1. Introduction
The uncertain prices of crude oil, the increasing awareness of carbon emissions, and the resulting desire to become gradually independent from fossil fuels constitute strong driving forces behind the search for sustainable alternatives to fossil fuel-based chemicals [
1]. An ever increasing number of processes are “going green”. The first and most obvious approach towards this end is the direct substitution of petrol-based chemicals by their bio-based counterparts. There are various examples of commercially available chemicals obtained from renewable resources, such as polyethylene [
2] and succinic acid [
3]. These “drop-in” products require no special adaptation before using them as they are essentially the same chemical, merely isolated from a different feedstock. The transition towards the use of green chemicals is not always smooth since obtaining these chemicals is hindered by a few complicating factors. Most predominantly, it is often cumbersome to obtain them in high purities as bio-based chemicals are often obtained from a mixture of usually very similar and hard-to-separate chemicals [
4]. This problem is particularly pressing for green monomers as these require very high purities in order to be able to undergo successful polymerization reactions [
5]. Fortunately, there are cases where a mixture of products has successfully been used as a green component. Indeed, up to 75% of phenol used in wood adhesives can be replaced by a mixture of various phenolic compounds obtained from lignin [
6], and polyols from vegetable oils have successfully been employed as crosslinkers in polyurethanes [
7]. However, these are rare exceptions rather than common practice. Nevertheless, despite these complications, several successful implementations of green chemicals as monomers have been reported [
8,
9,
10], reducing both the carbon footprint of the associated materials and their fossil fuel dependency. Unfortunately, despite these positive aspects, the simple substitution of oil-based chemicals with biomass-derived ones does not provide an exhaustive answer to the sustainability issue as waste streams (i.e., the waste produced at the end of the product’s life) remain virtually unaffected. Green thermoplastic materials might be recyclable in a “cradle-to-cradle” fashion as this is generally true for polymeric materials with physical (e.g., van der Waals) interactions between the chains [
11]. However, thermoset materials are notoriously and factually impossible to recycle according to the “cradle-to-cradle” approach [
12]. This is obviously true independent of their origin, i.e., whether oil- or bio-based. In the past decade, many efforts have been made in order to increase the recyclability of these materials, and a promising recent advancement is the use of thermoreversible crosslinking by means of the Diels–Alder reaction [
8,
9,
10]. The reversible nature of the Diels–Alder reaction has been successfully applied in self-healing polymers [
13,
14,
15,
16,
17,
18,
19,
20,
21]. Most popular is the reaction between a furan and maleimide [
11,
22], mainly due to fast kinetics and the wide availability of the reacting groups [
12], although systems based on cyanofumarate and fulvene [
23] or anthracene and maleimide [
24] have also been reported. The application of the Diels–Alder reaction in crosslinking has many advantages: as the formed bonds are covalent, the superior properties attributed to thermosets will be retained (e.g., due to their crosslinked structure, these usually possess superior barrier and mechanical properties, as well as higher chemical resistance than most thermoplastic ones). Furthermore, as the reaction is an equilibrium one and its extent can be easily influenced by changing the temperature, it constitutes an ideal candidate for use as a crosslinking reaction. Finally, the temperatures for bond formation and the reverse reaction, which yields the de-crosslinked product, are sufficiently far apart (namely, 50–80 °C and 110–170 °C, respectively, for furan and maleimide) to ensure that the obtained materials have a large application window.
The conceptual combination of thermal reversibility with the “green” character of available monomers constitutes a possible solution to the problems outlined above, thus providing in principle a lower carbon footprint as well as recyclability (cradle-to-cradle) of the end product. In this context, the presence of functional groups (e.g., -OH ones), is often considered a major drawback of green chemicals, when compared to fossil fuel-based ones, might be conveniently exploited to provide the chemicals with an added functionality. A paradigmatic example is constituted by diphenolic acid (DPA), a structural analog to bisphenol A [
25] obtained via a condensation reaction of levulinic acid [
26] with phenol. The only difference with bis-phenol A is the presence of an extra carboxylic acid moiety. Bisphenol A is widely used as monomer in polycarbonate and epoxy resin synthesis, but is also employed as a rigidifier in polyester resins. Apart from the fact that it is currently obtained from non-renewable resources, it is also a toxic chemical with shown estrogenic properties. Polycarbonates and polyesters have been successfully synthesized using unmodified DPA [
25,
27] or after protection of the acid group [
28]. However, the acidic group might also be employed for a chemical reaction with a furan-bearing molecule, thus enabling the possibility at later stage for a Diels–Alder crosslinking reaction.
