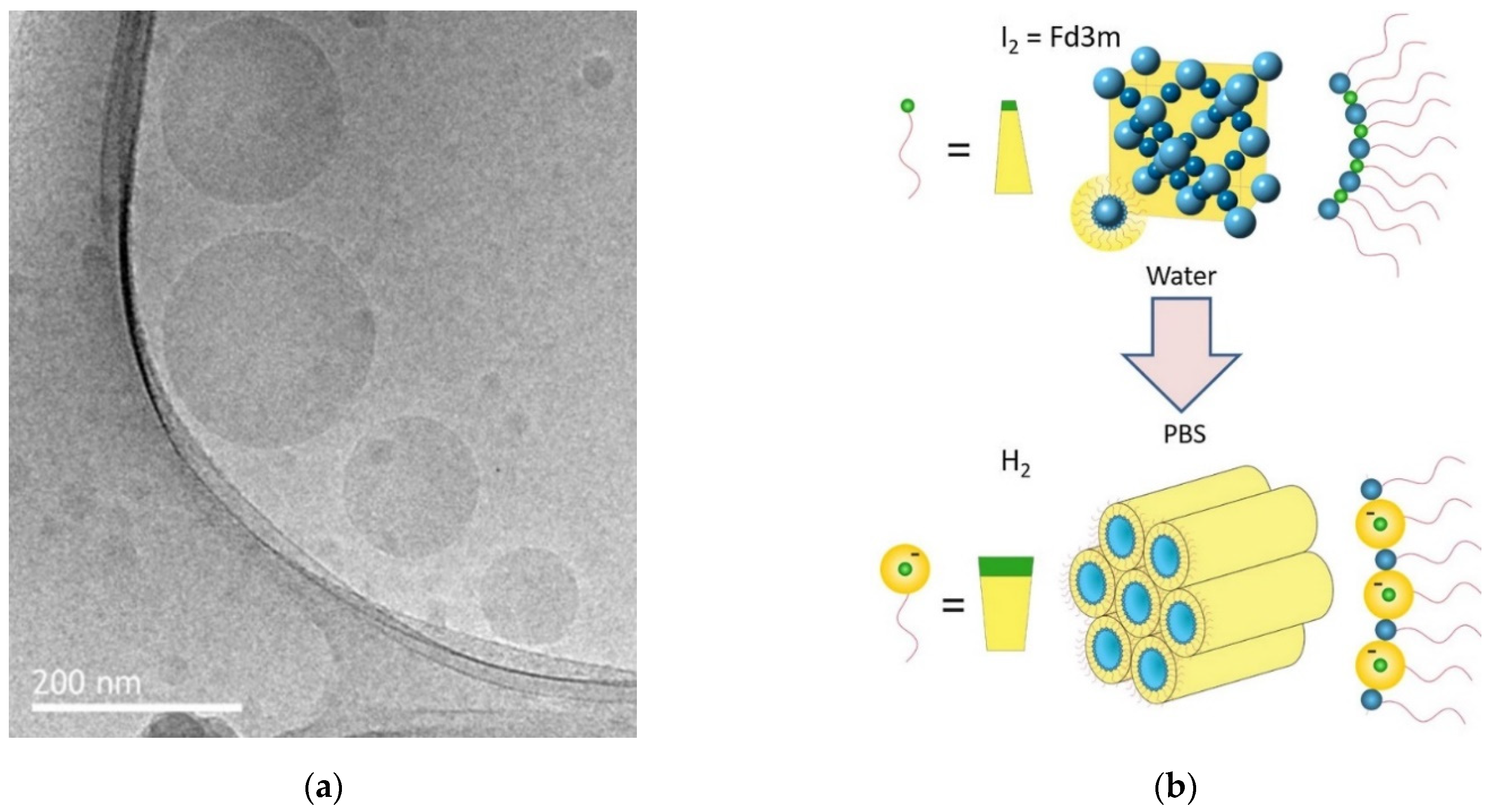
- Fong, C.; Zhai, J.; Drummond, C.J.; Tran, N. Micellar Fd3m cubosomes from monoolein—Long chain unsaturated fatty acid mixtures: Stability on temperaure and pH response. J. Colloid Interface Sci. 2020, 566, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Teshigawara, T.; Sugita, A.; Leesajakul, W.; Taniguchi, A.; Kamo, T.; Matsuoka, H.; Handa, T. Dispersions of Liquid Crystalline Phases of the Monoolein/Oleic Acid/Pluronic F127 System. Langmuir 2002, 18, 9283–9288. [Google Scholar] [CrossRef]
- Marsh, D. (Ed.) Phospholipids. In Handbook of Lipid Bilayers, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2013; Section II, subsection II.6; pp. 259–269. [Google Scholar]
- Arvidson, G.; Brentel, I.; Khan, A.; Lindblom, G.; Fontell, K. Phase equilibria in four lysophosphatidylcholine/water systems. Exceptional behavior of 1-palmitoyl-glycerophosphocholine. Eur. J. Biochem. 1985, 152, 753–759. [Google Scholar] [CrossRef]
- Inoue, K.; Suzuki, K.; Nojima, S. Morphology of Lipid Micelles Containing Lysolecithin. J. Biochem. 1977, 81, 1097–1106. [Google Scholar] [CrossRef]
- Mattai, J.; Shipley, G.G. The kinetics of formation and structure of the low-temperature phase of 1-stearoyl-lysophosphatidylcholine. Biochim. Biophys. Acta 1986, 859, 257–265. [Google Scholar] [CrossRef]
- Tristram-Nagle, S.; Petrache, H.I.; Nagle, J.F. Strucure and Interactions of Fully Hydrated Dioleoylphosphatidylcholine Bilayers. Biophys. J. 1998, 75, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Wiener, M.C.; White, S.H. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. III: Complete structure. Biophys. J. 1992, 61, 434–447. [Google Scholar] [CrossRef] [Green Version]
- Bergenståhl, B.A.; Stenius, P. Phase Diagrams of Dioleoylphosphatidylcholine with Formamide, Methylformamide, and Dimethylformamide. J. Phys. Chem. 1987, 91, 5944–5948. [Google Scholar] [CrossRef]
- Hoffmann, I.; Michel, R.; Sharp, M.; Holderer, O.; Appavou, M.-S.; Polzer, F.; Farago, B.; Gradzielski, M. Softening of phospholipid membranes by the adhesion of silica nanoparticles—As seen by neutron spin-echo (NSE). Nanoscale 2014, 6, 6945–6952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios, L.E.; Wang, T. Egg-Yolk Lipid Fractionation and Lecithin Characterization. J. Am. Oil Chem. Soc. 2005, 82, 571–578. [Google Scholar] [CrossRef]
- Kiełbowicz, G.; Gładkowski, W.; Chojnacka, A.; Wawrzeńczyk, C. A simple method for positional analysis of phosphatidylcholine. Food Chem. 2012, 135, 2542–2548. [Google Scholar] [CrossRef] [PubMed]
- Small, D. Phase equilibria and structure of dry and hydrated egg lecithin. J. Lipid Res. 1967, 8, 551–557. [Google Scholar] [CrossRef]
- Bangham, A.D.; Horn, R.W. Negative Staining of Phospholipids and their Structural Modification by Surface-active Agents as observed in the Electron Microsope. J. Mol. Biol. 1964, 8, 660–668. [Google Scholar] [CrossRef]
- Holzer, M.; Bernert, S.; Momm, J.; Schubert, R. Preparative size exclusion chromatography combined with detergent removal as a versatile tool to prepare unilamellar and spherical liposomes of highly uniform size distribution. J. Chromatogr. A 2009, 1216, 5838–5848. [Google Scholar] [CrossRef] [PubMed]
- Bergenståhl, B.; Fontell, K. Phase equilibria in the system soybean lecithin/water. Progr. Colloid Polym. Sci. 1983, 68, 48–52. [Google Scholar]
- Lynch, D.V.; Steponkus, P.L. Lyotropic phase behavior of unsaturated phosphatidylcholine species: Relevance to the mechanism of plasma membrane destabilization and freezing injury. Biochim. Biophys. Acta 1989, 984, 267–272. [Google Scholar] [CrossRef]
- Koster, K.L.; Webb, M.S.; Bryant, G.; Lynch, D.V. Interactions between soluble sugars and POPC (1-palmitoyl-2-oleoyphosphatidylcholine) during dehydration: Vitrification of sugars alters the phase behavior of the phospholipid. Biochim. Biophys. Acta 1994, 1193, 143–150. [Google Scholar] [CrossRef]
- Kučerka, N.; Tristram-Nagle, S.; Nagle, J.F. Structure of Fully Hydrated Fluid Phase Lipid Bilayers with Monounsaturated Chains. J. Membr. Biol. 2005, 208, 193–202. [Google Scholar] [CrossRef]
- Koynova, R.; Caffrey, M. Phases and phase transitions of the phosphatidylcholines. Biochim. Biophys. Acta 1998, 1376, 91–145. [Google Scholar] [CrossRef]
- O’Neill, S.D.; Leopold, A.D. An Assessment of Phase Transitions in Soybean Membranes. Plant Physiol. 1982, 70, 1405–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, P.G.; Gunstone, F.D. Hydrocarbon Chain Packing and Molecular Motion in Phospholipid Bilayers Formed from Unsaturated Lecithins. Synthesis and Properties of Sixteen Positional Isomers of 1,2-Dioctadecenoyl-sn-glycero-3-phosphorylcholine. J. Biol. Chem. 1975, 250, 4470–4476. [Google Scholar] [CrossRef]
- Marsh, D. Thermodynamic Analysis of Chain-Melting Transition Temperatures for Monounsaturated Phospholipid Membranes: Dependence on cis-Monoenoic Double Bond Position. Biophys. J. 1999, 77, 953–963. [Google Scholar] [CrossRef] [Green Version]
- Ipsen, J.H.; Mouritsen, O.G.; Zuckermann, M.J. Theory of thermal anomalies in the specific heat of lipid bilayers containing cholesterol. Biophys. J. 1989, 56, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Tieleman, D.P.; Berendsen, H.J.C. Molecular dynamics simulations of a fully hydrated dipalmitoylphosphatidylcholine bilayer with different macroscopic boundary conditions and parameters. J. Chem. Phys. 1996, 105, 4871–4880. [Google Scholar] [CrossRef] [Green Version]
- Rappolt, M.; Pabst, G.; Rapp, G.; Kriechbaum, M.; Amenitsch, H.; Krenn, C.; Bernstorff, S.; Laggner, P. New evidence for gel-liquid crystalline phase coexistence in the ripple phase of phosphatidylcholines. Eur. Biophys. J. 2000, 29, 125–133. [Google Scholar] [CrossRef] [PubMed]
- de Vries, A.H.; Yefimov, S.; Mark, A.E.; Marrink, S.J. Molecular structure of the lecithin ripple phase. Proc. Natl. Acad. Sci. USA 2005, 102, 5392–5396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Jastimi, R.; Edwards, K.; Lafleur, M. Characterization of Permeability and Morphological Perturbations Induced by Nisin on Phosphatidylcholine Membranes. Biophys. J. 1999, 77, 842–852. [Google Scholar] [CrossRef] [Green Version]
- Farkuh, L.; Hennies, P.T.; Nunes, C.; Reis, S.; Barreiros, L.; Segundo, M.A.; Oseliero Filho, P.L.; Oliveira, C.L.P.; Cassago, A.; Portugal, R.V.; et al. Characterization of phospholipid vesicles containing lauric acid: Physicochemical basis for process and product development. Heliyon 2019, 5, e02648. [Google Scholar] [CrossRef] [Green Version]
- Matviykiv, S.; Deyhle, H.; Kohlbrecher, J.; Neuhaus, N.; Zumbuehl, A.; Müller, B. Small-Angle Neutron Scattering Study of Temperature-Induced Structural Changes in Liposomes. Langmuir 2019, 35, 11210–11216. [Google Scholar] [CrossRef] [PubMed]
- Doskocz, J.; Dałek, P.; Foryś, A.; Trzebicka, B.; Przybyło, M.; Mesarec, L.; Iglič, A.; Langner, M. The effect of lipid phase on liposome stability upon exposure to the mechanical stress. Biochim. Biophys. Acta 2020, 1862, 183361. [Google Scholar] [CrossRef] [PubMed]
- Linblom, G.; Rilfors, L.; Hauksson, J.B.; Brentel, I.; Sjölund, M.; Bergenståhl, B. Effect of Head-Group Structure and Counterion Condensation on Phase Equilibria in Anionic Phospholipid-Water Systems Studied by 2H, 23Na, and 31P NMR and X-ray Diffraction. Biochemistry 1991, 30, 10938–10948. [Google Scholar] [CrossRef] [PubMed]
- Tocanne, J.-F.; Teissié, J. Ionization of phospholipids and phospholipid-supported interfacial lateral diffusion of protons in membrane model systems. Biochim. Biophys. Acta 1990, 1031, 111–142. [Google Scholar] [CrossRef]
- Faraudo, J.; Travesset, A. Phosphatidic Acid Domains in Membranes. Effect of Divalent Counterions. Biophys. J. 2007, 92, 2806–2818. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Cathcart, G.; Vidalis, A.S.; Allen, H.C. Cation effects on phosphatidic acid monolayers at various pH conditions. Chem. Phys. Lipids 2016, 200, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Hauser, H. Mechanism of spontaneous vesiculation. Proc. Natl. Acad. Sci. USA 1989, 86, 5351–5355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, M.; Fukuda, M.; Kudo, T.; Matsuzaki, N.; Azuma, T.; Sekine, K.; Endo, H.; Handa, T. Flip-Flop of Phospholipids in Vesicles: Kinetic Analysis with Time-Resolved Small-Angle Neutron Scattering. J. Phys. Chem. B 2009, 113, 6745–6748. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, M.M.; Leikin, S.; Rand, R.P. Bending, Hydration and Interstitial Energies Quantitatively Account for the Hexagonal-Lamellar-Hexagonal Reentrant Phase Transition in Dioleoylphosphatidylethanolamine. Biophys. J. 1994, 67, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Shyamsunder, E.; Gruner, S.M.; Tate, M.W.; Turner, D.C.; So, P.T.C.; Tilcock, C.P.S. Observation of Inverted Cubic Phase in Hydrated Dioleoylphosphatidylethanolamine Membranes. Biochemistry 1988, 27, 2332–2336. [Google Scholar] [CrossRef] [PubMed]
- Erbes, J.; Czeslik, C.; Hahn, W.; Winter, R.; Rappolt, M.; Rapp, G. On the Existence of Bicontinuous Cubic Phases in Dioleoylphosphatidylethanolamine. Ber. Bunsenges. Phys. Chem. 1994, 98, 1287–1293. [Google Scholar] [CrossRef]
- Gawrisch, K.; Parsegain, V.A.; Hajduk, D.A.; Tate, M.W.; Gruner, S.M.; Fuller, N.L.; Rand, R.P. Energetics of the Hexagonal-Lamellar-Hexagonal-Phase Transition Sequence in Dioleoylphosphatidylethanolamine Membranes. Biochemistry 1992, 31, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Cistola, D.P.; Small, D.M. On Micelle Formation and Phase Separation. J. Am. Chem. Soc. 1990, 112, 3214–3215. [Google Scholar] [CrossRef]
- Romsted, L. Introduction to Surfactant Self-Assembly. In Supramolecular Chemitry: From Molecules to Nanomaterials; Gale, P.A., Steed, J.W., Eds.; Wiley: New York, NY, USA, 2012; Volume 1, 23p. [Google Scholar]
- Israelachvili, J.N.; Mitchell, D.J.; Ninham, B.W. Theory of self-assembly of lipid bilayers and vesicles. Biochim. Biophys. Acta 1977, 470, 185–201. [Google Scholar] [CrossRef]
- Namani, T.; Walde, P. From Decanoate Micelles to Decanoic Acid/Dodecylbenzenesulfonate Vesicles. Langmuir 2005, 21, 6210–6219. [Google Scholar] [CrossRef]
- Smith, R.; Tanford, C. The Critical Micelle Concentration of L-α-dipalmitoylphosphatidylcholine in Water and Water/methanol Solutions. J. Mol. Biol. 1972, 67, 75–83. [Google Scholar] [CrossRef]
- Buboltz, J.T.; Feigenson, G.W. Phospholipid Solubility Determined by Equilibrium Distribution between Surface and Bulk Phase. Langmuir 2005, 21, 6296–6301. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, R.G. The Aqueous Phase Behavior of Surfactants; Academic Press: London, UK, 1994. [Google Scholar]
- Alfutimie, A.; Curtis, R.; Tiddy, G.J.T. Lyotropic Surfactant Liquid Crystals: Micellar Systems. In Handbook of Liquid Crystals, 2nd ed.; Goodby, J.W., Collings, P.J., Kato, T., Tschierske, C., Gleeson, H.J., Rynes, P., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; Volume 6, Chapter 12; pp. 1–44. [Google Scholar]
- Kékicheff, P.; Grabielle-Mandelmont, C.; Ollivon, M. Phase Diagram of Sodium Dodecyl Sulfate-Water System. 1. A Calorimetric Study. J. Colloid Interface Sci. 1989, 131, 112–132. [Google Scholar] [CrossRef]
- Varade, D.; Aramaki, K.; Stubenrauch, C. Phase diagrams of water–alkyltrimethylammonium bromide systems. Colloids Surf. A 2008, 315, 205–209. [Google Scholar] [CrossRef]
- Corcoran, J.; Fuller, S.; Rahman, A.; Shinde, N.; Tiddy, G.J.T.; Attard, G.S. Amphitropic Liquid Crystals. Part 1.—Effect of a Thermotropic Mesogen on Lyotropic Mesomorphism, and of a Surfactant on Thermotropic Mesomorphism. The C16EO8–5-CB−Water System. J. Mater. Chem. 1992, 2, 695–702. [Google Scholar] [CrossRef]
- Strey, R.; Schomäcker, R.; Roux, D.; Nallet, F.; Olsson, U. Dilute Lamellar and L3 Phases in the Binary Water–C12E5 System. J. Chem. Soc. Faraday Trans. 1990, 86, 2253–2261. [Google Scholar] [CrossRef]
- Riaz, M.K.; Riaz, M.A.; Zhang, X.; Lin, C.; Wong, K.H.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Surface Functionalization and Targeting Strategies of Liposomes in Solid Tumor Therapy: A Review. Int. J. Mol. Sci. 2018, 19, 195. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.S.; Hussein, S.A.; Ali, A.H.; Korma, S.A.; Lipeng, Q.; Jinghua, C. Liposome: Composition, characterisation, preparation, and recent innovation in clinical applications. J. Drug Target. 2019, 27, 742–761. [Google Scholar] [CrossRef]
- Filipczak, N.; Pan, J.; Yalamarty, S.S.K.; Torchilin, V.P. Recent advancements in liposome technology. Adv. Drug Deliv. Rev. 2020, 156, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Crommelin, D.J.A.; van Hoogevest, P.; Stom, G. The role of liposomes in clinical nanomedicine development. What now? Now what? J. Control. Release 2020, 318, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Barriga, H.M.G.; Holme, M.N.; Stevens, M.M. Cubosomes: The Next Generation of Smart Lipid Nanoparticles? Angew. Chem. Int. Ed. 2019, 58, 2958–2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barauskas, J.; Johnsson, M.; Joabsson, F.; Tiberg, F. Cubic Phase Nanoparticles (Cubosomes): Principles for Controlling Size, Structure, and Stability. Langmuir 2005, 21, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Barauskas, J.; Johnsson, M.; Tiberg, F. Self-Assembled Lipid Superstructures: Beyond Vesicles and Liposomes. Nano Lett. 2005, 5, 1615–1619. [Google Scholar] [CrossRef]
