
Light-Activating PROTACs in Cancer: Chemical Design, Challenges, and Applications



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
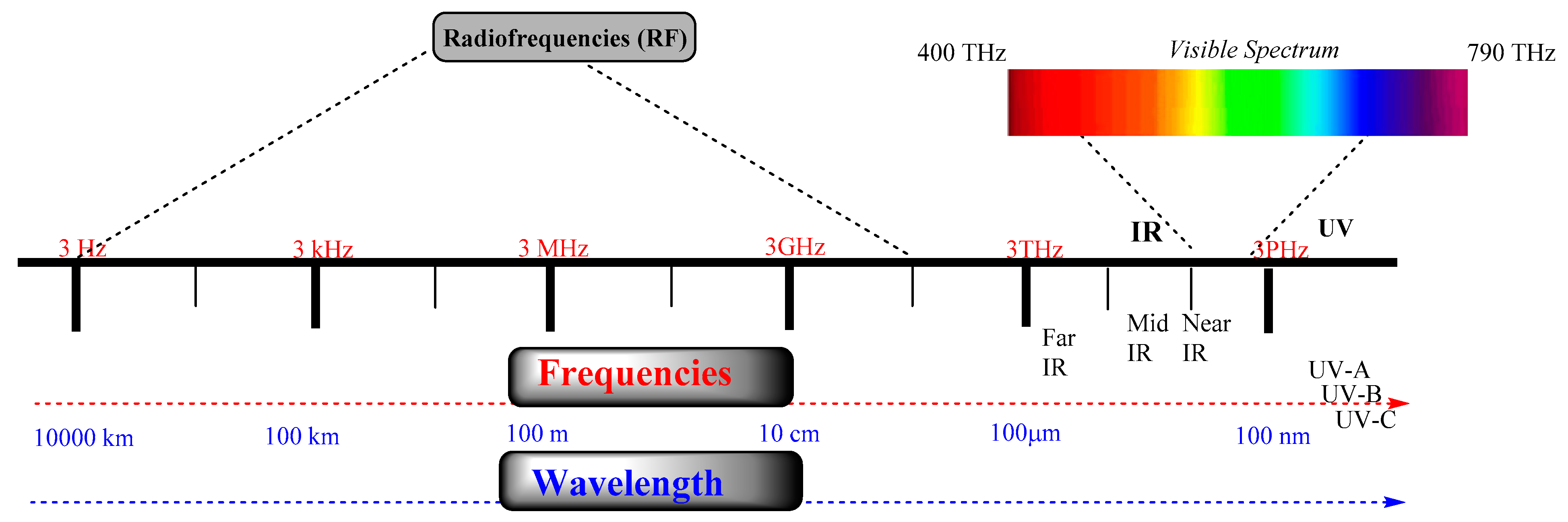
:1. Introduction
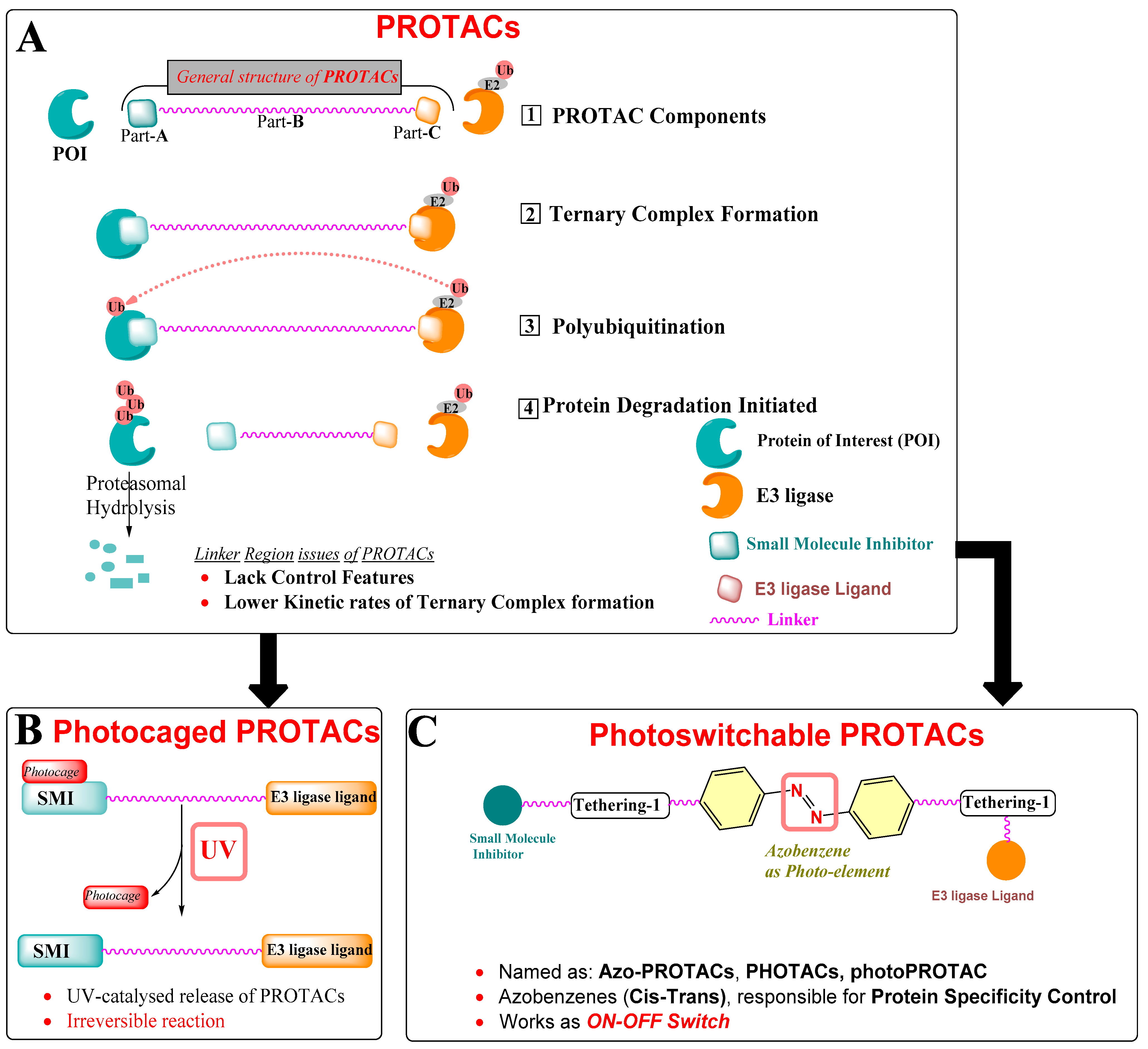
2. Protein Degradation Strategies
3. Intracellular Delivery and Controlling Activation/Deactivation PROTACs
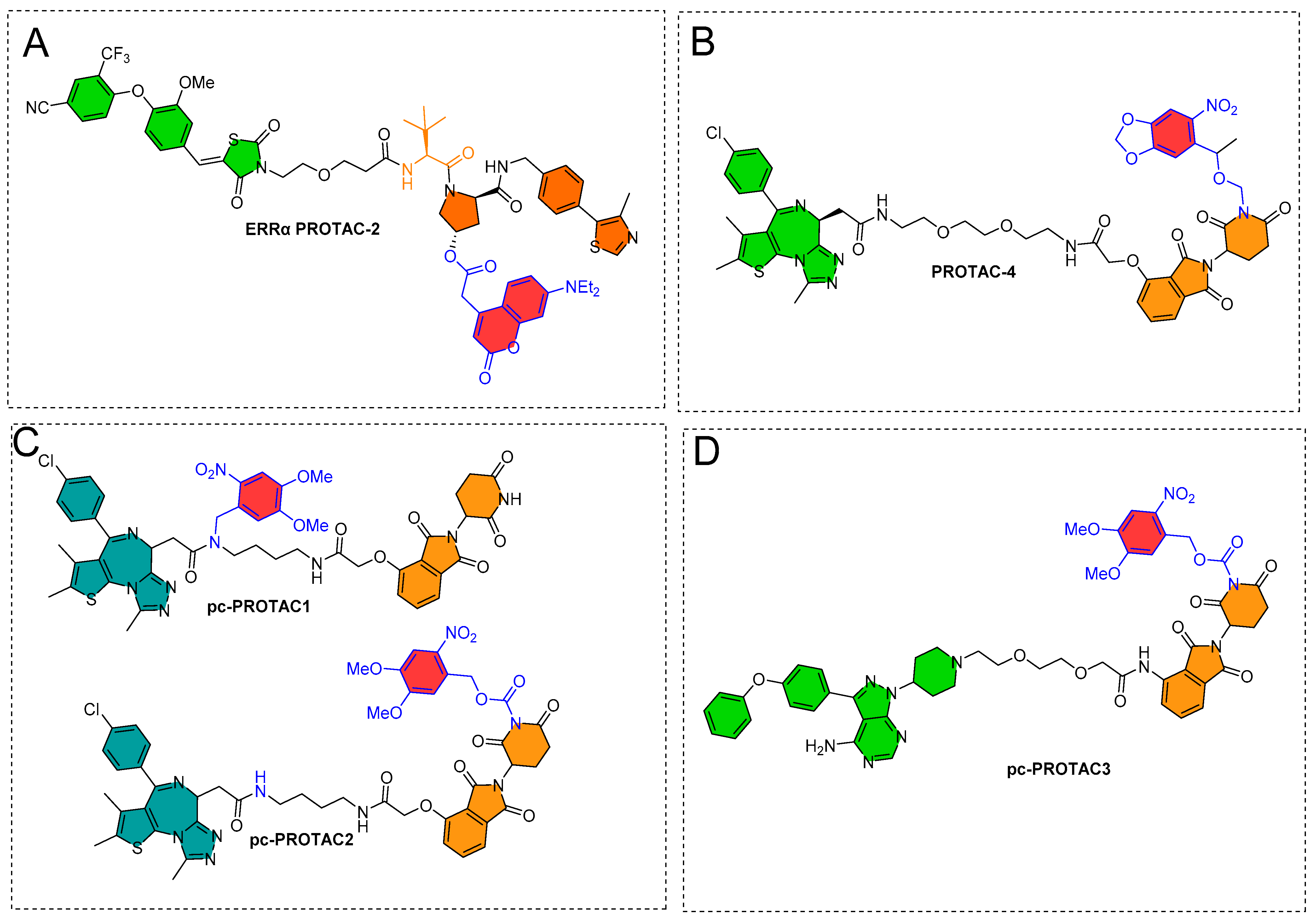
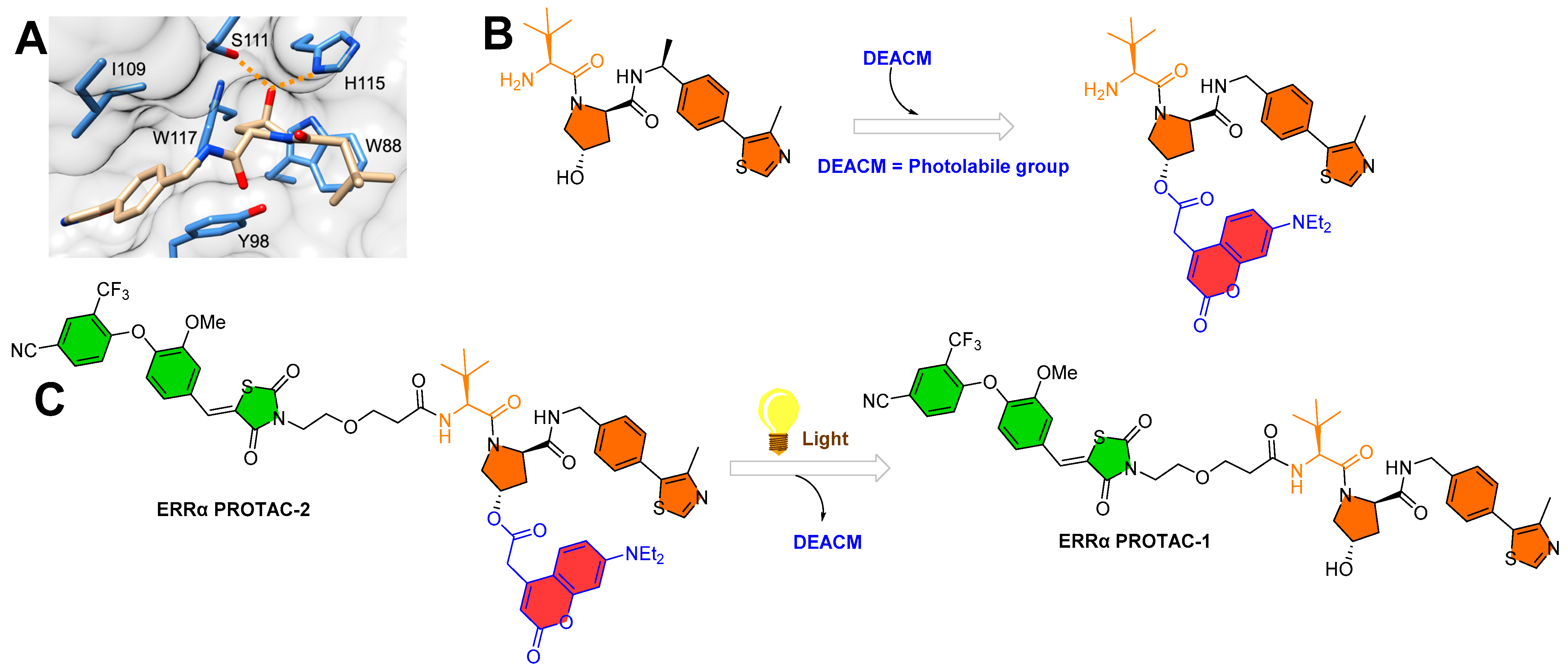
3.1. Photocaging PROTACs of Estrogen Receptor
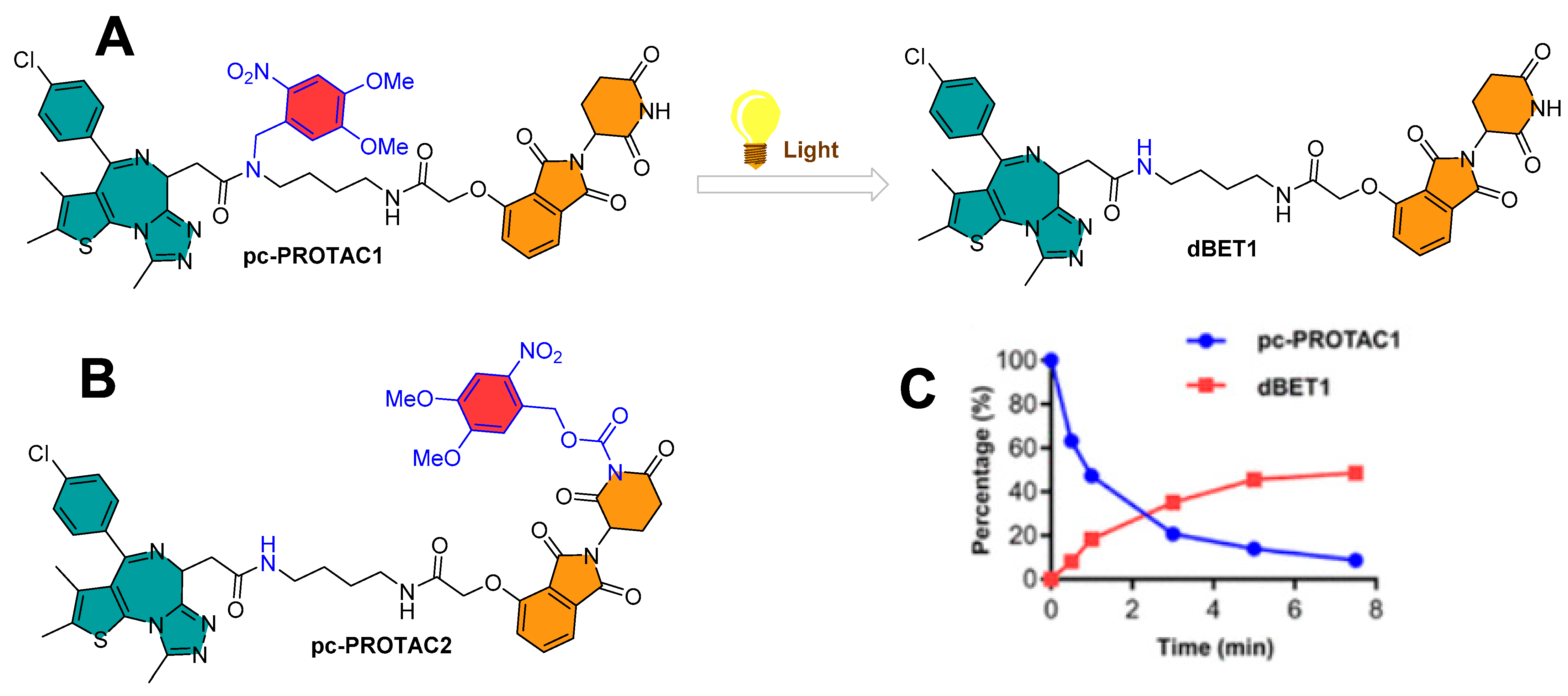
3.2. Photocaging PROTACs of Bromodomain Proteins
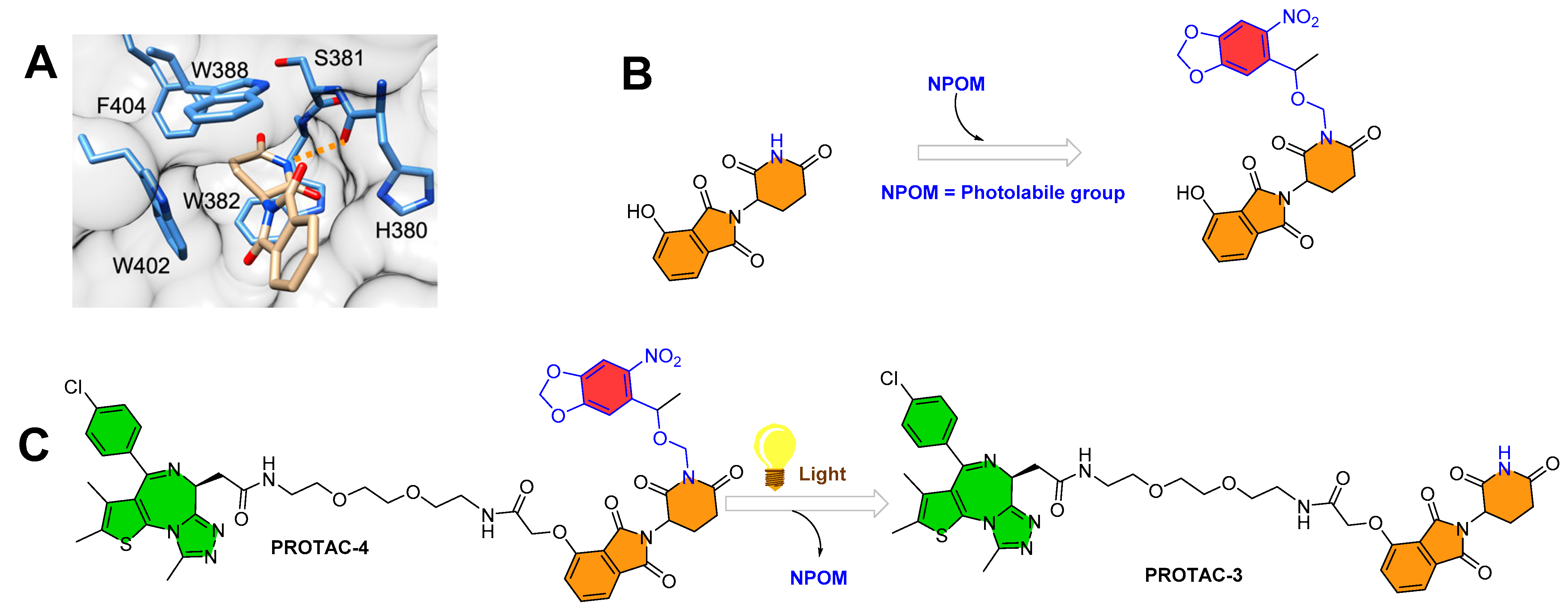
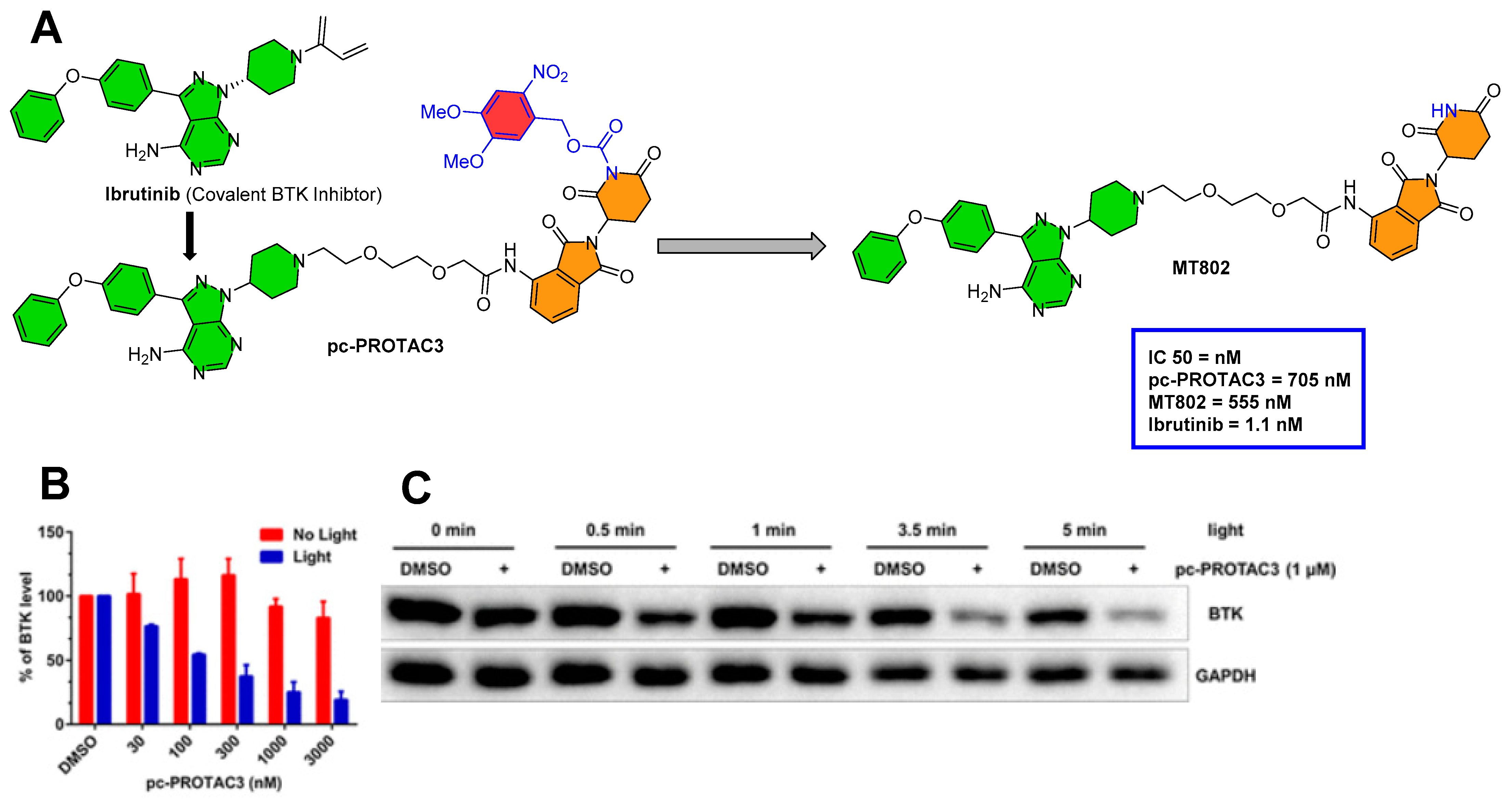
3.3. Photocaging PROTACs of BTK Kinase Proteins
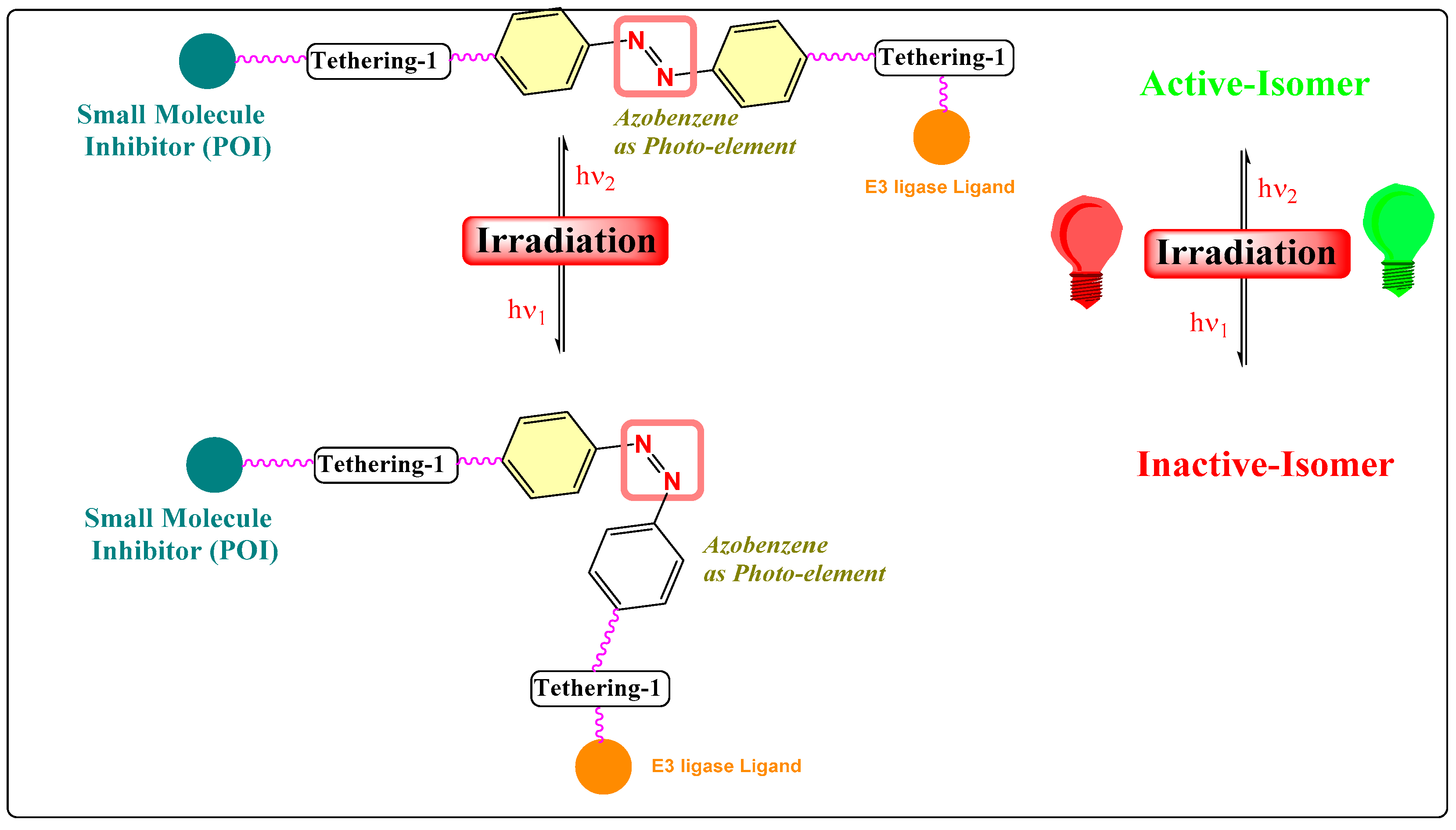
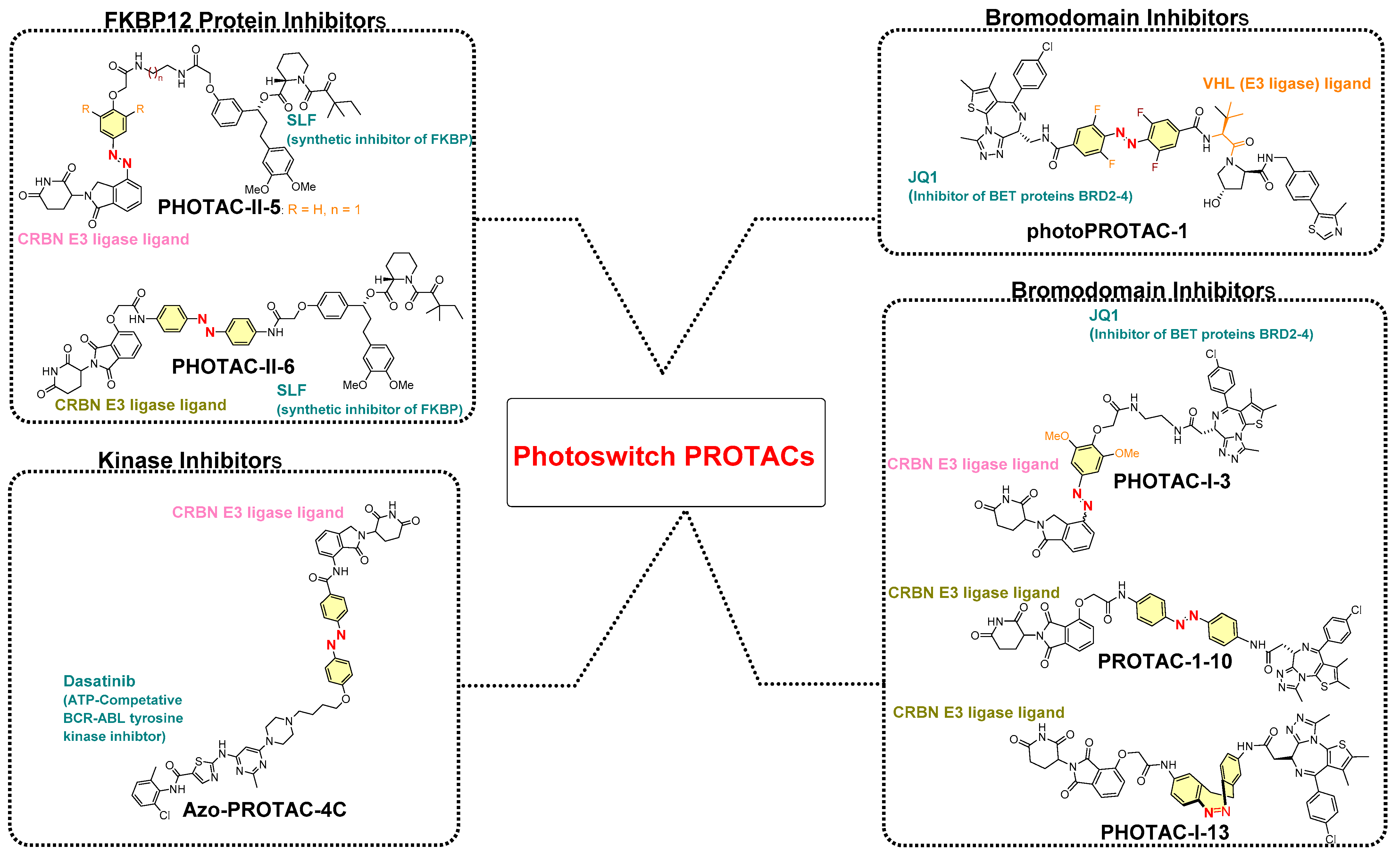