The aim of this work is to ideally combine the two strategies towards sustainability: i.e., the bio-based character and the possibility for thermally reversible crosslinking and. thus. The possibility for recycling. To this end, DPA was modified with furfuryl amine to obtain a furan-functionalized diol. The incorporation of this monomer into a fully aromatic novel polyester is described based on known polymerization concepts. Finally, the obtained polymer was crosslinked by employing the Diels–Alder reaction with a bismaleimide in order to provide a preliminary proof of principle for the possibility of recycling the end product.
2. Materials and Methods
2.1. Chemicals
4,4′bis(hydroxyphenyl)valeric acid (Diphenolic acid, DPA) 95%, 2-methyltetrahydrofuran (MeTHF) anhydrous, N,N′Carbonyldiimidazole (CDI) 97%, Terephtaloylchloride 99+%, and 1,1(methylenedi-4,1-phenylene)bismaleimide 95% were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used as received. Furfurylamine 99+% was purchased from Sigma-Aldrich and distilled prior to use. Dodecyl bismaleimide was synthesized as described in the literature [
29].
2.2. Equipment
The 1H-NMR spectra were recorded on a Varian Mercury Plus 400 MHz using DMSO-d6 as a solvent. The 1H-NMR spectra of the samples at elevated temperatures were recorded on a Varian Mercury Plus 500 MHz using DMSO-d6 as a solvent. The PPM values are given relative to tetramethylsilane (TMS). Thermogravimetric analysis was performed on a Mettler Toledo TGA. The samples were weighed (ca. 10 mg) and placed in the analyzer. Subsequently, the temperature was raised from 25 °C to 900 °C at a heating rate of 10 °C per minute under N2 atmosphere. Differential scanning calorimetry (DSC) was performed on a Perkin Elmer differential scanning calorimeter Pyris 1 under N2 atmosphere. Before DSC, the sample was weighed (ca. 12 mg) and subsequently heated from 25 °C to 180 °C. Multiple cycles were performed at a heating rate of 10 °C/min throughout the measurements. The DMTA measurements were performed using a Rheometrics scientific solid analyzer (RSA II) under air using the dual cantilever mode at an oscillation frequency of 1 Hz and a heating rate of 5 °C/min. The GPC measurements were performed on a HP1100 equipped with three 300 × 7.5 mm PLgel 3 μm MIXED-E columns in a series using a GBC LC 1240 RI detector. The average molecular weight calculations were performed with the PSS WinGPC Unity software from Polymer Standards Service.
The following conditions were used: THF as eluent at a flow rate of 1 mL min−1; 140 bar, a column temperature of 42 °C, 20 μL injection volume, and a 10 mg mL−1 sample concentration. Toluene was used as a flow marker, and polystyrene samples with different molecular weights were used as the calibration standard. The pressing of samples was performed in a Taunus-Ton press type VS up to 150 A.
2.3. GPC
GPC measurements were performed to determine the average chain length, and, while absolute values cannot be given due to the absence of a suitable reference calibration, the spectra obtained across various samples confirm a consistent distribution of chain lengths between batches.
2.4. Synthesis of DPA-Fur
Diphenolic acid (1, 5 g, 17.5 mmol), 2-methylhydrofuran (30 mL) and N,N′-carbonyldiimidazole (1.2 equivalent, 3.398 g, 21 mmol) were charged to a dry three-neck round-bottomed flask connected to a cooler under N2. The reaction was stirred under reflux conditions (90 °C) for 2 h. Furfurylamine (1.2 equivalent, 1.85 mL, 21 mmol) was added, and the reaction was stirred at 90 °C for an additional 5 h. The reaction mixture was then allowed to cool to room temperature and diluted with additional 2-methylhydrofuran (45 mL). The solution was washed with 1M HCl (2 × 37 mL), 0.1 M NaOH (2 × 47 mL) and brine (1 × 15 mL) successively. Evaporation of the organic layer yields a yellow oil. The final purification by recrystallization from ethanol gave 3.26 g (51.1%) of white solid.