- Larsson, K.; Quinn, P.; Sato, K.; Tiberg, F. Lipids: Structure, Physical Properties and Functionality; The Oily Press, PJ Barnes & Associates: Bridgwater, UK, 2006. [Google Scholar]
- Mertins, O.; Mathews, P.D.; Angelova, A. Advances in the Design of pH-Sensitive Cubosome Liquid Crystalline Nanocarriers for Drug Delivery Applications. Nanomaterials 2020, 10, 963. [Google Scholar] [CrossRef]
- Azmi, I.D.M.; Moghimi, S.M.; Yaghmur, A. Cubosomes and hexosomes as versatile platforms for drug delivery. Ther. Deliv. 2015, 6, 1347–1364. [Google Scholar] [CrossRef] [PubMed]
- Amar-Yuli, I.; Wachtel, E.; Ben Shoshan, E.; Danino, D.; Aserin, A.; Garti, N. Hexosome and Hexagonal Phases Mediated by Hydration and Polymeric Stabilizer. Langmuir 2007, 23, 3637–3645. [Google Scholar] [CrossRef]
- Conn, C.E.; Seddon, J.M. Nonlamellar Lipid Aggregates. In Liposomes, Lipid Bilayers and Model Membranes. From Basic Research to Applications; Pabst, G., Kučerka, N., Nieh, M.-P., Katsaras, J., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2014; Chapter 2; pp. 31–47. [Google Scholar]
- Treyer, M.; Walde, P.; Oberholzer, T. Permeability Enhancement of Lipid Vesicles to Nucleotides by Use of Sodium Cholate: Basic Studies and Application to an Enzyme-Catalyzed Reaction Occurring inside the Vesicles. Langmuir 2002, 18, 1043–1050. [Google Scholar] [CrossRef]
- Heerklotz, H. Interactions of surfactants with lipid membranes. Q. Rev. Biophys. 2008, 41, 205–264. [Google Scholar] [CrossRef] [PubMed]
- Heerklotz, H.; Blume, A. Detergent Interactions with Lipid Bilayers and Membrane Proteins. In Comprehensive Biophysics; Egelman, E.H., Ed.; Academic Press, Elsevier: Amsterdam, The Netherlands, 2012; Volume 5, pp. 63–91. [Google Scholar]
- Lichtenberg, D.; Ahyayauch, H.; Goñi, F.M. The Mechanism of Detergent Solubilization of Lipid Bilayers. Biophys. J. 2013, 105, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arouri, A.; Mouritsen, O.G. Membrane-perturbing effect of fatty acids and lysolipids. Prog. Lipid Res. 2013, 52, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Motlaq, V.F.; Ortega-Holmberg, M.; Edwards, K.; Gedda, L.; Lyngsø, J.; Pedersen, J.S.; Bergström, L.M. Investigation of the enhanced ability of bile salt surfactants to solubilize phospholipid bilayers and form mixed micelles. Soft Matter 2021, 17, 7769–7780. [Google Scholar] [CrossRef]
- Grit, M.; Crommelin, D.J.A. The effect of aging on the physical stability of liposome dispersions. Chem. Phys. Lipids 1992, 62, 113–122. [Google Scholar] [CrossRef]
- Sabín, J.; Prieto, G.; Ruso, J.M.; Hildago-Álvarez, R.; Sarimento, F. Size ad stability of liposomes: A possible role of hydration and osmotic forces. Eur. Phys. J. E 2006, 20, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Walde, P.; Koine, N.; Ichikawa, S.; Ishikawa, T.; Nagahama, R.; Ishihara, T.; Tsujii, T.; Shudou, M.; Omokawa, Y.; et al. Temperature-Sensitive Nonionic Vesicles Prepared from Span 80 (Sorbitan Monooleate). Langmuir 2008, 24, 10762–10770. [Google Scholar] [CrossRef]
- Ong, S.G.M.; Chitneni, M.; Lee, K.S.; Ming, L.C.; Yuen, K.H. Evaluation of Extrusion Technique for Nanosizing Liposomes. Pharmaceutics 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Kanásova, M.; Nesměrák, K. Systematic reviews of liposome’s characterization methods. Monatsh. Chem. 2017, 148, 1581–1593. [Google Scholar] [CrossRef]
- Fan, Y.; Marioli, M.; Zhang, K. Analytical characterization of liposomes and other lipid nanoparticles for drug delivery. J. Pharm. Biomed. Anal. 2021, 192, 113642. [Google Scholar] [CrossRef] [PubMed]
- Rand, P.R.; Fuller, N.; Parsegian, V.A.; Rau, D.C. Variation in Hydration Forces between Neutral Phospholipid Bilayers: Evidence for Hydration Attraction. Biochemistry 1988, 27, 7711–7722. [Google Scholar] [CrossRef]
- Smirnova, Y.G.; Aeffner, S.; Risselada, H.J.; Salditt, T.; Marrink, S.J.; Müller, M.; Knecht, V. Interbilayer repulsion forces between tension-free lipid bilayers from simulation. Soft Matter 2013, 9, 10705–10718. [Google Scholar] [CrossRef] [Green Version]
- New, R.R.C. (Ed.) Preparation of liposomes. In Liposomes. A practical Approach; Oxford University Press: Oxford, UK, 1990; Chapter 2; pp. 33–104. [Google Scholar]
- Zuidam, N.J.; van Winden, E.; de Vrueh, R.; Crommelin, D.J.A. Stability, storage, and sterilization of liposomes. In Liposomes. A Practical Approach, 2nd ed.; Torchilin, V., Weissig, V., Eds.; Oxford University Press: Oxford, UK, 2003; Chapter 5; pp. 149–165. [Google Scholar]
- Crommelin, D.J.A. Influence of Lipid Composition and Ionic Strength on the Physical Stability of Liposomes. J. Pharm. Sci. 1984, 73, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Carrión, F.J.; de la Maza, A.; Parra, J.L. The Influence of Ionic Strength and Lipid Bilayer Charge on the Stability of Liposomes. J. Colloid Interface Sci. 1994, 164, 78–87. [Google Scholar] [CrossRef]
- du Plessis, J.; Ramachandran, C.; Weiner, N.; Müller, D.G. The influence of lipid composition and lamellarity of liposomes on the physical stability of liposomes upon storage. Int. J. Pharm. 1996, 127, 273–278. [Google Scholar] [CrossRef]
- Armengo, X.; Estelrich, J. Physical stability of different liposome compositions obtained by extrusion method. J. Microencapsulation 1995, 12, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Traïkia, M.; Warschawski, D.E.; Recouvreur, M.; Cartaud, J.; Devaux, P.F. Formation of unilamellar vesicles by repetitive freeze-thaw cycles: Characterization by electron microsocpy and 31P-nuclear magnetic resonance. Eur. Biophys. J. 2000, 29, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Heurtault, B.; Saulnier, P.; Pech, B.; Proust, J.-E.; Benoit, J.-P. Physico-chemical stability of colloidal lipid particles. Biomaterials 2003, 24, 4283–4300. [Google Scholar] [CrossRef]
- Nielsson, C.; Østergaard, J.; Weng Larsen, S.; Larsen, C.; Urtti, A.; Yaghmur, A. PEGylation of Phytantriol-Based Lyotropic Liquid Crystalline Particles—The Effect of Lipid Composition, PEG Chain Length, and Temperature on the Internal Nanostructure. Langmuir 2014, 30, 6398–6407. [Google Scholar] [CrossRef] [PubMed]
- Kronberg, B.; Dahlman, A.; Carlfors, J.; Karlsson, J.; Artursson, P. Preparation and Evaluation of Sterically Stabilized Liposomes: Colloidal Stability, Serum Stability, Macrophage Uptake, and Toxicity. J. Pharm. Sci. 1990, 79, 667–671. [Google Scholar] [CrossRef]
- Allen, T.M.; Hansen, C.; Martin, F.; Redemann, C.; Yau-Young, A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged half-lives in vivo. Biochim. Biophys. Acta 1991, 1066, 29–36. [Google Scholar] [CrossRef]
- Lasic, D.D.; Needham, D. The “Stealth” Liposome: A Prototypical Biomaterial. Chem. Rev. 1995, 95, 2601–2628. [Google Scholar] [CrossRef]
- Čeh, B.; Winterhalter, M.; Frederik, P.M.; Vallner, J.J.; Lasic, D.D. Stealth® liposomes: From theory to product. Adv. Drug Deliv. Rev. 1997, 24, 165–177. [Google Scholar]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Li, M.; Jiang, S.; Simon, J.; Passlick, D.; Frey, M.-L.; Wagner, M.; Mailänder, V.; Crespy, D.; Landfester, K. Brush Conformation of Polyethylene Glycol Determines the Stealth Effect of Nanocarriers in the Low Protein Adsorption Regime. Nano Lett. 2021, 21, 1591–1598. [Google Scholar] [CrossRef]
- Larsson, K. Cubic Lipid–Water Phases: Structure and Biomembrane Aspects. J. Phys. Chem. 1989, 89, 7304–7314. [Google Scholar] [CrossRef]
- Pitto-Barry, A.; Barry, N.P.E. Pluronic® block-copolymers in medicine: From chemical and biological versatility to rationalisation and clinical advances. Polym. Chem. 2014, 5, 3291–3297. [Google Scholar] [CrossRef] [Green Version]
- Laughlin, R. Equilibrium vesicles: Fact or fiction? Colloids Surf. A 1997, 128, 27–38. [Google Scholar] [CrossRef]
- Lasic, D.D.; Joannic, R.; Keller, B.C.; Frederik, P.M.; Auvray, L. Spontaneous vesiculation. Adv. Colloid Interface Sci. 2001, 89–90, 337–349. [Google Scholar] [CrossRef]
- Guida, V. Thermodynamics and kinetics of vesicles formation processes. Adv. Colloid Interface Sci. 2010, 161, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Käs, J.; Sackmann, E. Shape transitions and shape stability of giant phospholipid vesicles in pure water induced by area-to-volume changes. Biophys. J. 1991, 60, 825–844. [Google Scholar] [CrossRef]
- Lipowsky, R. The morphology of lipid membranes. Curr. Opin. Struct. Biol. 1995, 5, 531–540. [Google Scholar] [CrossRef]
- Mui, B.L.-S.; Döbereiner, H.-G.; Madden, T.D.; Cullis, P.R. Influence of Transbilayer Area Asymmetry on the Morphology of Large Unilamellar Vesicles. Biophys. J. 1995, 69, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Svetina, S.; Žekš, B. Shape Behavior of Lipid Vesicles as the Basis of Some Cellular Processes. Anat. Rec. 2002, 268, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, F.; Mueller, D.; Tanasescu, R.; Balog, S.; Ishikawa, T.; Brezesinski, G.; Zumbuehl, A. Vesicle Origami: Cuboid Phospholipid Vesicles Formed by Template-Free Self-Assembly. Angew. Chem. Int. Ed. 2017, 56, 6515–6518. [Google Scholar] [CrossRef] [PubMed]
- Lasic, D.D. Liposomes: From Physics to Applications; Elsevier: Amsterdam, The Netherlands, 1993. [Google Scholar]
- Lasch, J.; Weissig, V.; Brandl, M. Preparation of liposomes. In Liposomes. A Practical Approach, 2nd ed.; Torchilin, V., Weissig, V., Eds.; Oxford University Press: Oxford, UK, 2003; Chapter 1; pp. 3–29. [Google Scholar]
- Szoka, F., Jr.; Papahadjopoulos, D. Comparative Properties and Methods of Preparation of Lipid Vesicles (Liposomes). Annu. Rev. Biophys. Bioeng. 1980, 9, 467–508. [Google Scholar] [CrossRef] [PubMed]
- Walde, P.; Ichikawa, S. Enzymes inside lipid vesicles: Preparation, reactivity and applications. Biomol. Eng. 2001, 18, 143–177. [Google Scholar] [CrossRef]
- Jesorska, A.; Orwar, P. Liposomes: Technologies and Analytical Applications. Annu. Rev. Anal. Chem. 2008, 1, 801–832. [Google Scholar] [CrossRef] [PubMed]
- Walde, P. Preparation of Vesicles (Liposomes). In Encyclopedia of Nanoscience and Nanotechnology; Nalwa, H.S., Ed.; American Scientific Publishers: Stevenson Ranch, CA, USA, 2004; Volume 9, pp. 43–79. [Google Scholar]
- Walde, P.; Cosentino, K.; Engel, H.; Stano, P. Giant Vesicles: Preparations and Applications. ChemBioChem 2010, 11, 848–865. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Sunthar, P. A comprehensive review on recent preparation techniques of liposomes. J. Liposome Res. 2020, 30, 336–365. [Google Scholar] [CrossRef] [PubMed]
- Dimova, R.; Stano, P.; Marques, C.M.; Walde, P. Preparation methods for giant unilamellar vesicles. In The Giant Vesicle Book; Dimova, R., Marques, C.M., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2020; Chapter 1; pp. 3–20. [Google Scholar]
- Laouini, A.; Jaafar-Maalej, C.; Limayem-Blouza, I.; Sfar, S.; Charcosset, C.; Fessi, H. Preparation, Characterization and Applications of Liposomes: State of the Art. J. Colloid Sci. Biotechnol. 2012, 1, 147–168. [Google Scholar] [CrossRef]
- Olson, F.; Hunt, C.A.; Szoka, F.C.; Vail, J.J.; Papahadjopoulos, D. Preparation of Liposomes of Defined Size Distribution by Extrusion Through Polycarbonate Membranes. Biochim. Biophys. Acta 1979, 557, 9–23. [Google Scholar] [CrossRef]
- Mayer, L.D.; Hope, M.J.; Cullis, P.R. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta 1986, 858, 161–168. [Google Scholar] [CrossRef]
- Cullis, P.R.; Mayer, L.D.; Bally, M.B.; Madden, T.D.; Hope, M.J. Generating and loading of liposomal systems for drug-delivery applications. Adv. Drug Deliv. Rev. 1989, 3, 267–282. [Google Scholar] [CrossRef]
- MacDonald, R.C.; MacDonald, R.I.; Menco, B.P.M.; Takeshita, K.; Subbarao, N.K.; Hu, L.-R. Small-volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim. Biophys. Acta 1991, 1061, 297–303. [Google Scholar] [CrossRef]
- Hinna, A.; Steiniger, F.; Hupfeld, S.; Stein, P.; Kuntsche, J.; Brandl, M. Filter-extruded liposomes revisited: A study into size distributions and morphologies in relation to lipid-composition and process parameters. J. Liposome Res. 2016, 26, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Jousma, H.; Talsma, H.; Spies, F.; Joosten, J.G.H.; Junginger, H.E.; Crommelin, D.J.A. Characterization of liposomes. The influence of extrusion of multilamellar vesicles through polycarbonate membranes on particle size, particle size distribution and number of bilayers. Int. J. Pharm. 1987, 35, 263–274. [Google Scholar] [CrossRef]
- Hunter, D.G.; Frisken, B.J. Effect of Extrusion Pressure and Lipid Properties on the Size and Polydispersity of Lipid Vesicles. Biophys. J. 1998, 74, 2996–3002. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, M.; Joós, B. Extrusion of small vesicles through nanochannels: A model for experiments and molecular dynamics simulations. Phys. Rev. E 2012, 85, 051910. [Google Scholar] [CrossRef] [PubMed]