4. Photoswitches in PROTACs
- (i)
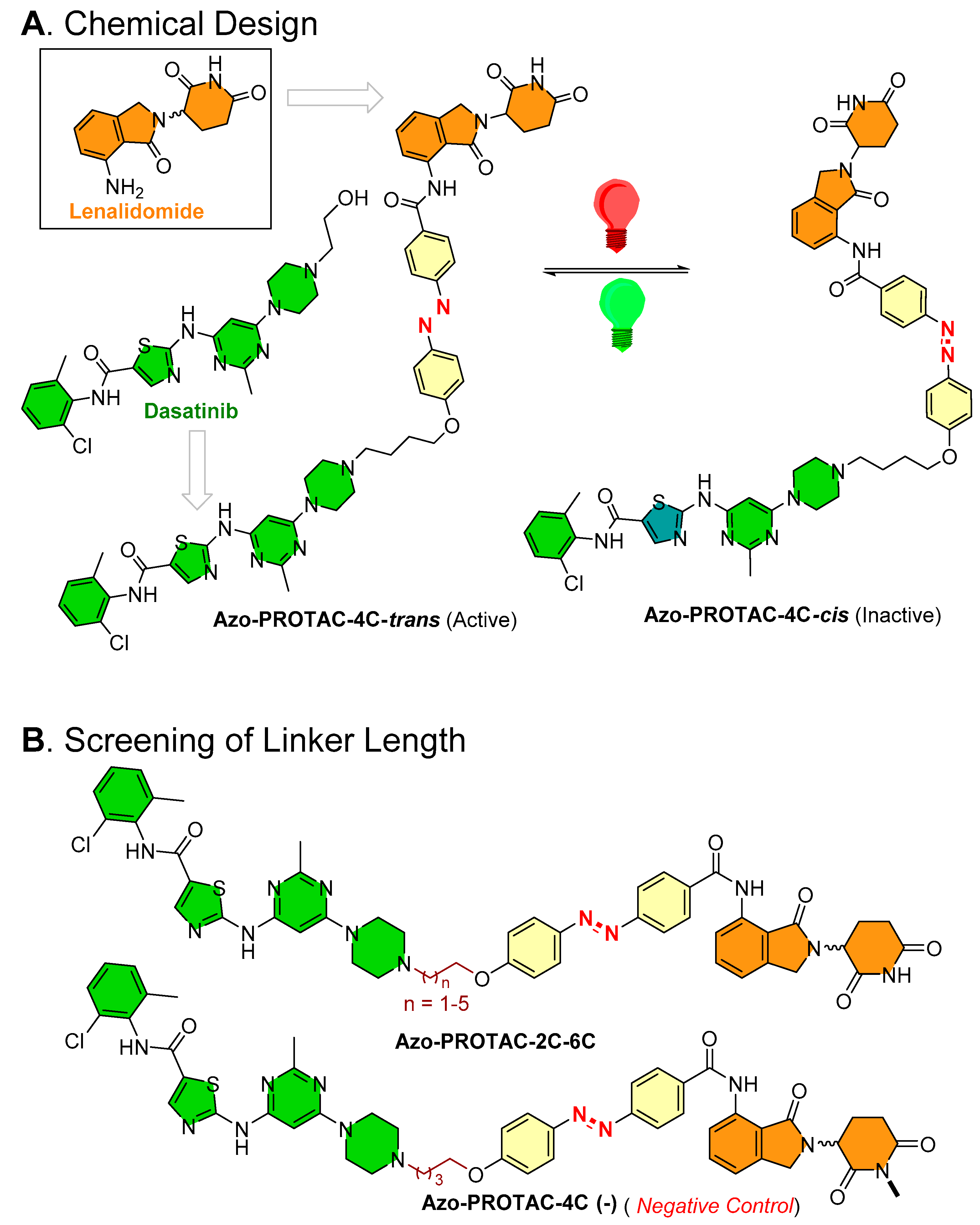
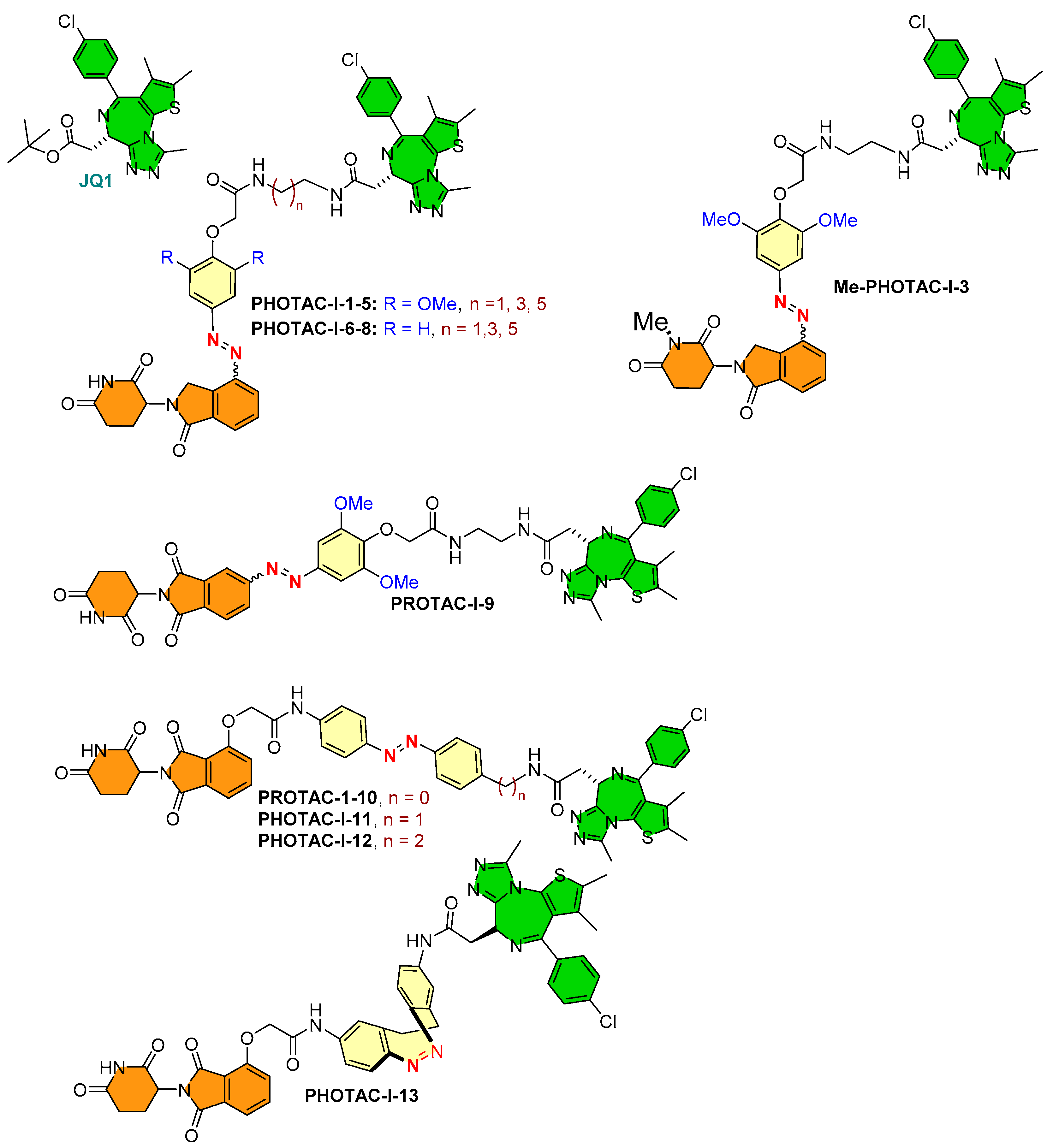
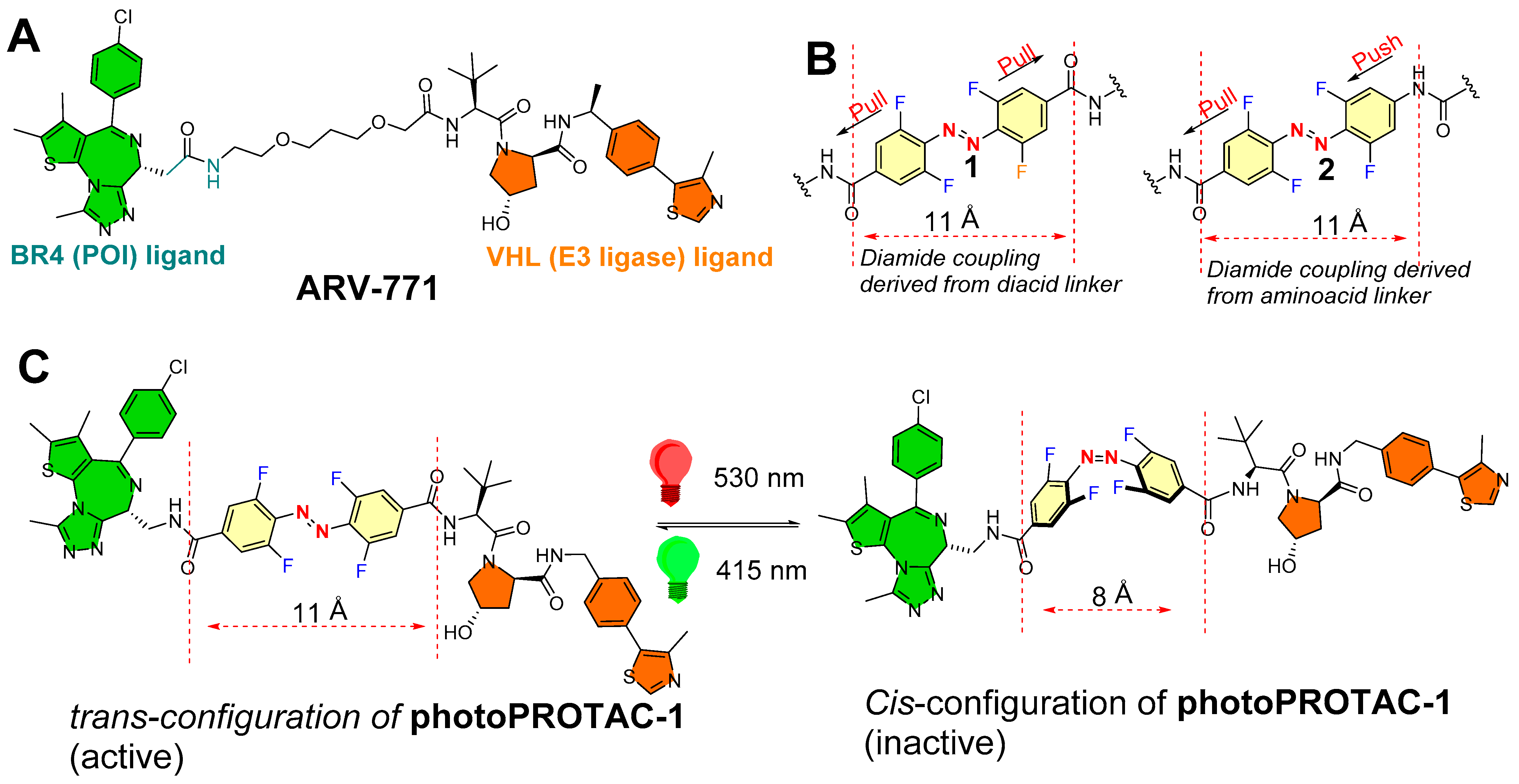
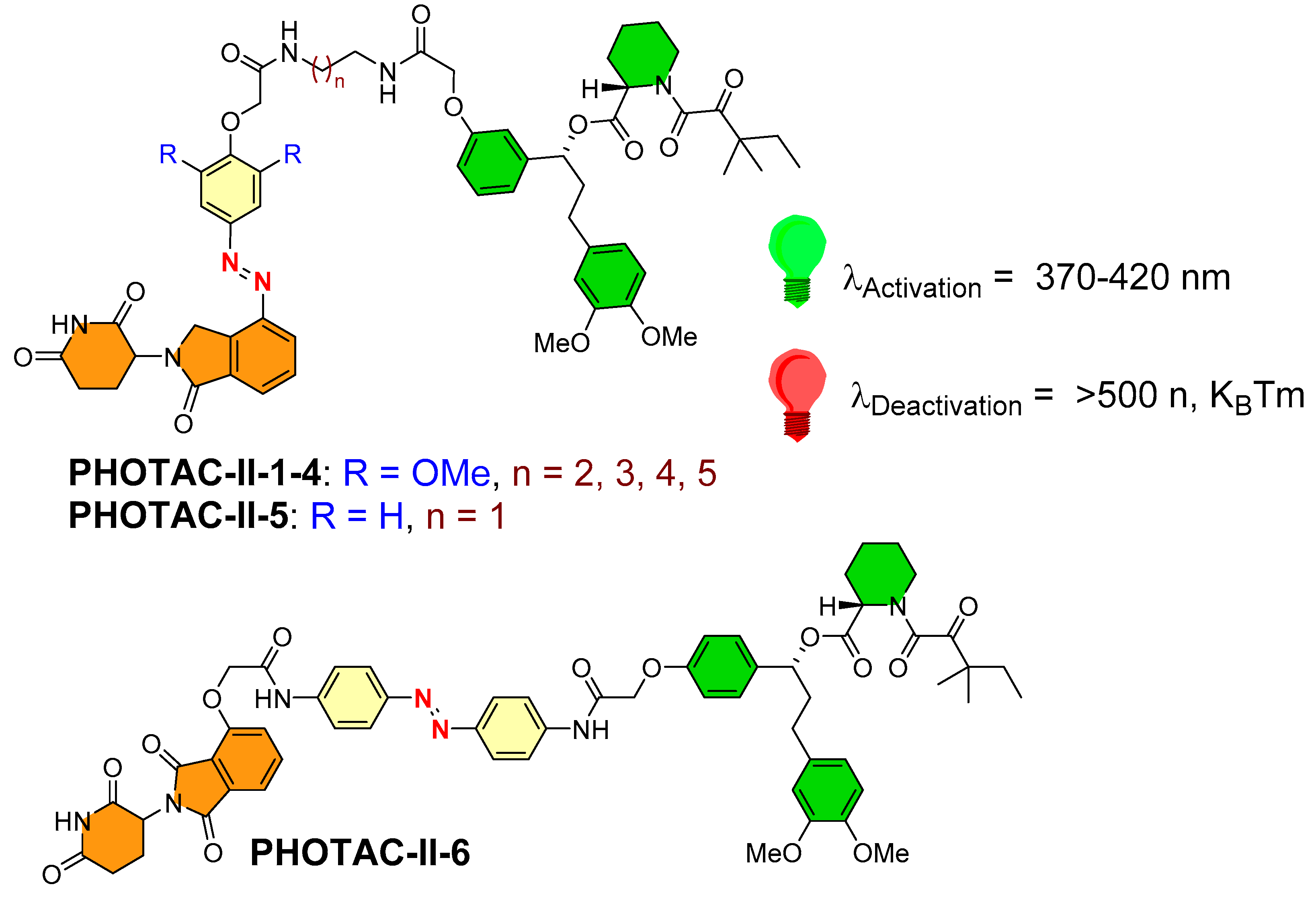
- Photoswitch positioning in the linker region, equidistant from both sides of the PROTAC structure. This is a typical strategy implemented to find an optimum length of the linker, besides improving the chemical nature and orientation of the PROTAC. Reported photoswitchable PROTACs with such positioning of azobenzene photoswitch are commonly tethered through linear hydrocarbon chains (polyether, aliphatic, or amide-aliphatic linkage) to both terminals (for example, Azo-PROTAC-2C-6C shown in Figure 11, PHOTAC-I-11 and 12 shown in Figure 12, Cis-/trans-PhotoPROTAC-1 shown in Figure 13, and PHOTAC-II-6 shown in Figure 14).