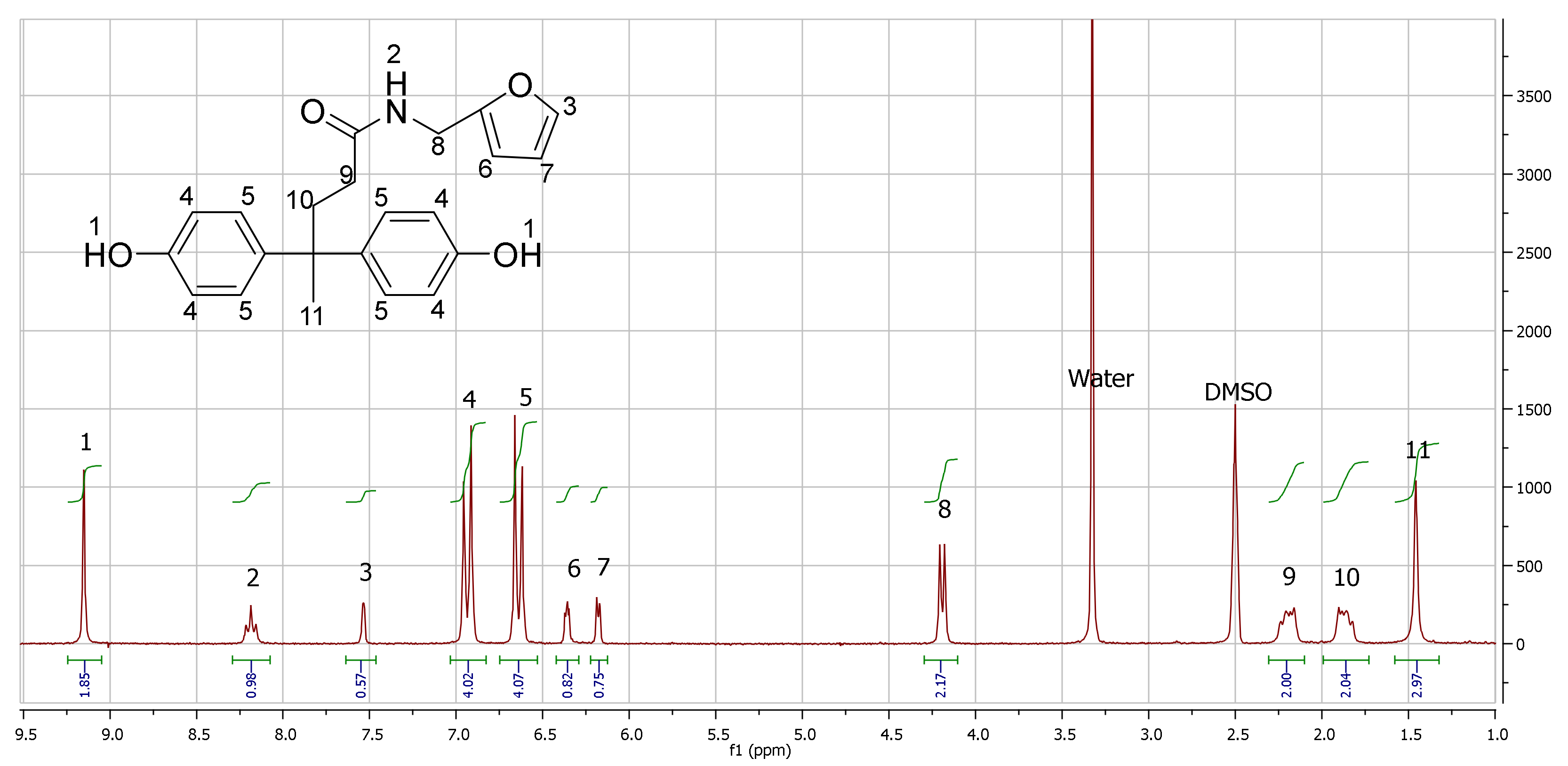
1H NMR (300 MHz, DMSO-d6)δ 9.16 (s, 2H, OH) 8.19 (t, 1H, NH) 7.53 (s, 1H, fur-p) 6.93 (d, 4H, benzene-o) 6.62 (d, 4H, benzene-m) 6.35 (t, 1H, fur-m) 6.18 (d, 1H, fur-o) 4.19 (d, 2H, N-CH2) 2.17 (t, 2H, -CH2-) 1.86 (t, 2H, -CH2-) 1.45 (s, 3H, -CH3).