- Mui, B.; Chow, L.; Hope, M.J. Extrusion Technique to Generate Liposomes of Defined Size. Methods Enzymol. 2003, 367, 3–14. [Google Scholar] [PubMed]
- Scott, H.L.; Skinkle, A.; Kelley, E.G.; Waxham, M.N.; Levental, I.; Heberle, F.A. On the Mechanism of Bilayer Separation by Extrusion, or Why Your LUVs Are Not Really Unilamellar. Biophys. J. 2019, 117, 1381–1386. [Google Scholar] [CrossRef]
- Huang, C.-H. Studies on Phosphatidylcholine Vesicles. Formation and Physical Characteristics. Biochemistry 1969, 8, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, W. The size of bilayer Vesicles generated by sonication. Phys. Lett. 1974, 50A, 115–116. [Google Scholar] [CrossRef]
- Szoka, F., Jr.; Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaportion. Proc. Natl. Acad. Sci. USA 1978, 75, 4194–4198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumbuehl, O.; Weder, H.-G. Liposomes of controllable size in the range of 40 to 180 nm by defined dialysis of lipid/detergent mixed micelles. Biochim. Biophys. Acta 1981, 640, 252–262. [Google Scholar] [CrossRef]
- Ueno, M.; Tanford, C.; Reynolds, J.A. Phospholipid Vesicle Formation Using Nonionic Detergents with Low Monomer Solubility. Kinetic Factors Determine Vesicle Size and Permeability. Biochemistry 1984, 23, 3070–3076. [Google Scholar] [CrossRef]
- Schubert, R. Liposome Preparation by Detergent Removal. Methods Enzymol. 2003, 367, 46–70. [Google Scholar]
- Batzri, S.; Korn, E.D. Single bilayer liposomes prepared without sonication. Biochim. Biophys. Acta 1973, 298, 1015–1019. [Google Scholar] [CrossRef]
- Kremer, J.M.H.; Van der Esker, M.W.J.; Pathmamanoharan, C.; Wiersema, P.H. Vesicles of Variable Diameter Prepared by a Modified Injection Method. Biochemistry 1977, 16, 3932–3935. [Google Scholar] [CrossRef] [PubMed]
- Pons, M.; Foradada, M.; Estelrich, J. Liposomes obtained by the ethanol injection method. Int. J. Pharm. 1993, 95, 51–56. [Google Scholar] [CrossRef]
- Wagner, A.; Vorauer-Uhl, K.; Kreismayr, G.; Katinger, H. The crossflow injection technique: An improvement of the ethanol injection method. J. Liposome Res. 2002, 12, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Jaafar-Maalej, C.; Diab, R.; Andrieu, V.; Elaissari, A.; Fessi, H. Ethanol injection method for hydrophilic and lipophilic drug-loaded liposome preparation. J. Liposome Res. 2010, 20, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Gouda, A.; Sakr, O.S.; Nasr, M.; Sammour, O. Ethanol injection technique for liposome formulation: An insight into development, influencing factors, challenges and applications. J. Drug Deliv. Sci. Technol. 2021, 61, 102174. [Google Scholar] [CrossRef]
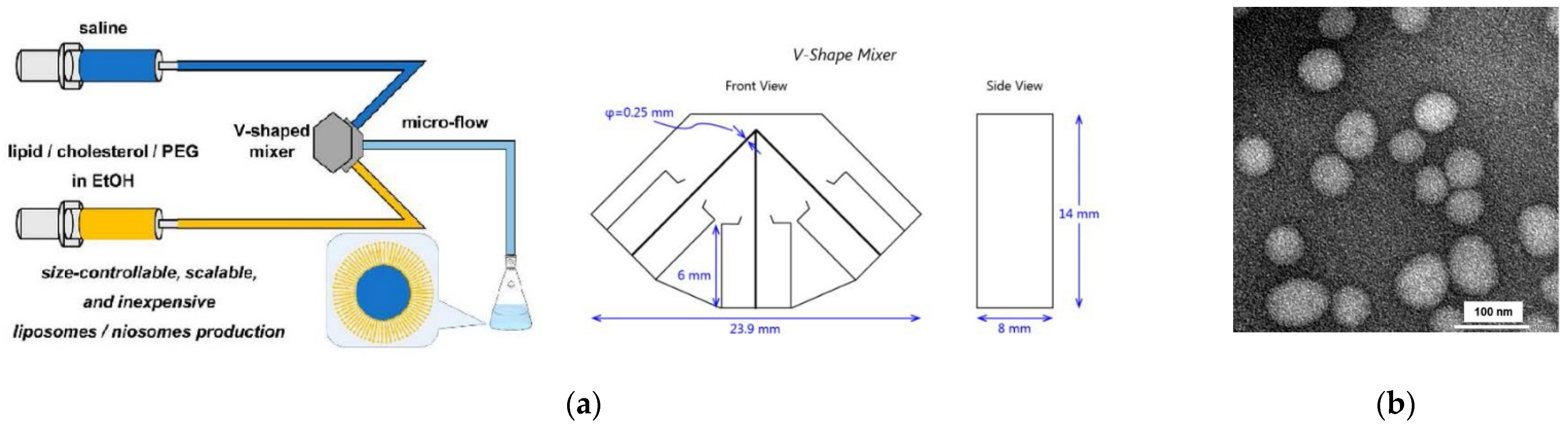
- Kawamura, J.; Kitamura, H.; Otake, Y.; Fuse, S.; Nakamura, H. Size-Controllable and Scalable Production of Liposomes Using a V-Shaped Mixer Micro-Flow Reactor. Org. Process Res. Dev. 2020, 24, 2122–2127. [Google Scholar] [CrossRef]
- Reeves, J.P.; Dowben, R.M. Formation and Properties of Thin-walled Phospholipid Vesicles. J. Cell. Physiol. 1969, 73, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Tsumoto, K.; Matsuo, H.; Tomita, M.; Yoshimura, T. Efficient formation of giant liposomes through the gentle hydration of phosphatidylcholine films doped with sugar. Colloids Surf. B 2009, 68, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.I.; Dimitrov, D.S. Liposome Electroformation. Faraday Discuss. Chem. Soc. 1986, 81, 303–311. [Google Scholar] [CrossRef]
- Angelova, M.I. Liposome Electroformation. In Giant Vesicles; Luisi, P.L., Walde, P., Eds.; Perspectives in Supramolecular Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2000; Volume 6, Chapter 3; pp. 27–43. [Google Scholar]
- Méléard, P.; Bagatolli, L.A.; Pott, T. Giant Unilamellar Vesicle Electroformation: From Lipid Mixtures to Native Membranes Under Physiological Conditions. Methods Enzymol. 2009, 465, 161–175. [Google Scholar] [PubMed]
- Shimanouchi, T.; Umakoshi, H.; Kuboi, R. Kinetic Study on Giant Vesicle Formation with Electroformation Method. Langmuir 2009, 25, 4835–4840. [Google Scholar] [CrossRef]
- Okumura, Y.; Shuuhei, O. Effect of Counter Electrode in Electroformation of Giant Vesicles. Membranes 2011, 1, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, X.; Ma, S.; Zhang, Y.; Han, X. Electroformation of giant unilamellar vesicles in saline solution. Colloids Surf. B 2016, 147, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Drabik, D.; Doskocz, J.; Przybyło, M. Effects of electroformation protocol parameters on quality of homogeneous GUV populations. Chem. Phys. Lipids 2018, 212, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Pautot, S.; Frisken, B.J.; Weitz, D.A. Engineering asymmetric vesicles. Proc. Natl. Acad. Sci. USA 2003, 100, 10718–10721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrara, P.; Stano, P.; Luisi, P.L. Giant Vesicles “Colonies”: A Model for Primitive Cell Communities. ChemBioChem 2012, 13, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Moga, A.; Yandrapalli, N.; Dimova, R.; Robinson, T. Optimization of the Inverted Emulsion Method for High-Yield Production of Biomimetic Giant Unilamellar Vesicles. ChemBioChem 2019, 20, 2674–2682. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Ding, J.; Xu, J.; Yomo, T. Giant Vesicles Produced with Phosphatidylcholines (PCs) and Phosphatidylethanolamines (PEs) by Water-in-Oil Inverted Emulsions. Life 2021, 11, 233. [Google Scholar] [CrossRef]
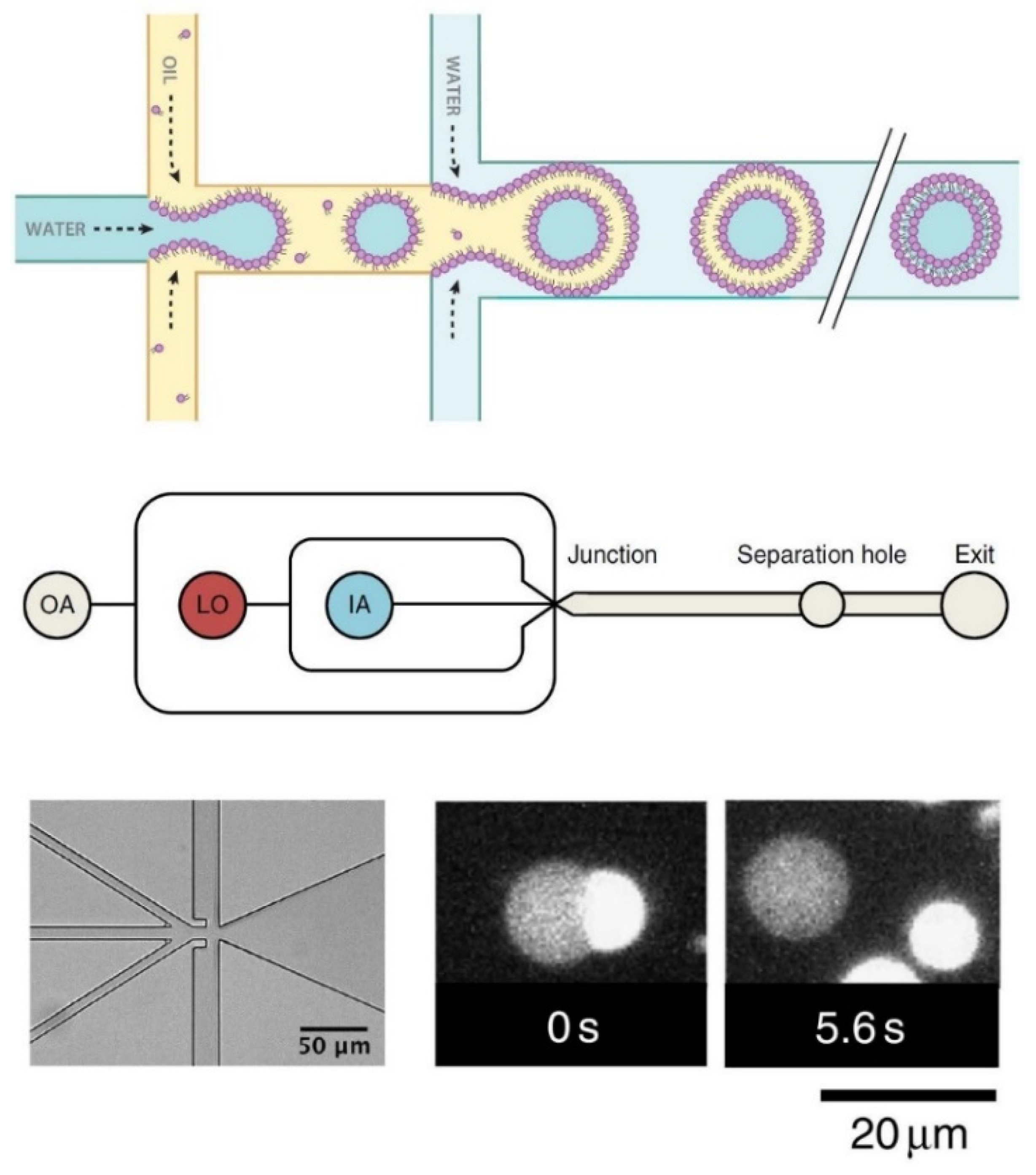
- Sugiura, S.; Kuroiwa, T.; Kagota, T.; Nakajima, M.; Sato, S.; Mukataka, S.; Walde, P.; Ichikawa, S. Novel Method for Obtaining Giant Vesicles from a Monodisperse Water-in-Oil Emulsion Prepared with a Microfluidic Device. Langmuir 2008, 24, 4581–4588. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, K.; Suzuki, H.; Takeuchi, S. Formation of Giant Lipid Vesiclelike Compartments from a Planar Lipid Membrane by a Pulsed Jet Flow. J. Am. Chem. Soc. 2007, 129, 12608–12609. [Google Scholar] [CrossRef] [PubMed]
- Stachowiak, J.C.; Richmond, D.L.; Li, T.H.; Liu, A.P.; Parekh, S.H.; Flecther, D.A. Unilamellar vesicle formation and encapsulation by microfluidic jetting. Proc. Natl. Acad. Sci. USA 2008, 105, 4697–4702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, K.; Kawano, R.; Osaki, T.; Akiysohi, K.; Takeuchi, S. Cell-sized asymmetric lipid vesicles facilitate the investigation of asymmetric membranes. Nat. Chem. 2016, 8, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Naeff, R. Feasibility of topical liposome drugs produced on an industrial scale. Adv. Drug Deliv. Rev. 1996, 18, 343–347. [Google Scholar] [CrossRef]
- Kriftner, R.W. Liposome Production: The Ethanol Injection Technique and the Development of the First Approved Liposome Dermatics. In Liposome Dermatics; Braun-Falco, O., Korting, H.C., Maibach, H.I., Eds.; Springer: Berlin, Germany, 1992; pp. 91–100. [Google Scholar]
- Jahn, A.; Vreeland, W.N.; DeVoe, D.L.; Locascio, L.E.; Gaitan, M. Microfluidic Directed Formation of Liposomes of Controlled Size. Langmuir 2007, 23, 6289–6293. [Google Scholar] [CrossRef] [PubMed]
- Jahn, A.; Stavis, S.M.; Hong, J.S.; Vreeland, W.N.; DeVoe, D.L.; Gaitan, M. Microfluidic Mixing and the Formation of Nanoscale Lipid Vesicles. ACS Nano 2010, 4, 2077–2087. [Google Scholar] [CrossRef] [PubMed]
- Jahn, A.; Lucas, F.; Wepf, R.A.; Dittrich, P.S. Freezing Continuous-Flow Self-Assembly in a Microfluidic Device: Toward Imaging of Liposome Formation. Langmuir 2013, 29, 1717–1723. [Google Scholar] [CrossRef]
- Bottaro, E.; Nastruzzi, C. “Off-the-shelf” microfluidic devices for the production of liposomes for drug delivery. Mater. Sci. Eng. C 2016, 64, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Malouff, T.D.; Seneviratne, D.S.; Ebner, D.K.; Stross, W.C.; Waddle, M.R.; Trifiletti, D.M.; Krishnan, S. Boron Neutron Capture Therapy: A Review of Clinical Applications. Front. Oncol. 2021, 11, 601820. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Liu, Z.; Simões, S.L.; Correia, A.; Rahikkala, A.; Seitsonen, J.; Ruokolainen, J.; Aguiar-Ricardo, A.; Santos, H.A.; Corvo, M.L. One-step microfluidics production of enzyme-loaded liposomes for the treatment of inflammatory diseases. Colloids Surf. B 2021, 199, 111556. [Google Scholar] [CrossRef] [PubMed]
- Keele, B.B., Jr.; McCord, J.M.; Fridovich, I. Further Characterization of Bovine Superoxide Dismutase and Its Isolation from Bovine Heart. J. Biol. Chem. 1971, 246, 2875–2880. [Google Scholar] [CrossRef]
- Hyslop, P.A.; Morel, B.; Sauerheber, R.D. Organization and Interaction of Cholesterol and Phosphatidylcholine in Model Bilayer Membranes. Biochemistry 1990, 29, 1025–1039. [Google Scholar] [CrossRef] [PubMed]
- Patil, Y.P.; Jadhav, S. Novel methods for liposome preparation. Chem. Phys. Lipids 2014, 177, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Santo, I.E.; Campardelli, R.; Albuquerque, E.C.; de Melo, S.V.; Della Porta, G.; Reverchon, E. Liposomes preparation using a supercritical fluid assisted continuous process. Chem. Eng. J. 2014, 249, 153–159. [Google Scholar] [CrossRef]
- Campardalli, R.; Trucillo, P.; Reverchon, E. A Supercritical Fluid-Based Process for the Production of Fluorescein-Loaded Liposomes. Ind. Eng. Chem. Res. 2016, 55, 5359–5365. [Google Scholar] [CrossRef]
- Bigazzi, W.; Penoy, N.; Evrard, B.; Piel, G. Supercritical fluid methods: An alternative to conventional methods to prepare liposomes. Chem. Eng. J. 2020, 383, 123106. [Google Scholar]
- Khadke, S.; Roces, C.B.; Donaghey, R.; Giacobbo, V.; Su, Y.; Perrie, Y. Scalable solvent-free production of liposomes. J. Pharm. Pharmacol. 2020, 72, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Leitgeb, M.; Knez, Ž.; Primožič, M. Sustainable technologies for liposome preparation. J. Supercrit. Fluids 2020, 165, 104984. [Google Scholar]
- Trucillo, P.; Reverchon, E. Production of PEG-coated liposomes using a continuous supercritical assisted process. J. Supercrit. Fluids 2021, 167, 105048. [Google Scholar] [CrossRef]
- Zhigaltsev, I.V.; Tam, Y.K.; Leung, A.K.K.; Cullis, P.R. Production of limit size nanoliposomal systems with potential utility as ultra-small drug delivery agents. J. Liposome Res. 2016, 26, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Ota, S.; Yoshizawa, S.; Takeuchi, S. Microfluidic Formation of Monodisperse, Cell-Sized, and Unilamellar Vesicles. Angew. Chem. Int. Ed. 2009, 48, 6533–6537. [Google Scholar] [CrossRef] [PubMed]
- Teh, S.-Y.; Khnouf, R.; Fan, H.; Lee, A.P. Stable, biocompatible lipid vesicle generation by solvent extraction-based droplet microfluidics. Biomicrofluidics 2011, 5, 044113. [Google Scholar] [CrossRef] [Green Version]
- Matosevic, S.; Paegel, B.M. Stepwise Synthesis of Giant Unilamellar Vesicles on a Microfluidic Assembly Line. J. Am. Chem. Soc. 2011, 133, 2798–2800. [Google Scholar] [CrossRef] [PubMed]
- van Swaay, D.; deMello, A. Microfluidic methods for forming liposomes. Lab Chip 2013, 13, 752–767. [Google Scholar] [CrossRef]
- Carugo, D.; Bottaro, E.; Owen, J.; Stride, E.; Nastruzzi, C. Liposome production by microfluidics: Potential and limiting factors. Sci. Rep. 2016, 6, 25876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, S.; Caspi, Y.; Meijering, A.E.C.; Dekker, C. Octanol-assisted liposome assembly on chip. Nat. Commun. 2016, 7, 10447. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, S.; Dekker, C. On-chip microfluidic production of cell-sized liposomes. Nat. Protoc. 2018, 13, 856–874. [Google Scholar] [CrossRef]
- Robinson, T. Microfluidics and giant vesicles: Creation, capture, and applications for biomembranes. In Multiresponsive Behavior of Biomembranes and Giant Vesicles. Biomembranes and Lipid Self-Assembly; Academic Press: London, UK, 2019; Volume 30, Chapter 3; pp. 271–315. [Google Scholar]
- Yandrapalli, N.; Seemann, T.; Robinson, T. On–Chip Inverted Emulsion Method for Fast Giant Vesicle Production, Handling, and Analysis. Micromachines 2020, 11, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaich, M.; Sobota, D.; Sleath, H.; Cam, J.; Keyser, U.F. Characterization of lipid composition and diffusivity in OLA generated vesicles. Biochim. Biophys. Acta, Biomembr. 2020, 1862, 183359. [Google Scholar] [CrossRef]
- Litschel, T.; Schwille, P. Protein Reconstitution Inside Giant Unilamellar Vesicles. Annu. Rev. Biophys. 2021, 50, 525–548. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, S.R.; Ohlinger, A.; Pfeiffer, T.; Urban, A.S.; Stefani, F.D.; Deak, A.; Lutich, A.A.; Feldmann, J. Membrane composition of jetted lipid vesicles: A Raman spectroscopy study. J. Biophotonics 2012, 5, 40–46. [Google Scholar] [CrossRef] [PubMed]
- New, R.R.C. (Ed.) Characterization of liposomes. In Liposomes. A practical Approach; Oxford University Press: Oxford, UK, 1990; Chapter 3; pp. 105–161. [Google Scholar]
- Zuidam, N.J.; de Vruh, R.; Crommelin, D.J.A. Characterization of liposomes. In Liposomes. A Practical Approach, 2nd ed.; Torchilin, V., Weissig, V., Eds.; Oxford University Press: Oxford, UK, 2003; Chapter 2; pp. 31–78. [Google Scholar]