- (ii)
- If the photoswitch is directly tethered to the POI warhead, it could restrict the binding conformational of respective photoswitchable PROTAC to its POI. However, it would also pose a synthetic challenge to an organic chemist in building such molecular structures where the azoswitch tethers POI warheads and could significantly reduce the targeting protein degradation; therefore, such attempts were not reported in the literature.
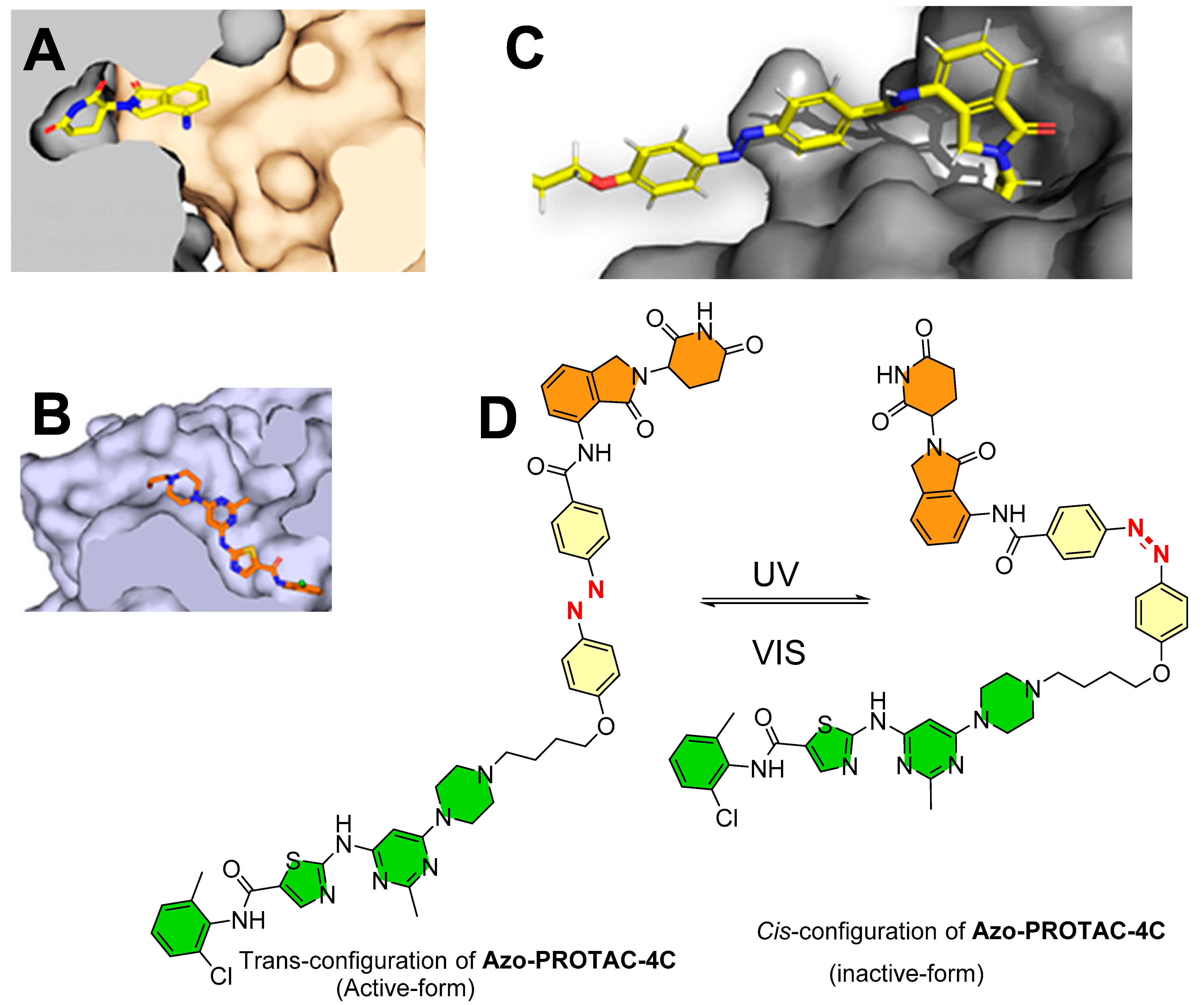
4.1. Photoswitchable PROTACs Targeting Kinase Protein
4.2. Photoswitchable PROTACs in Bromodomain Proteins
4.3. Photoswitchable PROTACs in Immunophilins
5. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Clift, D.; So, C.; McEwan, W.A.; James, L.C.; Schuh, M. Acute and rapid degradation of endogenous proteins by Trim-Away. Nat. Protoc. 2018, 13, 2149–2175. [Google Scholar] [CrossRef] [PubMed]
- Neklesa, T.K.; Tae, H.S.; Schneekloth, A.R.; Stulberg, M.J.; Corson, T.W.; Sundberg, T.B.; Raina, K.; Holley, S.A.; Crews, C.M. Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 2011, 7, 538–543. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar]
- Clift, D.; McEwan, W.A.; Labzin, L.I.; Konieczny, V.; Mogessie, B.; James, L.C.; Schuh, M. A method for the acute and rapid degradation of endogenous proteins. Cell 2017, 171, 1692–1706.e18. [Google Scholar] [CrossRef]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chem. Bio. 2018, 25, 67–77.e3. [Google Scholar] [CrossRef]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Negi, A.; Murphy, P.V. Natural products as Mcl-1 inhibitors: A comparative study of experimental and computational modelling data. Chemistry 2022, 4, 983–1009. [Google Scholar] [CrossRef]
- Wang, Z.; He, N.; Guo, Z.; Niu, C.; Song, T.; Guo, Y.; Cao, K.; Wang, A.; Zhu, J.; Zhang, X.; et al. Proteolysis targeting chimeras for the selective degradation of Mcl-1/Bcl-2 derived from nonselective target binding ligands. J. Med. Chem. 2019, 62, 8152–8163. [Google Scholar] [CrossRef] [PubMed]
- Negi, A.; Murphy, P.V. Development of Mcl-1 inhibitors for cancer therapy. Eur. J. Med. Chem. 2021, 210, 113038. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Bio. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Powell, C.E.; Gao, Y.; Tan, L.; Donovan, K.A.; Nowak, R.P.; Loehr, A.; Bahcall, M.; Fischer, E.S.; Jänne, P.A.; George, R.E.; et al. Chemically induced degradation of anaplastic lymphoma kinase (ALK). J. Med. Chem. 2018, 61, 4249–4255. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar]
- Silva, M.C.; Ferguson, F.M.; Cai, Q.; Donovan, K.A.; Nandi, G.; Patnaik, D.; Zhang, T.; Huang, H.T.; Lucente, D.E.; Dickerson, B.C.; et al. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 2019, 8, e45457. [Google Scholar] [CrossRef]
- Phase, I.A. Clinical Trial of ARV-110 in Patients With Metastatic Castration-resistant Prostate Cancer. Full Text View-Clinical Trials. Available online: https://clinicaltrials.gov/ct2/show/NCT03888612 (accessed on 29 May 2019).