2.5. Polymerization (DPA-Fur/Tereph)
DPA-fur (3, 1.755 g, 4.8 mmol), tert-butyl ammonium bromide (0.152 g, 8.7 wt.% on DPA-fur), sodium hydroxide (0.384 g, 9.6 mmol), and water (45 mL) were charged to a one-neck round-bottomed flask (250 mL). The mixture was stirred vigorously. Terephthaloyl chloride (0.975 g, 4.8 mmol) dissolved in chloroform (45 mL) was added to the reaction mixture. The reaction was stirred vigorously for 2 h. Precipitation in methanol (1800 mL) yielded 2.20 g (95.6%) of white solid.
Analysis:
1H NMR (300 MHz, DMSO-d6) δ 8.42–7.95 (m, 4H, aromatic not next to ester) 7.53 (s, 1H, fur-p) 7.42–7.12 (m, 8H, aromatic next to ester) 6.34 (s, 1H, fur-m) 6.19 (s, 1H, fur-o) 4.21 (d, 2H, N-CH2) 2,38 (broad s, 2H, -CH2-) 1.95 (broad s, 2H, -CH2-) 1.63 (s, 3H, -CH3).
2.6. DMTA Sample Preparation via Compression Moulding
First, a mixture of polymer and bismaleimide was prepared by dissolving the desired amount of bismaleimide in a minimal amount of chloroform (e.g., 0.25 g polymer, 0.09 g of bismaleimide in 1 mL of CHCl3). Mixing and successive solvent removal by rotary evaporation yielded a crosslinked film of polymer. This film was dried overnight in an oven at 60 °C, then frozen in liquid nitrogen and ground into particles using an IKA industrial hand grinder. This powder was pressed into bars (6 mm wide, 54 mm long, and 1 mm thick). Pressing was performed at 4 MPa at a temperature of 150 °C for 15 min. The material was expected to, and did, behave as a flowing thermoplastic polymer as this temperature is above the RDA decoupling temperature as well as above the Tg of the uncrosslinked polymer. The samples were cooled slowly inside the press (~30 min) to allow the formation of a rigid network through DA coupling. Afterwards, the samples were placed in an oven at 50 °C for 24 h to ensure complete crosslink formation.