- London, E. Membrane Structure-Function Insights from Asymmetric Lipid Vesicles. Acc. Chem. Res. 2019, 52, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; Thompson, T.E. Exchange and Flip-Flop of Dimyristoylphosphatidylcholine in Liquid-Crystalline, Gel, and Two-Component, Two-Phase Large Unilamellar Vesicles. Biochemistry 1990, 29, 1296–1303. [Google Scholar] [CrossRef]
- Gurtovenko, A.A.; Vattulainen, I. Molecular Mechanism for Lipid Flip-Flop. J. Phys. Chem. B 2007, 111, 13554–13559. [Google Scholar] [CrossRef] [Green Version]
- John, K.; Schreiber, S.; Kubelt, J.; Herrmann, A.; Müller, P. Transbilayer Movement of Phospholipids at the Main Phase Transition of Lipid Membranes: Implications for Rapid Flip. Biophys. J. 2002, 83, 3315–3323. [Google Scholar] [CrossRef] [Green Version]
- Marquardt, D.; Heberle, F.A.; Miti, T.; Eicher, B.; London, E.; Katsaras, J.; Pabst, G. 1H NMR Shows Phospholipid Flip-Flop in Gel and Fluid Bilayers. Langmuir 2017, 33, 3731–3741. [Google Scholar] [CrossRef] [PubMed]
- Hope, M.J.; Redelmeier, T.E.; Wong, K.F.; Rodrigueza, W.; Cullis, P.R. Phospholipid Asymmetry in Large Unilamellar Vesicles Induced by Transmembrane pH Gradients. Biochemistry 1989, 28, 4181–4187. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-T.; Megha; London, E. Preparation and Properties of Asymmetric Vesicles That Mimic Cell Membranes. Effect upon Lipid Raft Formation and Transmembrane Helix Orientation. J. Biol. Chem. 2009, 284, 6079–6092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiantia, S.; Schwille, P.; Klymchenko, A.S.; London, E. Asymmetric GUVs Prepared by MβCD-Mediated Lipid Exchange: An FCS Study. Biophys. J. 2011, 100, L01–L03. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.C.; Li, S.; Malmstadt, N. Microfluidic Fabrication of Asymmetric Giant Lipid Vesicles. ACS Appl. Mater. Interfaces 2011, 3, 1434–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doktorova, M.; Heberle, F.A.; Eicher, B.; Standaert, R.F.; Katsaras, J.; London, E.; Pabst, G.; Marquardt, D. Preparation of asymmetric phospholipid vesicles for use as cell membrane models. Nat. Protoc. 2018, 13, 2086–2101. [Google Scholar] [CrossRef]
- Markones, M.; Drechsler, C.; Kaiser, M.; Kalie, L.; Heerklotz, H.; Fiedler, S. Engineering Asymmetric Lipid Vesicles: Accurate and Convenient Control of the Outer Leaflet Lipid Composition. Langmuir 2018, 34, 1999–2005. [Google Scholar] [CrossRef]
- Li, B.; London, E. Preparation and Drug Entrapment Properties of Asymmetric Liposomes Containing Cationic and Anionic Lipids. Langmuir 2020, 36, 12521–12531. [Google Scholar] [CrossRef] [PubMed]
- Kisak, E.T.; Coldren, B.; Zasadzinski, J.A. Nanocompartments Enclosing Vesicles, Colloids, and Macromolecules via Interdigitated Lipid Bilayers. Langmuir 2002, 18, 284–288. [Google Scholar] [CrossRef]
- Giuliano, C.B.; Cvjetan, N.; Ayache, J.; Walde, P. Multivesicular Vesicles: Preparation and Applications. ChemSystemsChem 2021, 3, e2000049. [Google Scholar]
- Sunamoto, J.; Sato, T.; Hirota, M.; Fukushima, K.; Hiratani, K.; Hara, K. A newly developed immunoliposome—An egg phosphatidylcholine liposome coated with pullulan bearing both a cholesterol moiety and an IgMs fragment. Biochim. Biophys. Acta 1987, 898, 323–330. [Google Scholar] [CrossRef]
- Mastrobattista, E.; Koning, G.A.; Storm, G. Immunoliposomes for the targeted delivery of antitumor drugs. Adv. Drug Deliv. Rev. 1999, 40, 103–127. [Google Scholar] [CrossRef]
- Maruyama, K. PEG-Immunoliposome. Biosci. Rep. 2002, 22, 251–266. [Google Scholar] [CrossRef]
- Eloy, J.O.; Petrilli, R.; Trevizan, L.N.F.; Chorilli, M. Immunoliposomes: A review on functionalization strategies and targets for drug delivery. Colloids Surf. B 2017, 159, 454–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, M.; Zalba, S.; Garrido, M.J. Immunoliposomes in clinical oncology: State of the art and future perspective. J. Control. Release 2018, 275, 162–176. [Google Scholar] [CrossRef] [PubMed]
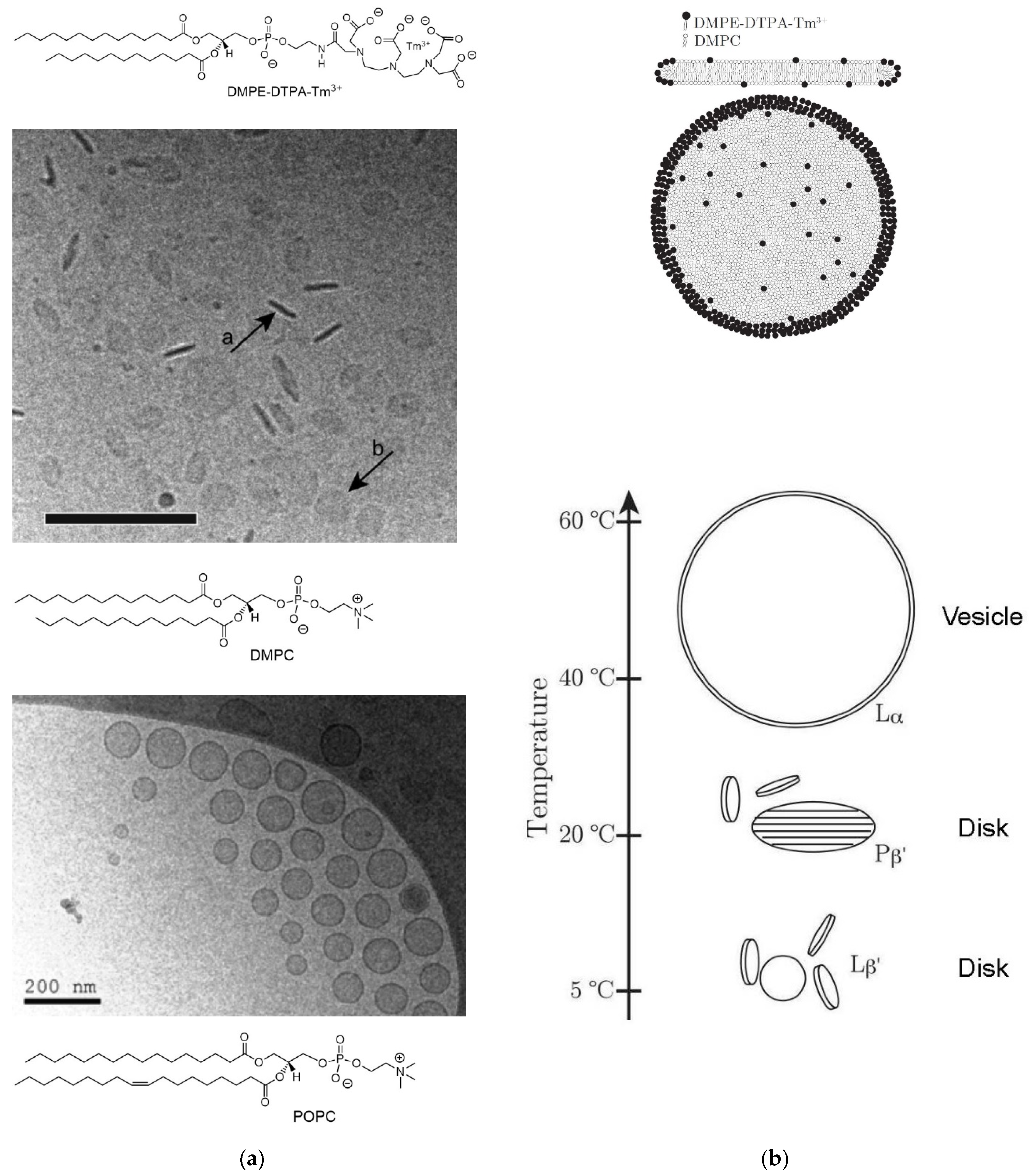
- Beck, P.; Liebi, M.; Kohlbrecher, J.; Ishikawa, T.; Rüegger, H.; Fischer, P.; Walde, P.; Windhab, E. Novel Type of Bicellar Disks from a Mixture of DMPC and DMPE-DTPA with Complexed Lanthanides. Langmuir 2010, 26, 5382–5387. [Google Scholar] [CrossRef] [PubMed]
- Liebi, M. Tailored Phospholipid Bicelles to Generate Magnetically Switchable Material. Ph.D. Thesis, ETH Zürich, Zürich, Switzerland, 2013. (no. 21048). [Google Scholar]
- Beck, P.; Liebi, M.; Kohlbrecher, J.; Ishikawa, T.; Rüegger, H.; Zepik, H.; Fischer, P.; Walde, P.; Windhab, E. Magnetic Field Alignable Domains in Phospholipid Vesicle Membranes Containing Lanthanides. J. Phys. Chem. B 2010, 114, 174–186. [Google Scholar] [CrossRef]
- Beck, P. Magnetic Field Assisted Biomaterials Processing. Ph.D. Thesis, ETH Zürich, Zürich, Switzerland, 2009. (no. 18292). [Google Scholar]
- Isabettini, S.; Massabni, S.; Kohlbrecher, J.; Schuler, L.D.; Walde, P.; Sturm, M.; Windhab, E.J.; Fischer, P.; Kuster, S. Understanding the Enhanced Magnetic Response of Aminocholesterol Doped Lanthanide-Ion-Chelating Phospholipid Bicelles. Langmuir 2017, 33, 8533–8544. [Google Scholar] [CrossRef] [PubMed]
- Sanders, C.R.; Schwonek, J.P. Characterization of Magnetically Orientable Bilayers in Mixtures of Dihexanoylphosphatidycholine and Dimyristoylphosphatidylcholine by Solid-State NMR. Biochemistry 1992, 31, 8898–8905. [Google Scholar] [CrossRef]
- Luchette, P.A.; Vetman, T.N.; Prosser, R.S.; Hancock, R.E.W.; Nieh, M.-P.; Glinka, C.J.; Krueger, S.; Katsaras, J. Morphology of fast-tumbling bicelles: A small angle neutron scattering and NMR study. Biochim. Biophys. Acta 2001, 1513, 83–94. [Google Scholar] [CrossRef]
- Triba, M.N.; Warschawski, D.E.; Devaux, P.F. Reinvestigation by Phosphorous NMR of Lipid Distribution in Bicelles. Biophys. J. 2005, 88, 1887–1901. [Google Scholar] [CrossRef] [Green Version]
- Suga, K.; Kitagawa, K.; Taguchi, S.; Okamoto, Y.; Umkoshi, H. Evaluation of Molecular Ordering in Bicelle Bilayer Membranes Based on Induced Circular Dichroism Spectra. Langmuir 2020, 36, 3242–3250. [Google Scholar] [CrossRef] [PubMed]
- Veatch, S.L.; Keller, S.L. Separation of Liquid Phases in Giant Vesicles of Ternary Mixtures of Phospholipids and Cholesterol. Biophys. J. 2003, 85, 3074–3083. [Google Scholar] [CrossRef] [Green Version]
- de Almeida, R.F.M.; Fedorov, A.; Prieto, M. Sphingomyelin/Phosphatidylcholine/Cholesterol Phase Diagram: Boundaries and Composition of Lipid Rafts. Biophys. J. 2003, 85, 2406–2416. [Google Scholar] [CrossRef] [Green Version]
- Veatch, S.L.; Polozov, I.V.; Gawrisch, K.; Keller, S.L. Liquid Domains in Vesicles Investigated by NMR and Fluorescence Microscopy. Biophys. J. 2004, 86, 2910–2922. [Google Scholar] [CrossRef] [Green Version]
- Polozov, I.V.; Gawrisch, K. Characterization of the Liquid-Ordered State by Proton MAS NMR. Biophys. J. 2006, 90, 2051–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- M’Baye, G.; Mély, Y.; Duportail, G.; Klymchenko, A.S. Liquid Ordered and Gel Phases of Lipid Bilayers: Fluorescent Probes Reveal Close Fluidity but Different Hydration. Biophys. J. 2008, 95, 1217–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feigenson, G.W. Phase diagrams and lipid domains in multicomponent lipid bilayer mixtures. Biochim. Biophys. Acta 2009, 1788, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Honerkamp-Smith, A.R.; Veatch, S.L.; Keller, S.L. An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes. Biochim. Biophys. Acta 2009, 1788, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konyakhina, T.M.; Wu, J.; Mastroianni, J.D.; Heberle, F.A.; Feigenson, G.W. Phase diagram of a 4-component lipid mixture: DSPC/DOPC/POPC/chol. Biochim. Biophys. Acta 2013, 1828, 2204–2214. [Google Scholar] [CrossRef] [Green Version]
- Suga, K.; Umakoshi, H. Detection of Nanosized Ordered Domains in DOPC/DPPC and DOPC/Ch Binary Lipid Mixture Systems of Large Unilamellar Vesicles Using TEMPO Quenching Method. Langmuir 2013, 29, 4830–4838. [Google Scholar] [CrossRef] [PubMed]
- Rheinstädter, M.C.; Mouritsen, O.G. Small-scale structure in fluid cholesterol-lipid bilayers. Curr. Opin. Colloid Interface Sci. 2013, 18, 440–447. [Google Scholar] [CrossRef]
- Klymchenko, A.S.; Kreder, R. Fluorescent Probes for Lipid Rafts: From Model Membranes to Living Cells. Chem. Biol. 2014, 21, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Feigenson, G.W. Pictures of the Substructure of Liquid-Ordered Domains. Biophys. J. 2015, 109, 854–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engberg, O.; Hautala, V.; Yasuda, T.; Dehio, H.; Murata, M.; Slotte, J.P.; Nyholm, T.K.M. The Affinity of Cholesterol for Different Phospholipids Affects Lateral Segregation in Bilayers. Biophys. J. 2016, 111, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cebecauer, M.; Amaro, M.; Jurkiewicz, P.; Sarmento, M.J.; Šachl, R.; Cwiklik, L.; Hof, M. Membrane Lipid Nanodomains. Chem. Rev. 2018, 118, 11259–11297. [Google Scholar] [CrossRef] [PubMed]
- Bolmatov, D.; Soloviov, D.; Zhernenkov, M.; Zav’yalov, D.; Mamontov, E.; Suvorov, A.; Cai, Y.Q.; Katsaras, J. Molecular picture of the transient nature of lipid rafts. Langmuir 2020, 36, 4887–4896. [Google Scholar] [CrossRef]
- Cornell, C.E.; Mileant, A.; Thakkar, N.; Lee, K.K.; Keller, S.L. Direct imaging of liquid domains in membranes by cryo-electron tomography. Proc. Natl. Acad. Sci. USA 2020, 117, 19713–19719. [Google Scholar] [CrossRef] [PubMed]
- Blosser, M.C.; Cornell, C.E.; Rayermann, S.P.; Keller, S.L. Phase diagrams and tie lines in giant unilamellar vesicles. In The Giant Vesicle Book; Dimova, R., Marques, C.M., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2020; Chapter 18; pp. 401–416. [Google Scholar]
- Feigenson, G.W. Phase behavior of lipid mixtures. Nat. Chem. Biol. 2006, 2, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Walde, P.; Blöchliger, E. Circular Dichroic Properties of Phosphatidylcholine Liposomes. Langmuir 1997, 13, 1668–1671. [Google Scholar] [CrossRef]
- Wagner, A.; Vorauer-Uhl, K.; Kreismayr, G.; Katinger, H. Enhanced protein loading into liposomes by the multiple crossflow injection technique. J. Liposome Res. 2002, 12, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, T.; Fujita, R.; Kobayashi, I.; Uemura, K.; Nakajima, M.; Sato, S.; Walde, P.; Ichikawa, S. Efficient Preparation of Giant Vesicles as Biomimetic Compartment Systems with High Entrapment Yields for Biomacromolecules. Chem. Biodiv. 2012, 9, 2453–2472. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.W.; Barchfeld, G.L. Encapsulation of Macromolecules by Lipid Vesicles under Simulated Prebiotic Conditions. J. Mol. Evol. 1982, 18, 203–206. [Google Scholar] [CrossRef]
- Kirby, C.; Gregoriadis, G. Dehydration-Rehydration Vesicles: A Simple Method for High Yield Drug Entrapment in Liposomes. Bio/Technology 1984, 2, 979–984. [Google Scholar] [CrossRef]
- Fenske, D.B.; Cullis, P.R. Medical Applications of Lipid Nanoparticles. In Liposomes, Lipid Bilayers and Model Membranes. From Basic Research to Applications; Pabst, G., Kučerka, N., Nieh, M.-P., Katsaras, J., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2014; Chapter 15; pp. 291–315. [Google Scholar]
- Mayer, L.D.; Bally, M.B.; Cullis, P.R. Uptake of adriamycin into large unilamellar vesicles in response to a pH gradient. Biochim. Biophys. Acta 1986, 857, 123–126. [Google Scholar] [CrossRef]
- Haran, G.; Cohen, R.; Bar, L.K.; Barenholz, Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim. Biophys. Acta 1993, 1151, 201–215. [Google Scholar] [CrossRef]
- Barenholz, Y. Liposome applications: Problems and prospects. Curr. Opin. Colloid Interface Sci. 2001, 6, 66–77. [Google Scholar] [CrossRef]
- Abraham, S.A.; Waterhouse, D.N.; Mayer, L.D.; Cullis, P.R.; Madden, T.D.; Bally, M.B. The Liposomal Formulation of Doxorubicin. Methods Enzymol. 2005, 391, 71–97. [Google Scholar] [PubMed]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Zucker, D.; Marcus, D.; Barenholz, Y.; Goldblum, A. Liposome drugs’ loading efficiency: A working model based on loading conditions and drug’s physicochemical properties. J. Control. Release 2009, 139, 73–80. [Google Scholar] [CrossRef]
- Gubernator, J. Active methods of drug loading into liposomes: Recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin. Drug Deliv. 2011, 8, 565–580. [Google Scholar] [CrossRef]
- Li, T.; Cipolla, D.; Rades, T.; Boyd, B.J. Drug nanocrystallisation within liposomes. J. Control. Release 2018, 288, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Fritze, A.; Hens, F.; Kimpfler, A.; Schubert, R.; Peschka-Süss, R. Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient. Biochim. Biophys. Acta 2006, 1758, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Schilt, Y.; Berman, T.; Wei, X.; Barenholz, Y.; Raviv, U. Using solution X-ray scattering to determine the high-resolution structure and morphology of PEGylated liposomal doxorubicin nanodrugs. Biochim. Biophys. Acta 2016, 1860, 108–119. [Google Scholar] [CrossRef]