- Broichhagen, J.; Frank, J.A.; Trauner, D. A roadmap to success in photopharmacology. Acc. Chem. Res. 2015, 48, 1947–1960. [Google Scholar] [CrossRef]
- Weinstain, R.; Slanina, T.; Kand, D.; Klán, P. Visible-to-NIR-light activated release: From small molecules to nanomaterials. Chem. Rev. 2020, 120, 13135–13272. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar]
- Negi, A.; Ramarao, P.; Kumar, R. Recent advancements in small molecule inhibitors of insulin–like growth factor-1 receptor (IGF-1R) tyrosine kinase as anticancer agents. Mini Rev. Med. Chem. 2013, 13, 653–681. [Google Scholar] [CrossRef] [PubMed]
- Kramer, C.; Klasmeyer, K.; Bojar, H.; Schulz, W.A.; Ackermann, R.; Grimm, M.O. Heparin-binding epidermal growth factor-like growth factor isoforms and epidermal growth factor receptor/ErbB1 expression in bladder cancer and their relation to clinical outcome. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2007, 109, 2016–2024. [Google Scholar] [CrossRef] [PubMed]
- Stavreus-Evers, A.; Aghajanova, L.; Brismar, H.; Eriksson, H.; Landgren, B.-M.; Hovatta, O. Co-existence of heparin-binding epidermal growth factor-like growth factor and pinopodes in human endometrium at the time of implantation. MHR Basic Sci. Reprod. Med. 2002, 8, 765–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klán, P.; Solomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable protecting groups in chemistry and biology: Reaction mechanisms and efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef]
- Ang, J.M.; Riaz, I.B.; Kamal, M.U.; Paragh, G.; Zeitouni, N.C. Photodynamic therapy and pain: A systematic review. Photodiagnosis Photodyn. Ther. 2017, 19, 308–344. [Google Scholar] [CrossRef]
- Galdeano, C.; Gadd, M.S.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-guided design and optimization of small molecules targeting the protein–protein interaction between the von Hippel–Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 2014, 57, 8657–8663. [Google Scholar] [CrossRef]
- Yu, F.; White, S.B.; Zhao, Q.; Lee, F.S. HIF-1α binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. USA 2001, 98, 9630–9635. [Google Scholar] [CrossRef]
- Min, J.-H.; Yang, H.; Ivan, M.; Gertler, F.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of an HIF-1α-pVHL complex: Hydroxyproline recognition in signaling. Science 2002, 296, 1886–1889. [Google Scholar]
- Hon, W.-C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL. Nature 2002, 417, 975–978. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef]
- Ranhotra, H.S. The estrogen-related receptor alpha: The oldest, yet an energetic orphan with robust biological functions. J. Recept. Signal Transduct. 2010, 30, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Geissler, D.; Hagen, V.; Bendig, J. Mechanism of photocleavage of (coumarin-4-yl) methyl esters. J. Phys. Chem. A 2007, 111, 5768–5774. [Google Scholar] [PubMed]
- Naro, Y.; Darrah, K.; Deiters, A. Optical control of small molecule-induced protein degradation. J. Am. Chem. Soc. 2020, 142, 2193–2197. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, J.M.; Wang, E.S.; Johannessen, L.; Kwiatkowski, N.; Sim, T.; Gray, N.S. Development of highly potent and selective steroidal inhibitors and degraders of CDK8. ACS Med. Chem. Lett. 2018, 9, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Mayer, A.; Buckley, D.L.; Erb, M.A.; Roderick, J.E.; Vittori, S.; Reyes, J.M.; di Iulio, J.; Souza, A.; Ott, C.J. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 2017, 67, 5–18.e19. [Google Scholar] [CrossRef]
- Lusic, H.A.; Deiters, A. A new photocaging group for aromatic N-heterocycles. Synthesis 2006, 2006, 2147–2150. [Google Scholar]
- Young, D.D.; Lively, M.O.; Deiters, A. Activation and deactivation of DNAzyme and antisense function with light for the photochemical regulation of gene expression in mammalian cells. J. Am. Chem. Soc. 2010, 132, 6183–6193. [Google Scholar] [CrossRef]
- Ankenbruck, N.; Courtney, T.; Naro, Y.; Deiters, A. Optochemical control of biological processes in cells and animals. Angew. Chem. Int. Ed. 2018, 57, 2768–2798. [Google Scholar] [CrossRef]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-induced protein degradation with photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Toyama, R.; Rebbert, M.L.; Dey, A.; Ozato, K.; Dawid, I.B. Brd4 associates with mitotic chromosomes throughout early zebrafish embryogenesis. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2008, 237, 1636–1644. [Google Scholar] [CrossRef]
- Buhimschi, A.D.; Armstrong, H.A.; Toure, M.; Jaime-Figueroa, S.; Chen, T.L.; Lehman, A.M.; Woyach, J.A.; Johnson, A.J.; Byrd, J.C.; Crews, C.M. Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry 2018, 57, 3564–3575. [Google Scholar] [CrossRef]
- Reynders, M.; Trauner, D. Optical control of targeted protein degradation. Cell Chem. Bio. 2021, 28, 969–986. [Google Scholar] [CrossRef] [PubMed]
- Negi, A.; Kieffer, C.; Voisin-Chiret, A.S. Azobenzene Photoswitches in Proteolysis Targeting Chimeras: Photochemical Control Strategies and Therapeutic Benefits. ChemistrySelect 2022, 7, e202200981. [Google Scholar] [CrossRef]
- Jin, Y.H.; Lu, M.C.; Wang, Y.; Shan, W.X.; Wang, X.Y.; You, Q.D.; Jiang, Z.Y. Azo-PROTAC: Novel light-controlled small-molecule tool for protein knockdown. J. Med. Chem. 2020, 63, 4644–4654. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, P.; Samarasinghe, K.T.; Crews, C.M.; Carreira, E.M. Reversible spatiotemporal control of induced protein degradation by bistable PhotoPROTACs. ACS Cent. Sci. 2019, 5, 1682–1690. [Google Scholar] [CrossRef]
- Reynders, M.; Matsuura, B.S.; Bérouti, M.; Simoneschi, D.; Marzio, A.; Pagano, M.; Trauner, D. PHOTACs enable optical control of protein degradation. Sci. Adv. 2020, 6, eaay5064. [Google Scholar] [CrossRef]
- Negi, A.; Reilly, C.O.; Jarikote, D.V.; Zhou, J.; Murphy, P.V. Multi-targeting protein-protein interaction inhibitors: Evolution of macrocyclic ligands with embedded carbohydrates (MECs) to improve selectivity. Eur. J. Med. Chem. 2019, 176, 292–309. [Google Scholar] [CrossRef]
- Chamberlain, P.P.; Lopez-Girona, A.; Miller, K.; Carmel, G.; Pagarigan, B.; Chie-Leon, B.; Rychak, E.; Corral, L.G.; Ren, Y.J.; Wang, M.; et al. Structure of the human Cereblon–DDB1–lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 2014, 21, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Tokarski, J.S.; Newitt, J.A.; Chang, C.Y.; Cheng, J.D.; Wittekind, M.; Kiefer, S.E.; Kish, K.; Lee, F.Y.; Borzillerri, R.; Lombardo, L.J.; et al. The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006, 66, 5790–5797. [Google Scholar] [CrossRef]
- Kitagawa, D.; Nakahama, T.; Nakai, Y.; Kobatake, S. 1, 2-Diarylbenzene as fast T-type photochromic switch. J. Mater. Chem. C 2019, 7, 2865–2870. [Google Scholar] [CrossRef]
- Zorba, A.; Nguyen, C.; Xu, Y.; Starr, J.; Borzilleri, K.; Smith, J.; Zhu, H.; Farley, K.A.; Ding, W.; Schiemer, J.; et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. USA 2018, 115, E7285–E7292. [Google Scholar] [CrossRef] [PubMed]
- Douglass, E.F., Jr.; Miller, C.J.; Sparer, G.; Shapiro, H.; Spiegel, D.A. A comprehensive mathematical model for three-body binding equilibria. J. Am. Chem. Soc. 2013, 135, 6092–6099. [Google Scholar]