3. Results and Discussion
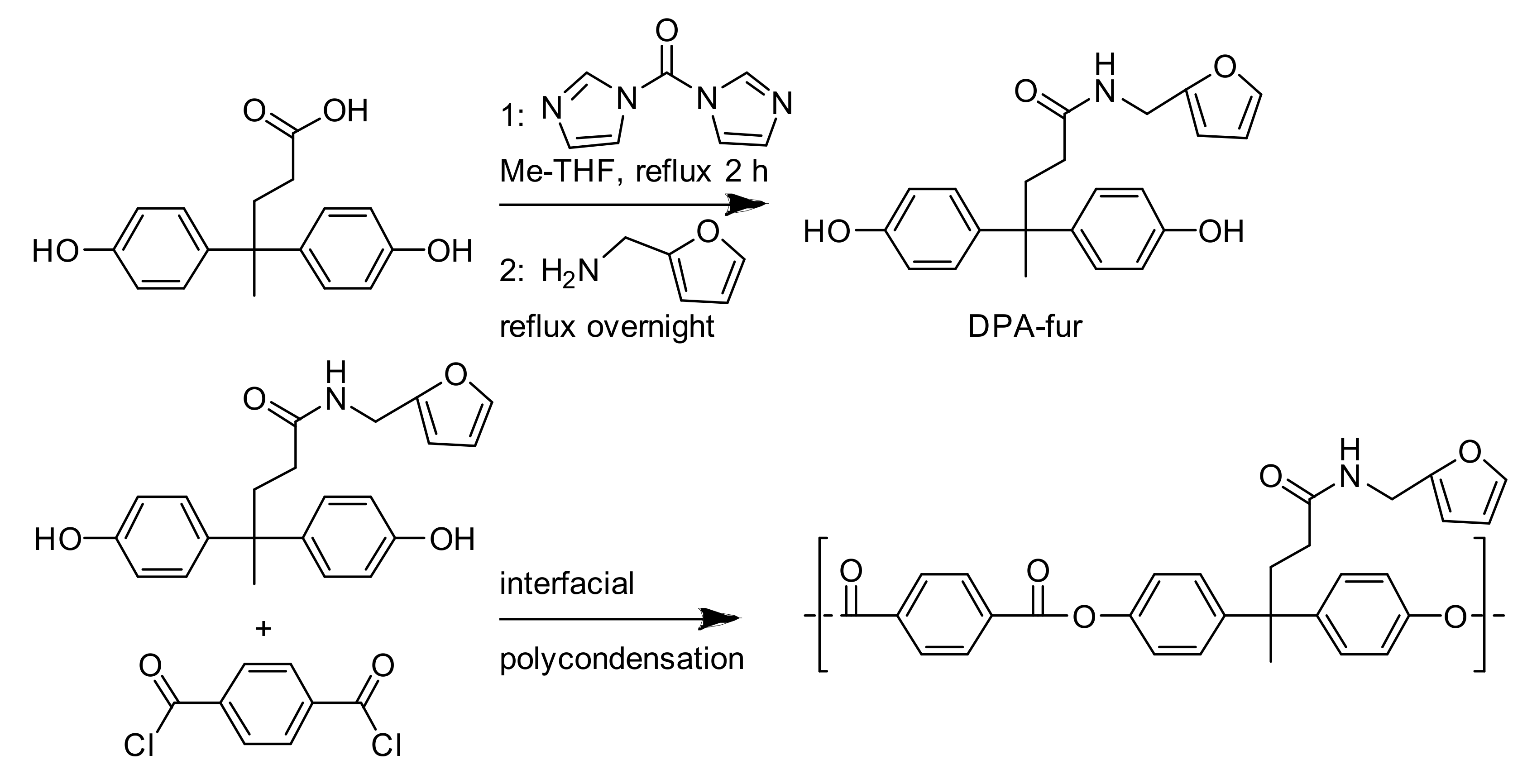
The overall strategy for the synthesis of the desired polymers relies on the possibility of modifying DPA and functionalizing it (
Scheme 1, top) with a Diels–Alder reactive group (in this case, furan). The employed reaction conditions, based on similar ones for amidation reactions [
30], entail the use of an intermediate (the imidazole peptide), subsequently reacted with fufurylamine to yield the desired product.
This synthetic strategy allows for very controlled reaction conditions and a high purity and acceptable yield of the end product. Indeed, after crystallization, the DPA-fur was obtained in a 51% yield as an off-white powder characterized by a
1H NMR spectrum with no relevant traces of impurities (
Figure 1). DPA-fur was then subsequently polymerized with terephtaloyl chloride in a two-phase system (
Scheme 1, bottom) as described in the literature [
31]. Clean polymer formation was evident from the complete disappearance of the signals at δ 9.4 ppm in
1H NMR (corresponding to the OH group on the DPA-fur monomer). Furthermore, aromatic protons from both terephtaloyl chloride and the DPA-fur shifted significantly. Two distinct groups of peaks can be identified, one at δ 7.42–7.12 ppm belonging to the aromatic protons next to an ester bond containing eight protons (4
o-from the DPA-fur, and 4 from the terephtaloyl monomer), and another group at δ 8.42–7.95 ppm containing four protons (the
o-protons from DPA-fur away from the ester bond).