- Lasic, D.D. Applications of Liposomes. In Structure and Dynamics of Membranes: From Cells to Vesicles; Lipowsky, R., Sackmann, E., Eds.; Handbook of Biological Physics; Elsevier Science B.V.: Amsterdam, The Netherlands, 1995; Volume 1A, Chapter 10; pp. 491–519. [Google Scholar]
- Martí, M.; de la Maza, A.; Parra, J.L.; Coderch, L. Role of Liposomes in Textile Dyeing. In Liposomes, Lipid Bilayers and Model Membranes. From Basic Research to Applications; Pabst, G., Kučerka, N., Nieh, M.-P., Katsaras, J., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2014; Chapter 20; pp. 401–414. [Google Scholar]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Kapoor, B.; Gupta, R.; Gulati, M.; Singh, S.K.; Khursheed, R.; Gupta, M. The Why, Where, Who, How, and What of the vesicular delivery systems. Adv. Colloid Interface Sci. 2019, 271, 101985. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-Gracia, E.; López-Camacho, A.; Higuera-Ciapara, I.; Velázquez-Fernández, J.B.; Vallejo-Cardona, A.A. Nanomedicine review: Clinical developments in liposomal applications. Cancer Nanotechnol. 2019, 10, 11. [Google Scholar] [CrossRef]
- Bisso, S.; Leroux, J.-C. Nanopharmaceuticals: A focus on their clinical translatability. Int. J. Pharm. 2020, 578, 119098. [Google Scholar] [CrossRef] [PubMed]
- Thi, T.T.H.; Suys, E.J.A.; Lee, J.S.; Nguyen, D.H.; Park, K.D.; Truong, N.P. Lipid-Based Nanoparticles in the Clinic and Clinical Trials: From Cancer Nanomedicine to COVID-19 Vaccines. Vaccines 2021, 9, 359. [Google Scholar] [CrossRef] [PubMed]
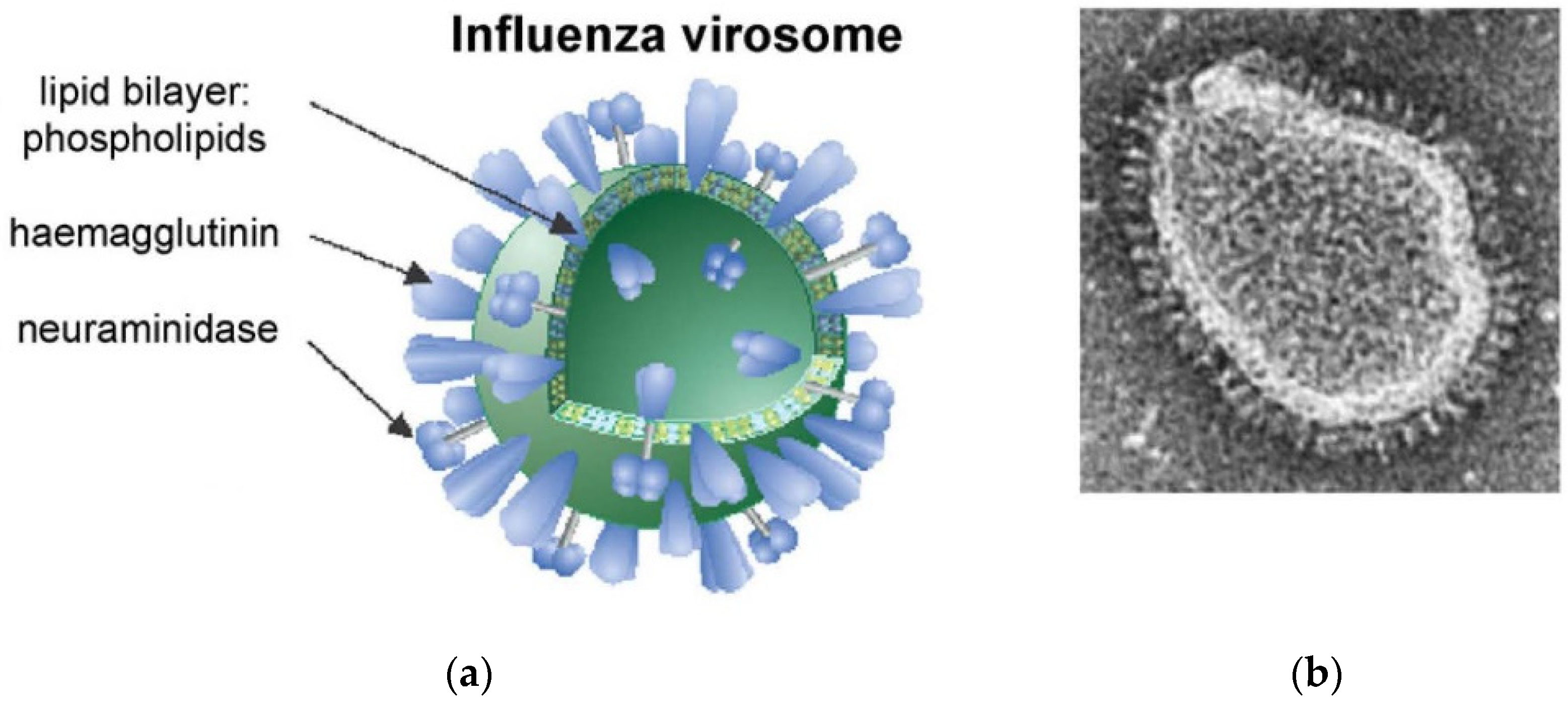
- Huckriede, A.; Bungener, L.; Stegmann, T.; Daemen, T.; Medema, J.; Palache, A.M.; Wilschut, J. The virosome concept for influenza vaccines. Vaccine 2005, 23, S26–S38. [Google Scholar] [CrossRef] [PubMed]
- Herzog, C.; Hartmann, K.; Künzi, V.; Kürsteiner, O.; Mischler, R.; Lazar, H.; Glück, R. Eleven years of Inflexal®V—A virosomal adjuvanted influenza vaccine. Vaccine 2009, 27, 4381–4387. [Google Scholar] [CrossRef]
- Schwendener, R.A. Liposomes as vaccine delivery systems: A review of the recent advances. Ther. Adv. Vaccines 2014, 2, 159–182. [Google Scholar] [CrossRef] [PubMed]
- De Serrano, L.O.; Burkhart, D.J. Liposomal vaccine formulations as prophylactic agents: Design considerations for modern vaccines. J. Nanobiotechnol. 2017, 15, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meybeck, A. Past, Present and Future of Liposome Cosmetics. In Liposome Dermatics; Braun-Falco, O., Korting, H.C., Maibach, H.I., Eds.; Springer: Berlin, Germany, 1992; pp. 341–352. [Google Scholar]
- Lautenschläger, H. Liposomes. In Handbook of Cosmetic Science and Technology, 2nd ed.; Paye, M., Barel, A.O., Maubach, H.I., Eds.; CRC Press: Boca Raton, FL, USA, 2006; pp. 155–163. [Google Scholar]
- Rahimpor, Y.; Hamishehkar, H. Liposomes in cosmeceutics. Expert Opin. Drug Deliv. 2012, 9, 443–455. [Google Scholar] [CrossRef] [PubMed]
- van Hoogevest, P.; Fahr, A. Phospholipids in Cosmetic Carriers. In Nanocosmetics. From Ideas to Products; Cornier, J., Keck, C.M., Van de Voorde, M., Eds.; Springer Nature Switzerland AG: Cham, Switzerland, 2019; Chapter 6; pp. 95–140. [Google Scholar]
- Chen, C.; Han, D.; Cai, C.; Tang, X. An overview of liposome lyophilization and its future potential. J. Control. Release 2010, 142, 299–311. [Google Scholar] [CrossRef]
- Franzé, S.; Selmin, F.; Samaritani, F.; Minghetti, P.; Cilurzo, F. Lyophilization of Liposomal Formulations: Still Necessary, Still Challenging. Pharmaceutics 2018, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, L.; Saunders, D.N.; Ranson, M.; Thurecht, K.J.; Storm, G.; Vine, K.L. Towards clinical translation of ligand-functionalized liposomes in targeted cancer therapy: Challenges and opportunities. J. Control. Release 2018, 277, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sackmann, E. Physical Basis of Self-Organization and Function of Membranes: Physics of Vesicles. In Structure and Dynamics of Membranes: From Cells to Vesicles; Lipowsky, R., Sackmann, E., Eds.; Handbook of Biological Physics; Elsevier Science B.V.: Amsterdam, The Netherlands, 1995; Volume 1A, Chapter 5; pp. 213–304. [Google Scholar]
- Seifert, U.; Lipowsky, R. Morphology of Vesicles. In Structure and Dynamics of Membranes: From Cells to Vesicles; Lipowsky, R., Sackmann, E., Eds.; Handbook of Biological Physics; Elsevier Science B.V.: Amsterdam, The Netherlands, 1995; Volume 1A, Chapter 8; pp. 403–463. [Google Scholar]
- Jimbo, T.; Sakuma, Y.; Urakami, N.; Ziherl, P.; Imai, M. Role of Inverse-Cone-Shape Lipids in Temperature-Controlled Self-Reproduction of Binary Vesicles. Biophys. J. 2016, 110, 1551–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méléard, P.; Gerbeaud, C.; Pott, T.; Fernandez-Puente, L.; Bivas, I.; Mitov, M.D.; Dufourcq, J.; Bothorel, P. Bending Elasticity of Model Membranes: Influences of Temperature and Sterol Content. Biophys. J. 1997, 72, 2616–2629. [Google Scholar] [CrossRef] [Green Version]
- Helfrich, W. Bending Elasticity of Fluid Membranes. In Giant Vesicles; Luisi, P.L., Walde, P., Eds.; Perspectives in Supramolecular, Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2000; Volume 6, Chapter 6; pp. 51–70. [Google Scholar]
- Döbereiner, H.-G.; Käs, J.; Noppl, D.; Sprenger, I.; Sackmann, E. Budding and Fission of Vesicles. Biophys. J. 1993, 65, 1396–1403. [Google Scholar] [CrossRef] [Green Version]
- Morowitz, H.J.; Heinz, B.; Deamer, D.W. The chemical logic of a minimum protocell. Origins Life Evol. Biospheres 1988, 18, 281–287. [Google Scholar]
- Svetina, S. Vesicle Budding and the Origin of Cellular Life. ChemPhysChem 2009, 10, 2769–2776. [Google Scholar] [CrossRef]
- Damer, B.; Deamer, D. Coupled Phases and Combinatorial Selection in Fluctuating Hydrothermal Pools: A Scenario to Guide Experimental Approaches to the Origin of Cellular Life. Life 2015, 5, 872–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.A.; Salehi-Ashtiani, K.; Szostak, J.W. RNA Catalysis in Model Protocell Vesicles. J. Am. Chem. Soc. 2005, 127, 13213–13219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansy, S.S.; Schrum, J.P.; Krishnamurthy, M.; Tobé, S.; Treco, D.A.; Szostak, J.W. Template-directed synthesis of a genetic polymer in a model protocell. Nature 2008, 454, 122–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deamer, D. First Life. Discovering the Connections Between Stars, Cells, and How Life Began; University of California Press: Berkeley/Los Angeles, CA, USA, 2011. [Google Scholar]
- Luisi, P.L. The Emergence of Life. From Chemical Origins to Synthetic Biology, 2nd ed.; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Chen, I.A.; Walde, P. From Self-Assembled Vesicles to Protocells. Cold Spring Harb. Perspect. Biol. 2010, 2, a002170. [Google Scholar] [CrossRef] [PubMed]
- Monnard, P.-A.; Walde, P. Current Ideas about Prebiological Compartmentalization. Life 2015, 5, 1239–1263. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.-C.; Chen, I.A. Protocells. Curr. Biol. 2020, 30, R484–R485. [Google Scholar] [CrossRef] [PubMed]
- Uchegbu, I.F.; Florence, A.T. Non-ionic Surfactant Vesicles (Niosomes): Physical and Pharmaceutical Chemistry. Adv. Colloid Interface Sci. 1995, 58, 1–55. [Google Scholar] [CrossRef]
- Bartelds, R.; Nematollahi, M.H.; Pols, T.; Stuart, M.C.A.; Pardakhty, A.; Asadikaram, G.; Poolman, B. Niosomes, an alternative for liposomal delivery. PLoS ONE 2018, 13, e0194179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Hanning, S.; Falconer, J.; Locke, M.; Wen, J. Recent advances in non-ionic surfactant vesicles (niosomes): Fabrication, characterization, pharmaceutical and cosmetic applications. Eur. J. Pharm. Biopharm. 2019, 144, 18–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fendler, J.H. Surfactant Vesicles as Membrane Mimetic Agents: Characterization and Utilization. Acc. Chem. Res. 1980, 13, 7–13. [Google Scholar] [CrossRef]
- Kunitake, T.; Okahata, Y. Synthetic Bilayer Membranes with Anionic Head Groups. Bull. Chem. Soc. Jpn. 1978, 51, 1877–1879. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Hauser, N.; Moreno, A.; Ishikawa, T.; Walde, P. AOT vesicles as templates for the horseradish peroxidase-triggered polymerization of aniline. Soft Matter 2011, 7, 180–193. [Google Scholar] [CrossRef]
- Junker, K.; Zandomeneghi, G.; Guo, Z.; Kissner, R.; Ishikawa, T.; Kohlbrecher, J.; Walde, P. Mechanistic aspects of the horseradish peroxidase-catalysed polymerisation of aniline in presence of AOT vesicles as templates. RSC Adv. 2012, 2, 6478–6495. [Google Scholar] [CrossRef]
- Gosh, S.; Roy, A.; Banik, D.; Kundu, N.; Kuchlyan, J.; Dhir, A.; Sarkar, N. How Does the Surface Charge of Ionic Surfactant and Cholesterol Forming Vesicles Control Rotational and Translational Motion of Rhodamine 6G Perchlorate (R6G ClO4)? Langmuir 2015, 31, 2310–2320. [Google Scholar] [CrossRef]
- Viseu, M.I.; Edwards, K.; Campos, C.S.; Costa, S.M.B. Spontaneous Vesicles Formed in Aqueous Mixtures of Two Cationic Amphiphiles. Langmuir 2000, 16, 2105–2114. [Google Scholar] [CrossRef]
- Kunitake, T.; Okahata, Y. A Totally Synthetic Bilayer Membrane. J. Am. Chem. Soc. 1977, 99, 3860–3861. [Google Scholar] [CrossRef]
- Sreejith, L.; Parathakkat, S.; Nair, S.M.; Kumar, S.; Varma, G.; Hassan, P.A.; Talmon, Y. Octanol-Triggered Self-Assemblies of the CTAB/KBr System: A Microstructural Study. J. Phys. Chem. B. 2011, 115, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Tasies, L.; Moreno-Calvo, E.; Cano-Sarabia, M.; Aguilella-Arzo, M.; Angelova, A.; Lesieur, S.; Ricart, S.; Faraudo, J.; Ventosa, N.; Veciana, J. Quatsomes: Vesicles Formed by Self-Assembly of Sterols and Quaternary Ammonium Surfactants. Langmuir 2013, 29, 6519–6528. [Google Scholar] [CrossRef] [PubMed]
- Marques, E.M.; Regev, O.; Khan, A.; Miguel, M.d.G.; Lindman, B. Vesicle Formation and General Phase Behavior in the Catanionic Mixture SDS-DDAB-Water. The Anionic-Rich Side. J. Phys. Chem. B 1998, 102, 6746–6758. [Google Scholar] [CrossRef] [Green Version]
- Marques, E.F.; Regev, O.; Khan, A.; Miguel, M.dG.; Lindman, B. Vesicle Formation and General Phase Behavior in the Cationic Mixture SDS-DDAB-Water. The Cationic-Rich Side. J. Phys. Chem. B 1999, 103, 8353–8363. [Google Scholar] [CrossRef]
- Kaler, E.W.; Murthy, A.K.; Rodriguez, B.E.; Zasadzinski, J.A.N. Spontaneous Vesicle Formation in Aqueous Mixtures of Single-Tailed Surfactants. Science 1989, 245, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Ahmed, F. Polymersomes. Annu. Rev. Biomed. Eng. 2006, 8, 323–341. [Google Scholar] [CrossRef] [PubMed]
- Kita-Tokarczyk, K.; Grumelard, J.; Haefele, T.; Meier, T. Block copolymer vesicles—Using concepts from polymer chemistry to mimic biomembranes. Polymer 2005, 46, 3540–3563. [Google Scholar] [CrossRef] [Green Version]
- LoPresti, C.; Lomas, H.; Massignani, M.; Smart, T.; Battaglia, G. Polymersomes: Nature inspired nanometer sized compartments. J. Mater. Chem. 2009, 19, 3576–3590. [Google Scholar] [CrossRef]
- Tanner, P.; Baumann, P.; Enea, R.; Onaca, O.; Palivan, C.; Meier, W. Polymeric Vesicles: From Drug Carriers to Nanoreactors and Artificial Organelles. Acc. Chem. Res. 2011, 44, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Palivan, C.G.; Goers, R.; Najer, A.; Zhang, X.; Car, A.; Meier, W. Bioinspired polymer vesicles and membranes for biological and medical applications. Chem. Soc. Rev. 2016, 45, 377–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rideau, E.; Dimova, R.; Schwille, P.; Wurm, F.R.; Landfester, K. Liposomes and polymersomes: A comparative review towards cell mimicking. Chem. Soc. Rev. 2018, 47, 8572–8610. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zhang, Y.; Xie, Z.; Jing, X.; Bellotti, A.; Gu, Z. Stimuli-Responsive Polymersomes for Biomedical Applications. Biomacromolecules 2017, 18, 649–673. [Google Scholar] [CrossRef]
- Einfalt, T.; Witzigmann, D.; Edlinger, C.; Sieber, S.; Goers, R.; Najer, A.; Spulber, M.; Onaca-Fischer, O.; Huwyler, J.; Palivan, C.G. Biomimetic artificial organelles with in vitro and in vivo activity triggered by reduction in microenvironment. Nat. Commun. 2018, 9, 1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Oppen, L.M.P.E.; Abdelmohsen, L.K.E.A.; van Emst-de Vries, S.E.; Welzen, P.L.W.; Wilson, D.A.; Smeitink, J.A.M.; Koopman, W.J.H.; Brock, R.; Willems, P.H.G.M.; Williams, D.S.; et al. Biodegradable Synthetic Organelles Demonstrate ROS Shielding in Human-Complex-I-Deficient Fibroplasts. ACS Cent. Sci. 2018, 4, 917–928. [Google Scholar] [CrossRef]
- Paprocki, D.; Koszelewski, D.; Walde, P.; Ostaszewski, R. Efficient Passerini reactions in an aqueous vesicle system. RSC Adv. 2015, 5, 102828–102835. [Google Scholar] [CrossRef]
- Luginbühl, S.; Bertschi, L.; Willeke, M.; Schuler, L.D.; Walde, P. How Anionic Vesicles Steer the Oligomerization of Enzymatically Oxidized p-Aminodiphenylamine (PADPA) toward a Polyaniline Emeraldine Salt (PANI-ES)-Type Product. Langmuir 2016, 32, 9765–9779. [Google Scholar] [CrossRef]