- Brand, M.; Jiang, B.; Bauer, S.; Donovan, K.A.; Liang, Y.; Wang, E.S.; Nowak, R.P.; Yuan, J.C.; Zhang, T.; Kwiatkowski, N.; et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306.e9. [Google Scholar] [CrossRef]
- Rullo, A.; Reiner, A.; Reiter, A.; Trauner, D.; Isacoff, E.Y.; Woolley, G.A. Long wavelength optical control of glutamate receptor ion channels using a tetra-ortho-substituted azobenzene derivative. Chem. Commun. 2014, 50, 14613–14615. [Google Scholar] [CrossRef]
- Borowiak, M.; Nahaboo, W.; Reynders, M.; Nekolla, K.; Jalinot, P.; Hasserodt, J.; Rehberg, M.; Delattre, M.; Zahler, S.; Vollmar, A.; et al. Photoswitchable inhibitors of microtubule dynamics optically control mitosis and cell death. Cell 2015, 162, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Trads, J.B.; Hüll, K.; Matsuura, B.S.; Laprell, L.; Fehrentz, T.; Görldt, N.; Kozek, K.A.; Weaver, C.D.; Klöcker, N.; Barber, D.M.; et al. Sign inversion in photopharmacology: Incorporation of cyclic azobenzenes in photoswitchable potassium channel blockers and openers. Angew. Chem. 2019, 131, 15567–15574. [Google Scholar] [CrossRef]
- Hu, Z.; Crews, C.M. Recent Developments in PROTAC-Mediated Protein Degradation: From Bench to Clinic. ChemBioChem 2022, 23, e202100270. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018, 25, 78–87.e5. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Crew, A.P.; Raina, K.; Dong, H.; Qian, Y.; Wang, J.; Vigil, D.; Serebrenik, Y.V.; Hamman, B.D.; Morgan, A.; Ferraro, C.; et al. Identification and characterization of Von Hippel-Lindau-recruiting proteolysis targeting chimeras (PROTACs) of TANK-binding kinase 1. J. Med. Chem. 2018, 61, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Hu, J.; Xu, F.; Chen, Z.; Bai, L.; Fernandez-Salas, E.; Lin, M.; Liu, L.; Yang, C.Y.; Zhao, Y.; et al. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J. Med. Chem. 2018, 61, 462–481. [Google Scholar] [CrossRef]
- Bléger, D.; Schwarz, J.; Brouwer, A.M.; Hecht, S. o-Fluoroazobenzenes as readily synthesized photoswitches offering nearly quantitative two-way isomerization with visible light. J. Am. Chem. Soc. 2012, 134, 20597–20600. [Google Scholar] [CrossRef] [PubMed]
- Knie, C.; Utecht, M.; Zhao, F.; Kulla, H.; Kovalenko, S.; Brouwer, A.M.; Saalfrank, P.; Hecht, S.; Bléger, D. ortho-Fluoroazobenzenes: Visible light switches with very long-lived z isomers. Chem. Eur. J. 2014, 20, 16492–16501. [Google Scholar] [CrossRef]
- Passlick, S.; Richers, M.T.; Ellis-Davies, G.C. Thermodynamically stable, photoreversible pharmacology in neurons with one-and two-photon excitation. Angew. Chem. Int. Ed. 2018, 130, 12734–12737. [Google Scholar] [CrossRef]
- Velema, W.A.; Szymanski, W.; Feringa, B.L. Photopharmacology: Beyond proof of principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. [Google Scholar] [CrossRef] [Green Version]
- Vilella-Bach, M.; Nuzzi, P.; Fang, Y.; Chen, J. The FKBP12-rapamycin-binding domain is required for FKBP12-rapamycin-associated protein kinase activity and G1 progression. J. Biol. Chem. 1999, 274, 4266–4272. [Google Scholar] [CrossRef]
- Li, J.; Wen, P.-C.; Moradi, M.; Tajkhorshid, E. Computational characterization of structural dynamics underlying function in active membrane transporters. Curr. Opin. Struct. Biol. 2015, 31, 96–105. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, W. Ligand-mediated active targeting for enhanced oral absorption. Drug Discov. Today 2014, 19, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Anand, B.S.; Dey, S.; Mitra, A.K. Current prodrug strategies via membrane transporters/receptors. Expert Opin. Biol. Ther. 2002, 2, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Negi, A.; Voisin-Chiret, A.S. Strategies to reduce the On-target Platelet Toxicity of Bcl-xL Inhibitors: PROTACs, SNIPERs and Prodrug-based approaches. ChemBioChem 2022, e202100689. [Google Scholar]
- Zhang, C.; Zeng, Z.; Cui, D.; He, S.; Jiang, Y.; Li, J.; Huang, J.; Pu, K. Semiconducting polymer nano-PROTACs for activatable photo-immunometabolic cancer therapy. Nat. Commun. 2021, 12, 2934. [Google Scholar] [CrossRef]
- Negi, A.; Kesari, K.K. Chitosan Nanoparticle Encapsulation of Antibacterial Essential Oils. Micromachines 2022, 13, 1265. [Google Scholar] [CrossRef]
- Profio, A.E. Light transport in tissue. Appl. Opt 1989, 28, 2216–2222. [Google Scholar] [CrossRef]
- Zhang, H.; Salo, D.C.; Kim, D.M.; Komarov, S.; Tai, Y.C.; Berezin, M.Y. Penetration depth of photons in biological tissues from hyperspectral imaging in shortwave infrared in transmission and reflection geometries. J. Biomed. Opt. 2016, 21, 126006. [Google Scholar] [CrossRef]
- Cabré, G.; Garrido-Charles, A.; Moreno, M.; Bosch, M.; Porta-de-la-Riva, M.; Krieg, M.; Gascón-Moya, M.; Camarero, N.; Gelabert, R.; Lluch, J.M.; et al. Rationally designed azobenzene photoswitches for efficient two-photon neuronal excitation. Nat. Commun. 2019, 10, 907. [Google Scholar] [CrossRef]
- Helmy, S.; Leibfarth, F.A.; Oh, S.; Poelma, J.E.; Hawker, C.J.; Read de Alaniz, J. Photoswitching using visible light: A new class of organic photochromic molecules. J. Am. Chem. Soc. 2014, 136, 8169–8172. [Google Scholar] [CrossRef] [Green Version]
- Crespi, S.; Simeth, N.A.; König, B. Heteroaryl azo dyes as molecular photoswitches. Nat. Rev. Chem. 2019, 3, 133–146. [Google Scholar] [CrossRef]
- Vickerman, B.M.; Zywot, E.M.; Tarrant, T.K.; Lawrence, D.S. Taking phototherapeutics from concept to clinical launch. Nat. Rev. Chem. 2021, 5, 816–834. [Google Scholar] [CrossRef] [PubMed]
- Negi, A.; Alex, J.M.; Amrutkar, S.M.; Baviskar, A.T.; Joshi, G.; Singh, S.; Banerjee, U.C.; Kumar, R. Imine/amide–imidazole conjugates derived from 5-amino-4-cyano-N1-substituted benzyl imidazole: Microwave-assisted synthesis and anticancer activity via selective topoisomerase-II-α inhibition. Bioorganic Med. Chem. 2015, 23, 5654–5661. [Google Scholar] [CrossRef]
- Negi, A.; Mirallai, S.I.; Konda, S.; Murphy, P.V. An improved method for synthesis of non-symmetric triarylpyridines. Tetrahedron 2022, 121, 132930. [Google Scholar] [CrossRef]
- Fernández, I. Understanding the reactivity of polycyclic aromatic hydrocarbons and related compounds. Chem. Sci. 2020, 11, 3769–3779. [Google Scholar] [CrossRef] [PubMed]
- Velema, W.A.; Hansen, M.K.; Lerch, M.M.; Driessen, A.J.; Szymanski, W.; Feringa, B.L. Ciprofloxacin–photoswitch conjugates: A facile strategy for photopharmacology. Bioconjugate Chem. 2015, 26, 2592–2597. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negi, A.; Kesari, K.K.; Voisin-Chiret, A.S. Light-Activating PROTACs in Cancer: Chemical Design, Challenges, and Applications. Appl. Sci. 2022, 12, 9674. https://doi.org/10.3390/app12199674
Negi A, Kesari KK, Voisin-Chiret AS. Light-Activating PROTACs in Cancer: Chemical Design, Challenges, and Applications. Applied Sciences. 2022; 12(19):9674. https://doi.org/10.3390/app12199674
Chicago/Turabian StyleNegi, Arvind, Kavindra Kumar Kesari, and Anne Sophie Voisin-Chiret. 2022. "Light-Activating PROTACs in Cancer: Chemical Design, Challenges, and Applications" Applied Sciences 12, no. 19: 9674. https://doi.org/10.3390/app12199674
APA StyleNegi, A., Kesari, K. K., & Voisin-Chiret, A. S. (2022). Light-Activating PROTACs in Cancer: Chemical Design, Challenges, and Applications. Applied Sciences, 12(19), 9674. https://doi.org/10.3390/app12199674