- Okada, S.; Peng, S.; Spevak, W.; Charych, D. Color and Chromism of Polydiacetylene Vesicles. Acc. Chem. Res. 1998, 31, 229–239. [Google Scholar] [CrossRef]
- Lebègue, E.; Farre, C.; Jose, C.; Saulnier, J.; Lagarde, F.; Chevalier, Y.; Chaix, C.; Jaffrezic-Renault, N. Responsive Polydiacetylene Vesicles for Biosensing Microorganisms. Sensors 2018, 18, 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Städler, B. Recent Developments in Polydiacetylene-Based Sensors. Chem. Mater. 2019, 31, 1196–1222. [Google Scholar] [CrossRef]
- Wang, D.-E.; Gao, X.; Li, G.; Xue, T.; Yang, H.; Xu, H. Facile colorimetric assay of alkaline phosphatase activity using polydiacetylene liposomes with calcium ions and pyrophosphate. Sens. Actuators B 2019, 289, 85–92. [Google Scholar] [CrossRef]
- Handjani-Vila, R.M.; Ribier, A.; Rondot, B.; Vanlerberghie, G. Dispersions of lamellar phases of non-ionic lipids in cosmetic products. Int. J. Cosmet. Sci. 1979, 1, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Matoori, S.; Leroux, J.-C. Twenty-five years of polymersomes: Lost in translation? Mater. Horiz. 2020, 7, 1297–1309. [Google Scholar] [CrossRef]
- Dou, Y.; Hynynen, K.; Allen, C. To heat or not to heat: Challenges with clinical translation of thermosensitive liposomes. J. Control. Release 2017, 249, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, H.; Takechi, Y.; Tamai, N.; Matsuki, H.; Yomota, C.; Saito, H. Thermotropic Phase Behavior of Hydrogenated Soybean Phosphatidylcholine—Cholesterol Binary Liposome Membrane. Chem. Pharm. Bull. 2014, 62, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, G.M. The care and feeding of a commercial liposomal product: Liposomal amphotericin B (AmBisome®). J. Liposome Res. 2017, 27, 173–179. [Google Scholar] [CrossRef]
- Zurbriggen, R. Immunostimulating reconstituted influenza virosomes. Vaccine 2003, 21, 921–924. [Google Scholar] [CrossRef]
- Moser, C.; Amacker, M.; Zurbriggen, R. Influenza virosomes as a vaccine adjuvant and carrier system. Expert Rev. Vaccines 2011, 10, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Glück, R. Immunopotentiating Reconstituted Influenza Virosomes (IRIVs). In Vaccine Adjuvants. Preparation Methods and Research Protocols; O’Hagan, D.T., Ed.; Methods in Molecular Medicine; Humana Press: Totowa, NJ, USA, 2000; Volume 42, Chapter 9; pp. 151–178. [Google Scholar]
- Mischler, R.; Metcalfe, I.C. Inflexal®V a trivalent virosome subunit influenza vaccine: Production. Vaccine 2002, 20, B17–B23. [Google Scholar] [CrossRef]
- Gouma, S.; Anderson, E.M.; Hensley, S.E. Challenges of Making Effective Influenza Vaccines. Annu. Rev. Virol. 2020, 7, 495–512. [Google Scholar] [CrossRef] [PubMed]
- Bayat, F.; Hosseinpour-Moghadam, R.; Mehryab, F.; Fatahi, Y.; Shakeri, N.; Dinarvand, R.; Ten Hagen, T.L.M.; Haeri, A. potential application of liposomal nanodevices for non-cancer diseases: An update on design, characterization and biopharmaceutical evaluation. Adv. Colloid Interface Sci. 2020, 277, 102121. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.S.; Camilleri, J.P.; Goodfellow, R.M.; Williams, D.D. A single intra-articular injection of liposomally conjugated methotrexate suppresses joint inflammation in rat antigen-induced arthritis. J. Rheumatol. 1996, 35, 719–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera, P.; Blom, A.; van Lent, P.L.E.M.; van Bloois, L.; Beijnen, J.H.; van Rooijen, N.; de Waal Malefijt, M.C.; van de Putte, L.B.A.; Storm, G.; van den Berg, W.B. Synovial Macrophage Depletion with Clodronate-containing Liposomes in Rheumatoid Arthritis. Arthritis Rheum. 2000, 43, 1951–1959. [Google Scholar] [CrossRef] [Green Version]
- van den Hoven, J.M.; Van Tomme, S.R.; Metselaar, J.M.; Nuijen, B.; Beijnen, J.H.; Storm, G. Liposomal Drug Formulations in the Treatment of Rhematoid Arthritis. Mol. Pharm. 2011, 8, 1002–1015. [Google Scholar] [CrossRef]
- Kapoor, B.; Singh, S.K.; Gulati, M.; Gupta, R.; Vaidya, Y. Application of Liposomes in Treatment of Rheumatoid Arthritis: Quo Vadis. Sci. World J. 2014, 978351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, V.; Signorell, R.D.; Roveri, M.; Leroux, J.-C. Liposome-supported peritoneal dialysis for detoxification of drugs and endogenous metabolites. Sci. Transl. Med. 2014, 6, 258ra141. [Google Scholar] [CrossRef] [PubMed]
- Matoori, S.; Forster, V.; Agostini, V.; Bettschart-Wolfensberger, R.; Bektas, R.N.; Thöny, B.; Häberle, J.; Leroux, J.-C. Preclinical evaluation of liposome-supported peritoneal dialysis for the treatment of hyperammonemic crisis. J. Control. Release 2020, 328, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Landon, C.D.; Park, J.-Y.; Needham, D.; Dewhirst, M.W. Nanoscale Drug Delivery and Hyperthermia: The Materials Design and Preclinical and Clinical Testing of Low Temperature-Sensitive Liposomes Used in Combination with Mild Hyperthermia in the Treatment of Local Cancer. Open Nanomed. J. 2011, 3, 38–64. [Google Scholar] [CrossRef]
- May, J.P.; Li, S.-D. Hyperthermia-induced drug targeting. Expert Opin. Drug Deliv. 2013, 10, 511–527. [Google Scholar] [CrossRef] [PubMed]
- Hynynen, K. Hyperthermia-induced drug delivery in humans. Nat. Biomed. Eng. 2018, 2, 637–639. [Google Scholar] [CrossRef]
- Needham, D.; Anyarambhatla, G.; Kong, G.; Dewhirst, M.W. A New Temperature-sensitive Liposome for Use with Mild Hyperthermia: Characterization and Testing in a Human Tumor Xenograft Model. Cancer Res. 2000, 60, 1197–1201. [Google Scholar] [PubMed]
- Yatvin, M.B.; Weinstein, J.N.; Dennis, W.H.; Blumenthal, R. Design of Liposomes for Enhanced Local Release of Drugs by Hyperthermia. Science 1978, 202, 1290–1293. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Magin, R.L.; Yatvin, M.B.; Zaharko, D.S. Liposomes and Local Hyperthermia: Selective Delivery of Methotrexate to Heated Tumors. Science 1979, 204, 188–191. [Google Scholar] [CrossRef]
- Needham, D. Bringing Research to Clinical Application. Lessons from ThermoDox®: A Thermal-Sensitive Liposome for Treatment of Cancer. In Drug Delivery. Fundamentals & Applications, 2nd ed.; Hillery, A., Park, K., Eds.; CRC Press: Boca Raton, FL, USA, 2016; Chapter 23; pp. 523–585. [Google Scholar]
- Chander, N.; Morstein, J.; Bolten, J.S.; Shemet, A.; Cullis, P.R.; Trauner, D.; Witzigmann, D. Optimized Photoactivatable Lipid Nanoparticles Enable Red Light Triggered Drug Release. Small 2021, 17, 2008198. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur. J. Pharm. Biopharm. 2009, 71, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Lou, J.; Best, M.D. Strategies for altering lipid self-assembly to trigger liposome cargo release. Chem. Phys. Lipids 2020, 232, 104966. [Google Scholar] [CrossRef] [PubMed]
- Yuba, E. Development of functional liposomes by modification of stimuli-responsive materials and their biomedical applications. J. Mater. Chem. B 2020, 8, 1093–1107. [Google Scholar] [CrossRef] [PubMed]
- Davidsen, J.; Jørgensen, K.; Andresen, T.L.; Mouritsen, O.G. Secreted phospholipase A2 as a new enzymatic trigger mechanism for localised drug release and absorption in diseased tissue. Biochim. Biophys. Acta 2003, 1609, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Arouri, A.; Mouritsen, O.G. Phospholipase A2-susceptible liposomes of anticancer double lipid-prodrugs. Eur. J. Pharm. Sci. 2012, 45, 408–420. [Google Scholar] [CrossRef]
- Hansen, A.H.; Mouritsen, O.G.; Arouri, A. Enzymatic action of phospholipase A2 on liposomal drug delivery. Int. J. Pharm. 2015, 491, 49–57. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, J.A.; Witzigmann, D.; Leung, J.; van der Meel, R.; Zaifman, J.; Darjuan, M.M.; Grisch-Chan, H.M.; Thöny, B.; Tam, Y.Y.C.; Cullis, P.R. Fusion-dependent formation of lipid nanoparticles containing macromolecular payloads. Nanoscale 2019, 11, 9023–9031. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, M.J.; Alishetty, S.; Alameh, M.-G.; Said, H.; Wright, L.; Paige, M.; Soliman, O.; Weissman, D.; Cleveland IV, T.E.; Grishaev, A.; et al. Ionization and structural properties of mRNA lipid nanoparticles influence expression in intramuscular and intravascular administration. Commun. Biol. 2021, 4, 956. [Google Scholar] [CrossRef]
- Evers, M.J.W.; Kulkarni, J.A.; van der Meel, R.; Cullis, P.R.; Vader, P.; Schifflers, R.M. State-of-the Art Design and Rapid-Mixing Production Techniques of Lipid Nanoparticles for Nucleic Acid Delivery. Small Methods 2018, 2, 1700375. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticle Technology for Clinical Translation of siRNA Therapeutics. Acc. Chem. Res. 2019, 52, 2435–2444. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acids Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, J.A.; Witzigmann, D.; Leung, J.; Tam, Y.Y.C.; Cullis, P.R. On the role of helper lipids in lipid nanoparticle formulations of siRNA. Nanoscale 2019, 11, 21733–21739. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Kulkarni, J.A.; Huynh, A.; Chen, S.; van der Meel, R.; Tam, Y.Y.C.; Cullis, P.R. Characterization of Lipid Nanoparticles Containing Ionizable Cationic Lipids Using Design-of-Experiments Approach. Langmuir 2021, 37, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Wang, Y.; Gong, Y.; Lin, X.; Zhao, Y.; Zhi, D.; Zhou, Q.; Zhang, S. Correlation of the cytotoxic effects of cationic lipids with their headgroups. Toxicol. Res. 2018, 7, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Witzigmann, D.; Kulkarni, J.A.; Leung, J.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv. Rev. 2020, 159, 344–363. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Thomson, S.B.; Zaifman, J.; Leung, J.; Wagner, P.K.; Hill, A.; Tam, Y.Y.C.; Cullis, P.R.; Petkau, T.L.; Leavitt, B.R. Spontaneous, solvent-free entrapment of siRNA within lipid nanoparticles. Nanoscale 2020, 12, 23959–23966. [Google Scholar] [CrossRef] [PubMed]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivers using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Eygeris, Y.; Gupta, M.; Sahay, G. Self-assembled mRNA vaccines. Adv. Drug Deliv. Rev. 2021, 170, 83–112. [Google Scholar] [CrossRef] [PubMed]
- Viger-Gravel, J.; Schantz, A.; Pinon, A.C.; Rossini, A.J.; Schantz, S.; Emsley, L. Structure of Lipid Nanoparticles Containing siRNA or mRNA by Dynamic Nuclear Polarization-Enhanced NMR Spectroscopy. J. Phys. Chem. B 2018, 122, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Rozmanov, D.; Baoukina, S.; Tieleman, D.P. Density Based Visualization for Molecular Simulation. Faraday Discuss. 2014, 169, 225–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, A.K.K.; Hafez, I.M.; Baoukina, S.; Belliveau, N.M.; Zhigaltsev, I.V.; Afshinmanesh, E.; Tieleman, D.P.; Hansen, C.L.; Hope, M.J.; Cullis, P.R. Lipid nanoparticles containing siRNA synthesized by microfluidic mixing exhibit an electron-dense nanostructured core. J. Phys. Chem. C 2012, 116, 18440–18450. [Google Scholar] [CrossRef]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Brader, M.L.; Williams, S.J.; Banks, J.M.; Hui, W.H.; Zhou, Z.H.; Jin, L. Encapsulation state of messenger RNA inside lipid nanoparticles. Biophys. J. 2021, 120, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Noor, R. Development Status of the Potential Vaccines for the Mitigation of the COVID-19 Pandemic and a Focus on the Effectiveness of the Pfizer-BioNTech and Moderna mRNA Vaccines. Curr. Clin. Microbiol. Rep. 2021, 8, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, K.; Shelling, A.N.; Wu, Z. Nanotechnology-Enabled COVID-19 mRNA Vaccines. Encyclopedia 2021, 1, 773–780. [Google Scholar] [CrossRef]
- Cevc, G.; Schätzlein, A.G.; Richardsen, H.; Vierl, U. Overcoming Semipermeable Barriers, Such as the Skin, with Ultradeformable Mixed Lipid Vesicles, Transfersomes, Liposomes, or Mixed Lipid Micelles. Langmuir 2003, 19, 10753–10763. [Google Scholar] [CrossRef]
- Benson, H.A.E. Elastic Liposomes for Topical and Transdermal Drug Delivery. In Liposomes. Methods and Protocols; Weissig, V., Ed.; Volume 1: Pharmaceutical Nanocarriers; Humana Press: New York, USA, 2010; Chapter 4; pp. 77–86. [Google Scholar]
- Ashtikar, M.; Nagarsekar, K.; Fahr, A. Transdermal delivery from liposomal formulations—Evolution of the technology over the last three decades. J. Control. Release 2016, 242, 126–140. [Google Scholar] [CrossRef]
- Mirtaleb, M.S.; Shahraky, M.K.; Ekrami, E.; Mirtaleb, A. Advances in biological nano-phospholipid vesicles for transdermal delivery: A review on applications. J. Drug Deliv. Sci. Technol. 2021, 61, 102331. [Google Scholar] [CrossRef]
- Fernández-García, R.; Lalatsa, A.; Statts, L.; Bolás-Fernández, F.; Ballesteros, M.P.; Serrano, D.R. Transfersomes as nanocarriers for drugs across the skin: Quality by design from lab to industrial scale. Int. J. Pharm. 2020, 573, 118817. [Google Scholar] [CrossRef] [PubMed]
- Uhl, P.; Sauter, M.; Hertlein, T.; Witzigmann, D.; Laffleur, F.; Hofhaus, G.; Fidelj, V.; Tursch, A.; Özbek, S.; Hopke, E.; et al. Overcoming the Mucosal Barrier: Tetraether Lipid-Stabilized Liposomal Nanocarriers Decorated with Cell-Penetrating Peptides Enable Oral Delivery of Vancomycin. Adv. Therap. 2021, 4, 2000247. [Google Scholar] [CrossRef]
- Chong, P.L.-G.; Ayesa, U.; Daswani, V.P.; Hur, E.C. On the Physical Properties of Tetraether Lipid Membranes: Effects of Cyclopentane Rings. Archea 2012, 2012, 138439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhl, P.; Helm, F.; Hofhaus, G.; Brings, S.; Kaufman, C.; Leotta, K.; Urban, S.; Haberkorn, U.; Mier, W.; Fricker, G. A liposomal formulation for the oral application of the investigational hepatitis B drug Myrcludex B. Eur. J. Pharm. Biopharm. 2016, 103, 159–166. [Google Scholar] [CrossRef]
- Massing, U.; Cicko, S.; Ziroli, V. Dual asymmetric centrifugation (DAC)—A new technique for liposome preparation. J. Control. Release 2008, 125, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Uhl, P.; Pantze, S.; Storck, P.; Parmentier, J.; Witzigmann, D.; Hofhaus, G.; Huwyler, J.; Mier, W.; Fricker, G. Oral delivery of vancomycin by tetraether lipid liposomes. Eur. J. Pharm. Sci. 2017, 108, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Bohns Michalowski, C.; Beloqui, A. Advances in lipid carriers for drug delivery to the gastrointestinal tract. Curr. Opin. Colloid Interface Sci. 2021, 52, 101414. [Google Scholar] [CrossRef]
- Gomez, A.G.; Hosseinidoust, Z. Liposomes for Antibiotic Encapsulation and Delivery. ACS Infect. Dis. 2020, 6, 896–908. [Google Scholar] [CrossRef] [PubMed]
- Vamvakaki, V.; Chaniotakis, N.A. Pesticide detection with a liposome-based nano-biosensor. Biosens. Bioelectron. 2007, 22, 2848–2853. [Google Scholar] [CrossRef]
- Liu, Q.; Boyd, B.J. Liposomes in biosensors. Analyst 2013, 138, 391–409. [Google Scholar] [CrossRef]
- Mazur, F.; Bally, M.; Städler, B.; Chandrawati, R. Liposomes and lipid bilayers in biosensors. Adv. Colloid Interface Sci. 2017, 249, 88–99. [Google Scholar] [CrossRef]
- Hofmann, C.; Duerkop, A.; Baeumner, A.J. Nanocontainers for Analytical Applications. Angew. Chem. Int. Ed. 2019, 58, 12840–12860. [Google Scholar] [CrossRef]
- Sforzi, J.; Palagi, L.; Aime, S. Liposome-Based Bioassays. Biology 2020, 9, 202. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.A.; Bolduc, O.R.; Baeumner, A.J. Miniaturized bioanalytical systems: Enhanced performance through liposomes. Curr. Opin. Chem. Biol. 2012, 16, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Sforzi, J.; Ferrauto, G.; Aime, S.; Geninatti Crich, S. A Simple and Fast Assay Based on Carboxyfluorescein-Loaded Liposome for Quantitative DNA Detection. ACS Omega 2020, 5, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Mazur, F.; Tran, H.; Kuchel, R.P.; Chandrawati, R. Rapid Detection of Listeriolysin O Toxin Based on a Nanoscale Liposome-Gold Nanoparticle Platform. ACS Appl. Nano Mater. 2020, 3, 7270–7280. [Google Scholar] [CrossRef]
- Beigi, F.; Yang, Q.; Lundahl, P. Immobilized-liposome chromatographic analysis of drug partitioning into lipid bilayers. J. Chromatogr. A 1995, 704, 315–321. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, X.-Y.; Yoshimoto, M.; Kuboi, R.; Miyake, J. Covalent Immobilization of Unilamellar Liposomes in Gel Beads for Chromatography. Anal. Biochem. 1999, 268, 354–362. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Yang, Q.; Nakamura, C.; Miyake, J. Avidin-biotin-immobilized liposome column for chromatographic fluorescence on-line analysis of solute–membrane interactions. J. Chromatogr. A 2001, 750, 51–60. [Google Scholar] [CrossRef]
- Dusseillier, M.R.; Niederberger, B.; Städler, B.; Falconnet, D.; Textor, M.; Vörös, J. A novel crossed microfluidic device for the precise positioning of proteins and vesicles. Lab Chip 2005, 5, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Bally, M.; Bailey, K.; Sugihara, K.; Grieshaber, D.; Vörös, J.; Städler, B. Liposome and Lipid Bilayer Arrays Towards Biosensing Applications. Small 2010, 6, 2481–2497. [Google Scholar] [CrossRef] [PubMed]
- Hosta-Rigau, L.; Zhang, Y.; Teo, B.M.; Postma, A.; Städler, B. Cholesterol—a biological compound as a building block in bionanotechnology. Nanoscale 2013, 5, 89–109. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.F.; Knutson, J.R. Mechanism of Fluorescence Concentration Quenching of Carboxyfluorescein in Liposomes: Energy Transfer to Nonfluorescent Dimers. Anal. Biochem. 1988, 172, 61–77. [Google Scholar] [CrossRef]
- Nasr, G.; Greige-Gerges, H.; Elasissari, A.; Khreich, N. Liposomal membrane permeability assessment by fluorescent techniques: Main permeabilizing agents, applications and challenges. Int. J. Pharm. 2020, 580, 119198. [Google Scholar] [CrossRef]
- Schnitzer, E.; Lichtenberg, D.; Kozlov, M.M. Temperature-dependence of the solubilization of dipalmitoylphosphatidylcholine (DPPC) by the non-ionic surfactant Triton X-100, kinetic and structural aspects. Chem. Phys. Lipids 2003, 126, 55–76. [Google Scholar] [CrossRef]
- Schnitzer, E.; Kozlov, M.M.; Lichtenberg, D. The effect of cholesterol on the solubilization of phosphatidylcholine bilayers by the non-ionic surfactant Triton X-100. Chem. Phys. Lipids 2005, 135, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Dalgarno, P.A.; Juan-Cólas, J.; Hedley, G.J.; Piñeiro, L.; Novo, M.; Perez-Gonzalez, C.; Samuel, I.D.W.; Leake, M.C.; Johnson, S.; Al-Soufi, W.; et al. Unveiling the multi-step solubilization mechansim of sub-micron size vesicles by detergents. Sci. Rep. 2019, 9, 12897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Hens, A.; Fernández-Romero, J.M. The role of liposomes in analytical processes. Trends Anal. Chem. 2005, 24, 9–19. [Google Scholar] [CrossRef]
- Lin, C.; Guo, Y.; Zhao, M.; Sun, M.; Luo, F.; Guo, L.; Qiu, B.; Lin, Z.; Chen, G. Highly sensitive colorimetric immunosensor for influenza virus H5N1 based on enzyme-encapsulated liposome. Anal. Chim. Acta 2017, 963, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Josephy, P.D.; Eling, T.; Mason, R.P. Horseradish Peroxidase-catalyzed Oxidations of 3,5,3′,5′-Tetramethylbenzidine. J. Biol. Chem. 1982, 257, 3669–3675. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Higa, M. A kinetic analysis of catalytic production of oxygen in catalase-containing liposome dispersions for controlled transfer of oxygen in a bioreactor. J. Chem. Technol. Biotechnol. 2014, 89, 1388–1395. [Google Scholar] [CrossRef]
- Beck, S.M.; Brus, L.E. Transient Spontaneous Raman Study of Photoionization Kinetics at the Hydrocarbon/Water Interface in Micellar Solutions. J. Am. Chem. Soc. 1983, 105, 1106–1111. [Google Scholar] [CrossRef]
- Umena, Y.; Yorita, K.; Matsuoka, T.; Kita, A.; Fukui, K.; Morimoto, Y. The crystal structure of L-lactate oxidase from Aerococcus viridans at 2.1 Å resolution reveals the mechanism of strict substrate recognition. Biochem. Biophys. Res. Commun. 2006, 350, 249–256. [Google Scholar] [CrossRef]
- Maeda-Yorita, K.; Aki, K.; Sagai, H.; Misaki, H.; Massey, V. L-Lactate oxidase and L-lactate monooxygenase: Mechanistic variations on a common structural theme. Biochimie 1995, 77, 631–642. [Google Scholar] [CrossRef]
- Matoori, S.; Mooney, D.J. Near-Infrared Fluorescence Hydrogen Peroxide Assay for Versatile Metabolite Biosensing in Whole Blood. Small 2020, 16, 2000369. [Google Scholar] [CrossRef] [PubMed]
- Gabba, M.; Frallicciardi, J.; van’t Klooster, J.; Henderson, R.; Syga, Ł.; Mans, R.; van Maris, A.J.A.; Poolman, B. Weak Acid Permeation in Synthetic Lipid Vesicles and Across the Yeast Plasma Membrane. Biophys. J. 2020, 118, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, T.M.; Davidson, P.M.; Bruce, B.D.; Weiss, J. Liposomal Nanocapsules in Food Science and Agriculture. Crit. Rev. Food Sci. Nutr. 2005, 45, 587–605. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.C. Liposomes in nutrition. Trends Food Sci. Technol. 2001, 12, 25–31. [Google Scholar] [CrossRef]
- Singh, H.; Thompson, A.; Liu, W.; Corredig, M. Liposoms as food ingredients and nutraceutical delivery systems. In Encapsulation Technologies and Delivery Systems for Food Ingredients and Nutraceuticals; Garti, N., McClements, D.J., Eds.; Woodhead Publishing: Oxford, UK, 2012; Chapter 11; pp. 288–318. [Google Scholar]
- Khorasani, S.; Danaei, M.; Mozafari, M.R. Nanoliposome technology for the food and nutraceutical industries. Trends Food Sci. Technol. 2018, 79, 106–115. [Google Scholar] [CrossRef]
- Zhao, L.; Temelli, F.; Chen, L. Encapsulation of anthocyanin in liposomes using supercritical carbon dioxide: Effects of anthocyanin and sterol concentrations. J. Funct. Foods 2017, 34, 159–167. [Google Scholar] [CrossRef]
- Marín, D.; Alemán, A.; Montero, P.; Gómez-Guillén, M.C. Encapsulation of food waste compounds in soy phosphatidylcholine liposomes: Effect of freeze-drying, storage stability and functional aptitude. J. Food Eng. 2018, 223, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Kheadr, E.E.; Vuillemard, J.-C.; El Deeb, S.A. Accelerated Cheddar cheese ripening with encapsulated proteinases. Int. J. Food Sci. Technol. 2000, 35, 483–495. [Google Scholar] [CrossRef]
- Kheadr, E.E.; Vuillemard, J.-C.; El-Deeb, S.A. Impact of liposome-encapsulated enzyme cocktails on cheddar cheese ripening. Food Res. Int. 2003, 36, 241–252. [Google Scholar] [CrossRef]
- da Silva Malheiros, P.; Daroit, D.J.; Brandelli, A. Food applications of liposome-encapsulated antimicrobial peptides. Trends Food Sci. Technol. 2010, 21, 284–292. [Google Scholar] [CrossRef]
- Vergara, D.; López, O.; Bustamante, M.; Shene, C. An in vitro digestion study of encapsulated lactoferrin in rapeseed phospholipid-based liposomes. Food Chem. 2020, 321, 126717. [Google Scholar] [CrossRef] [PubMed]
- García-Toledo, J.A.; Torrestiana-Sánchez, B.; Martínez-Sánchez, C.E.; Tejero-Andrade, J.M.; García-Bórquez, A.; Mendoza-García, P.G. Nanoencapsulation of a Bacteriocin from Pediococcus acidilactici ITV26 by Microfluidization. Food Bioprocess Technol. 2019, 12, 88–97. [Google Scholar] [CrossRef]
- Liu, X.; Wang, P.; Zou, Y.-X.; Luo, Z.-G.; Tamer, T.M. Co-encapsulation of Vitamin C and β-Carotene in liposomes: Storage stability, antioxidant activity, and in vitro gastrointestinal digestion. Food Res. Int. 2020, 136, 109587. [Google Scholar] [CrossRef] [PubMed]
- Paolino, D.; Mancuso, A.; Cristiano, M.C.; Froiio, F.; Lammari, N.; Celia, C.; Fresta, M. Nanonutraceuticals: The New Frontier of Supplementary Food. Nanomaterials 2021, 11, 792. [Google Scholar] [CrossRef] [PubMed]
- Emani, S.; Azadmard-Damirichi, S.; Peighambardoust, S.H.; Valizadeh, H.; Hesari, J. Liposomes as carrier vehicles for functional compounds in food sector. J. Exp. Nanosci. 2016, 11, 737–759. [Google Scholar] [CrossRef]
- Shukla, S.; Haldorai, Y.; Hwang, S.K.; Bajpai, V.K.; Huh, Y.S.; Han, Y.-K. Current Demands for Food-Approved Liposome Nanoparticles in Food and Safety Sector. Front. Microbiol. 2017, 8, 02398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasibi, F.; Nasirouir, A.; Varshosaz, J.; García-Manrique, P.; Blanco-López, M.C.; Gutiérrez, G.; Matos, M. Formulation and Characterization of Taxifolin-Loaded Lipid Nanovesicles (Liposomes, Niosomes, and Transfersomes) for Beverage Fortification. Eur. J. Lipid Sci. Technol. 2020, 122, 1900105. [Google Scholar] [CrossRef]
- Subramani, T.; Ganapathyswamy, H. An overview of liposomal nano-encapsulation techniques and its applications in food and nutraceutical. J. Food Sci. Technol. 2020, 57, 3545–3555. [Google Scholar] [CrossRef] [PubMed]
- Zarrabi, A.; Abadi, M.A.A.; Khorasani, S.; Mohammadabadi, M.-R.; Jamshidi, A.; Torkaman, S.; Taghavi, E.; Mozafari, M.R.; Rasti, B. Nanoliposomes and Tocosomes as Multifunctional Nanocarriers for the Encapsulation of Nutraceutical and Dietary Molecules. Molecules 2020, 25, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blesso, C.N. Egg Phospholipids and Cardiovascular Health. Nutrients 2015, 7, 2731–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta 2017, 1859, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Couëdelo, L.; Vayasse, C.; Michalski, M.-C. Vegetable lecithins: A review of their compositional diversity, impact on lipid metabolism and potential in cardiometabolic disease prevention. Biochimie 2020, 169, 121–132. [Google Scholar] [CrossRef]
- Kim, I.-S.; Kim, C.-H.; Yang, W.-S. Physiologically Active Molecules and Functional Properties of Soybeans in Human Health—A Current Perspective. Int. J. Mol. Sci. 2021, 22, 4054. [Google Scholar] [CrossRef] [PubMed]
- Łukawski, M.; Dałek, P.; Borowik, T.; Foryś, A.; Langner, M.; Witkiewicz, W.; Przybło, M. New oral liposomal vitamin C formulation: Properties and bioavailability. J. Liposome Res. 2020, 30, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Prantl, L.; Eigenberger, A.; Gehmert, S.; Haerteis, S.; Aung, T.; Rachel, R.; Jung, E.M.; Felthaus, O. Enhanced Resorption of Liposomal Packed Vitamin C Monitored by Ultrasound. J. Clin. Med. 2020, 9, 1616. [Google Scholar] [CrossRef] [PubMed]
- van Hoogevest, P. Review—An update on the use of oral phospholipid excipients. Eur. J. Pharm. Sci. 2017, 108, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kayushin, L.P.; Skulachev, V.P. Bacteriorhodopsin as an Electrogenic Proton Pump: Reconstitution of Bacteriorhodopsin Proteoliposomes Generating Δψ and ΔpH. FEBS Lett. 1974, 39, 39–42. [Google Scholar] [CrossRef] [Green Version]
- Eytan, G.D. Use of Liposomes for Reconstitution of Biological Functions. Biochim. Biophys. Acta 1982, 694, 185–202. [Google Scholar] [CrossRef]
- Seddon, A.M.; Curnow, P.; Booth, P.J. Membrane proteins, lipids and detergents: Not just a soap opera. Biochim. Biophys. Acta 2004, 1666, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulik-Krzywicki, T.; Seigneuret, M.; Rigaud, J.L. Monomer-Oligomer Equilibrium of Bacteriorhodopsin in Reconstituted Proteoliposomes. A Freeze Fracture Electron Microscopy Study. J. Biol. Chem. 1987, 262, 15580–15588. [Google Scholar] [CrossRef]
- Rigaud, J.-L.; Lévy, D. Reconstitution of Membrane Proteins into Liposomes. Methods Enzymol. 2003, 372, 65–86. [Google Scholar]
- Walde, P.; Ichikawa, S.; Yoshimoto, M. The Fabrication and Applications of Enzyme-Containing Vesicles. In Bottom-Up Nanofabrication; Ariga, K., Nalwa, H.S., Eds.; American Scientific Publishers: Steveson Ranch, CA, USA, 2009; Volume 2, pp. 199–221. [Google Scholar]
- Lévy, D.; Gulik, A.; Bluzat, A.; Rigaud, J.-L. Reconstitution of the sarcoplasmic reticulum Ca2+-ATPase: Mechanisms of membrane protein insertion into liposomes during reconstitution procedures involving the use of detergents. Biochim. Biophys. Acta 1992, 1107, 283–298. [Google Scholar] [CrossRef]
- Rigaud, J.-L.; Pitard, B.; Levy, D. Reconstitution of membrane proteins into liposomes: Application to energy-transducing membrane proteins. Biochim. Biophys. Acta 1995, 1231, 223–246. [Google Scholar] [CrossRef] [Green Version]
- Girard, P.; Pécréaux, J.; Lenoir, G.; Falson, P.; Rigaud, J.-L.; Bassereau, P. A New Method for the Reconstitution of Membrane Proteins into Giant Unilamellar Vesicles. Biophys. J. 2004, 87, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Dezi, M.; Di Cicco, A.; Bassereau, P.; Lévy, D. Detergent-mediated incorporation of transmembrane proteins in giant unilamellar vesicles with controlled physiological contents. Proc. Natl. Acad. Sci. USA 2013, 110, 7276–7281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, I.L.; Kemmer, G.C.; Pomorski, T.G. Membrane protein reconstitution into giant unilamellar vesicles: A review on current techniques. Eur. Biophys. J. 2017, 46, 103–119. [Google Scholar] [CrossRef]
- Amati, A.M.; Graf, S.; Deutschmann, S.; Dolder, N.; von Ballmoos, C. Current problems and future avenues in proteoliposome research. Biochem. Soc. Trans. 2020, 48, 1473–1492. [Google Scholar] [CrossRef] [PubMed]
- Garten, M.; Lévy, D.; Bassereau, P. Protein reconstitution in giant vesicles. In The Giant Vesicle Book; Dimova, R., Marques, C.M., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2020; Chapter 3; pp. 37–51. [Google Scholar]
- Markones, M.; Fippel, A.; Kaiser, M.; Drechsler, C.; Hunte, C.; Heerklotz, H. Stairway to Asymmetry: Five Steps to Lipid-Asymmetric Proteoliposomes. Biophys. J. 2020, 118, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Fan, X.; Yan, N. Cryo-EM analysis of a membrane protein embedded in the liposome. Proc. Natl. Acad. Sci. USA 2020, 117, 18497–18503. [Google Scholar] [CrossRef]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Yamaguchi, A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 2002, 419, 587–593. [Google Scholar] [CrossRef]
- Baumgart, T.; Hammond, A.T.; Sengupta, P.; Hess, S.T.; Holowka, D.A.; Baird, B.A.; Webb, W.W. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc. Natl. Acad. Sci. USA 2007, 104, 3165–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levental, I.; Grzybek, M.; Simons, K. Raft domains of variable properties and compositions in plasma membrane vesicles. Proc. Natl. Acad. Sci. USA 2011, 108, 11411–11416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinkühler, J.; Sezgin, E.; Urbančič, I.; Eggeling, C.; Dimova, R. Mechanical properties of plasma membrane vesicles correlate with lipid order, viscosity and cell density. Commun. Biol. 2019, 2, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinkle, A.D.; Levental, K.R.; Levental, I. Cell-Derived Plasma Membrane Vesicles Are Permeable to Hydrophilic Macromolecules. Biophys. J. 2020, 118, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Lorent, J.H.; Levental, I. Preparation and properties of giant plasma membrane vesicles and giant unilamellar vesicles from natural membranes. In The Giant Vesicle Book; Dimova, R., Marques, C.M., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2020; Chapter 2; pp. 21–36. [Google Scholar]
- Srivatsav, A.T.; Kapoor, S. The Emerging World of Membrane Vesicles: Functional Relevance, Theranostic Avenues and Tools for Investigating Membrane Function. Front. Mol. Biosci. 2021, 8, 640355. [Google Scholar] [CrossRef]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 2012, 1820, 940–948. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Wiklander, O.P.B.; Brennan, M.Á.; Lötvall, J.; Breakefield, X.O.; Andaloussi, S.E.L. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, eaav8521. [Google Scholar] [CrossRef] [PubMed]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelery, R.M.; Andaloussi, S.E.L.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, 159, 332–343. [Google Scholar] [CrossRef]
- Gandham, S.; Su, X.; Wood, J.; Nocera, A.L.; Alli, S.C.; Milane, L.; Zimmerman, A.; Amiji, M.; Ivanov, A.R. Technologies and Standardization in Research on Extracellular Vesicles. Trends Biotechnol. 2020, 38, 1066–1098. [Google Scholar] [CrossRef] [PubMed]
- Prosser, R.S.; Evanics, F.; Kitevski, J.L.; Al-Abdul-Wahid, M.S. Current Applications of Bicelles in NMR Studies of Membrane-Associated Amphiphiles and Proteins. Biochemistry 2006, 45, 8453–8465. [Google Scholar] [CrossRef] [PubMed]
- Dürr, U.H.N.; Gildeberg, M.; Ramamoorthy, A. The Magic of Bicelles Lights Up Membrane Protein Structure. Chem. Rev. 2012, 112, 6054–6074. [Google Scholar] [CrossRef] [PubMed]
- Morrison, E.A.; Henzler-Wildman, K.A. Reconstitution of integral membrane proteins into isotropic bicelles with improved sample and expanded lipid composition profile. Biochim. Biophys. Acta 2012, 1818, 814–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberholzer, T.; Nierhaus, K.H.; Luisi, P.L. Protein Expression in Liposomes. Biochem. Biophys. Res. Commun. 1999, 261, 238–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, S.M.; Tsumoto, K.; Hamada, T.; Akiyoshi, K.; Nakatani, Y.; Yoshikawa, K. Gene Expression within Cell-Sized Lipid Vesicles. ChemBioChem 2003, 4, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Sato, K.; Shima, Y.; Urabe, I.; Yomo, T. Expression of a cascading genetic network within liposomes. FEBS Lett. 2004, 576, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luisi, P.L. Toward the Engineering of Minimal Living Cells. Anat. Rec. 2002, 268, 208–214. [Google Scholar] [CrossRef]
- Stano, P. Is Research on “Synthetic Cells” Moving to the Next Level? Life 2019, 9, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Hu, S.; Chen, X. Artificial cells: From basic science to applications. Mater. Today 2016, 19, 516–532. [Google Scholar] [CrossRef]
- Buddingh, B.C.; van Hest, J.C.M. Artificial Cells: Synthetic Compartments with Life-like Functionality and Adaptivity. Acc. Chem. Res. 2017, 50, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trantidou, T.; Friddin, M.; Elani, Y.; Brooks, N.J.; Law, R.V.; Seddon, J.M.; Ces, O. Engineering Compartmentalized Biomimetic Micro- and Nanocontainers. ACS Nano 2017, 11, 6549–6565. [Google Scholar] [CrossRef] [PubMed]
- Yewdall, N.A.; Mason, A.F.; van Hest, J.C.M. The hallmarks of living systems: Towards creating artificial cells. Interface Focus 2018, 8, 20180023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai, L.; Schwille, P. Cell-Free Protein Synthesis and Its Perspectives for Assembling Cells from the Bottom-Up. Adv. Biosys. 2019, 3, 1800322. [Google Scholar] [CrossRef] [PubMed]
- Lentini, R.; Martín, N.Y.; Forlin, M.; Belmonte, L.; Fontana, J.; Cornella, M.; Martini, L.; Tamburini, S.; Bentley, W.E.; Jousson, O.; et al. Two-Way Chemical Communication between Artificial and Natural Cells. ACS Cent. Sci. 2017, 3, 117–123. [Google Scholar] [CrossRef]
- Kuruma, Y.; Stano, P.; Ueda, T.; Luisi, P.L. A synthetic biology approach to the construction of membrane proteins in semi-synthetic minimal cells. Biochim. Biophys. Acta 2009, 1788, 567–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soga, H.; Fujii, S.; Yomo, T.; Kato, Y.; Watanabe, H.; Matsuura, T. In Vitro Membrane Protein Synthesis Inside Cell-Sized Vesicles Reveals the Dependence of Membrane Protein Integration on Vesicle Volume. ACS Synth. Biol. 2014, 3, 372–379. [Google Scholar] [CrossRef]
- Altamura, A.; Milano, F.; Tangorra, R.R.; Trotta, M.; Omar, O.H.; Stano, P.; Mavelli, F. Highly oriented photosynthetic reaction centers generate a proton gradient in synthetic protocells. Proc. Natl. Acad. Sci. USA 2017, 114, 3837–3842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altamura, E.; Albanese, P.; Marotta, R.; Milano, F.; Fiore, M.; Trotta, M.; Stano, P.; Mavelli, F. Chromatophores efficiently promote light-driven ATP synthesis and DNA transcription inside hybrid multicompartment artificial cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2012170118. [Google Scholar] [CrossRef] [PubMed]
- Gánti, T. Biogenesis Itself. J. Theor. Biol. 1997, 187, 583–593. [Google Scholar] [CrossRef]
- Gánti, T. The Principles of Life: With a Commentary by James Griesemer and Eörs Szathmáry; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Griesemer, J. The enduring value of Gánti’s chemoton model and life criteria: Heuristic pursuit of exact theoretical biology. J. Theor. Biol. 2015, 381, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Glass, J.I.; Lartigue, C.; Noskov, V.N.; Chuang, R.-Y.; Algire, M.A.; Benders, G.A.; Montague, M.G.; Ma, L.; Moodie, M.M.; et al. Creation of a Bacterial Cell Controlled by a Chemically Synthesized Genome. Science 2010, 329, 52–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. cell-free translation reconstitution with purified components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef]
- Kuruma, Y.; Ueda, T. The PURE system for the cell-free synthesis of membrane proteins. Nat. Protoc. 2015, 10, 1328–1344. [Google Scholar] [CrossRef] [PubMed]
- Luisi, P.L. About Various Definitions of Life. Origins Life Evol. Biospheres 1998, 28, 613–622. [Google Scholar] [CrossRef]
- Cleland, C.E.; Chyba, C.F. Defining ‘Life’. Origins Life Evol. Biospheres 2002, 32, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Mirazo, K.; Peretó, J.; Moreno, A. A Universal Definition of Life: Autonomy and Open-Ended Evolution. Origins Life Evol. Biospheres 2004, 34, 323–346. [Google Scholar] [CrossRef]
- Benner, S.A. Defining Life. Astrobiology 2010, 10, 1021–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bich, L.; Green, S. Is defining life pointless? Operational definitions at the frontiers of biology. Synthese 2018, 195, 3919–3946. [Google Scholar] [CrossRef]
- Amilburu, A.; Moreno, Á.; Ruiz-Mirazo, K. Definitions of life as epistemic tools that reflect and foster the advance of biological knowledge. Synthese 2021, 198, 10565–10585. [Google Scholar] [CrossRef]
- Varela, F.; Maturana, H.R.; Uribe, R. Autopoiesis: The organization of living systems, its characterization and a model. BioSystems 1974, 5, 187–196. [Google Scholar] [CrossRef]
- Luisi, P.L. Autopoiesis: A review and a reappraisal. Naturwissenschaften 2003, 90, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Luisi, P.L. The Minimal Autopoietic Unit. Origins Life Evol. Biospheres 2014, 44, 335–338. [Google Scholar] [CrossRef] [PubMed]







































| Acyl Chain (Fatty Acid) 2 | Egg PC (%) 3 | Soybean PC (%) 3 |
|---|---|---|
| C16:0 (palmitic acid) | 35.0 (37.0 ± 0.07) | 11.2 (13.6 ± 0.06) |
| C18:0 (stearic acid) | 13.4 (14.1 ± 0.03) | 11.9 (3.7 ± 0.02) |
| C18:1 (oleic acid) | 30.4 (22.1 ± 0.05) | 8.6 (10.6 ± 0.06) |
| C18:2 (linoleic acid) | 18.0 (21.2 ± 0.07) | 58.6 65.9 ± 0.09) |
| C18:3 (linolenic acid) | − | 9.9 (6.2 ± 0.04) |
| C20:4 (arachidonic acid) | 3.2 (3.9 ± 0.1) | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walde, P.; Ichikawa, S. Lipid Vesicles and Other Polymolecular Aggregates—From Basic Studies of Polar Lipids to Innovative Applications. Appl. Sci. 2021, 11, 10345. https://doi.org/10.3390/app112110345
Walde P, Ichikawa S. Lipid Vesicles and Other Polymolecular Aggregates—From Basic Studies of Polar Lipids to Innovative Applications. Applied Sciences. 2021; 11(21):10345. https://doi.org/10.3390/app112110345
Chicago/Turabian StyleWalde, Peter, and Sosaku Ichikawa. 2021. "Lipid Vesicles and Other Polymolecular Aggregates—From Basic Studies of Polar Lipids to Innovative Applications" Applied Sciences 11, no. 21: 10345. https://doi.org/10.3390/app112110345
APA StyleWalde, P., & Ichikawa, S. (2021). Lipid Vesicles and Other Polymolecular Aggregates—From Basic Studies of Polar Lipids to Innovative Applications. Applied Sciences, 11(21), 10345. https://doi.org/10.3390/app112110345