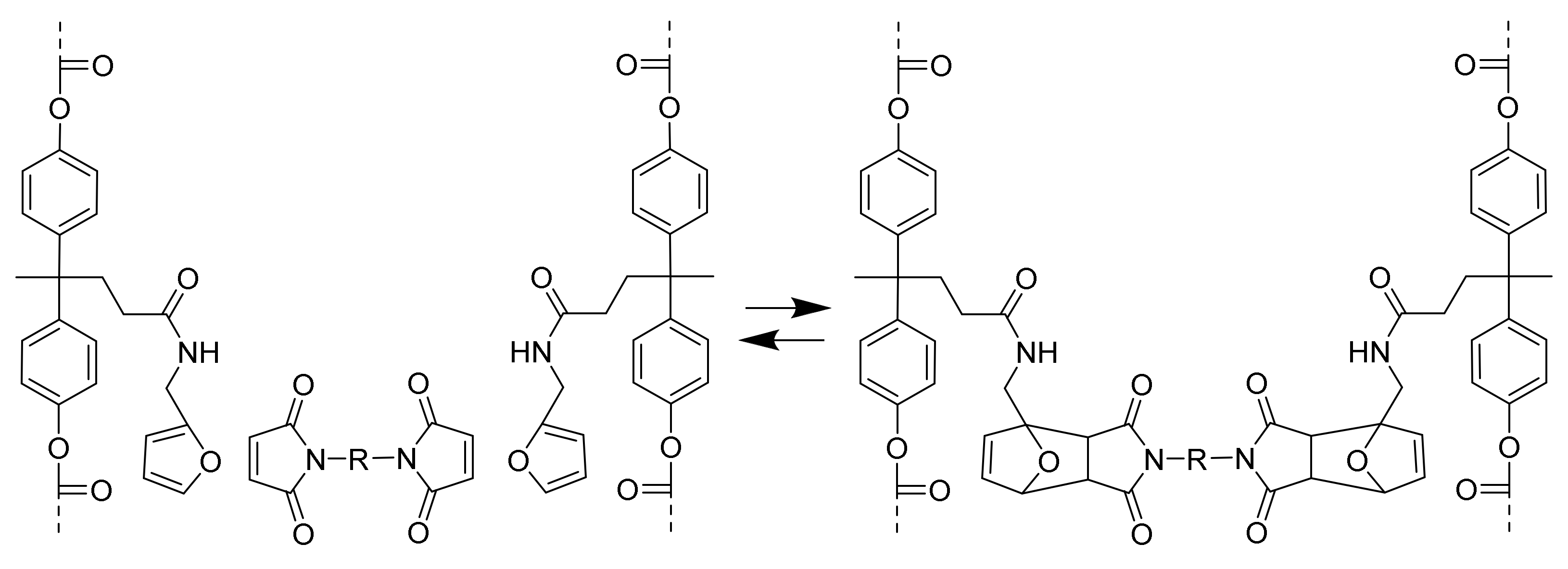
Crosslinking of the prepared polymer via the Diels–Alder reaction was achieved by mixing it with an equimolar amount of bismaleimide in DMSO. The system was expected to crosslink by a reaction of the pendant furan moieties with the maleimide groups (
Scheme 2).
The reaction was initially carried out in DMSO in order to visually observe gel formation (indicative of the formation of a network structure), as well as to preliminarily investigate its reversibility as a function of temperature. Two types of bismaleimide were added in various ratios, and the gelation time (defined as the time it took the stirrer to stop spinning), a rough indication for the gel formation kinetics, was determined (
Table 1). In the first instance, it is worth noting how both bismaleimides were able to induce gel formation, thus indicating network formation. It was also evident that the aromatic bismaleimide displayed faster kinetics than the aliphatic one. The significant difference in gelation times might be related to different reaction kinetics. On the other hand, this might also be due to the possibility of the aliphatic crosslinker (because of its flexible nature) to backbite coordination where both maleimide groups attach to the same polymer chain. The aromatic bismaleimide was expected to be far too rigid to be able to coordinate in this fashion [
32]. Furthermore, increasing the maleimide to furan ratio led to a decrease in gelation time. This has been observed for other polymeric systems crosslinked in the same way [
12] and is most probably related to the second-order kinetics (first-order in the bismaleimide) of the crosslinking reaction.
Generally speaking, the chemical reversibility of the furan–bismalimide systems has been already investigated and reported in previous studies (among others in our group [
12,
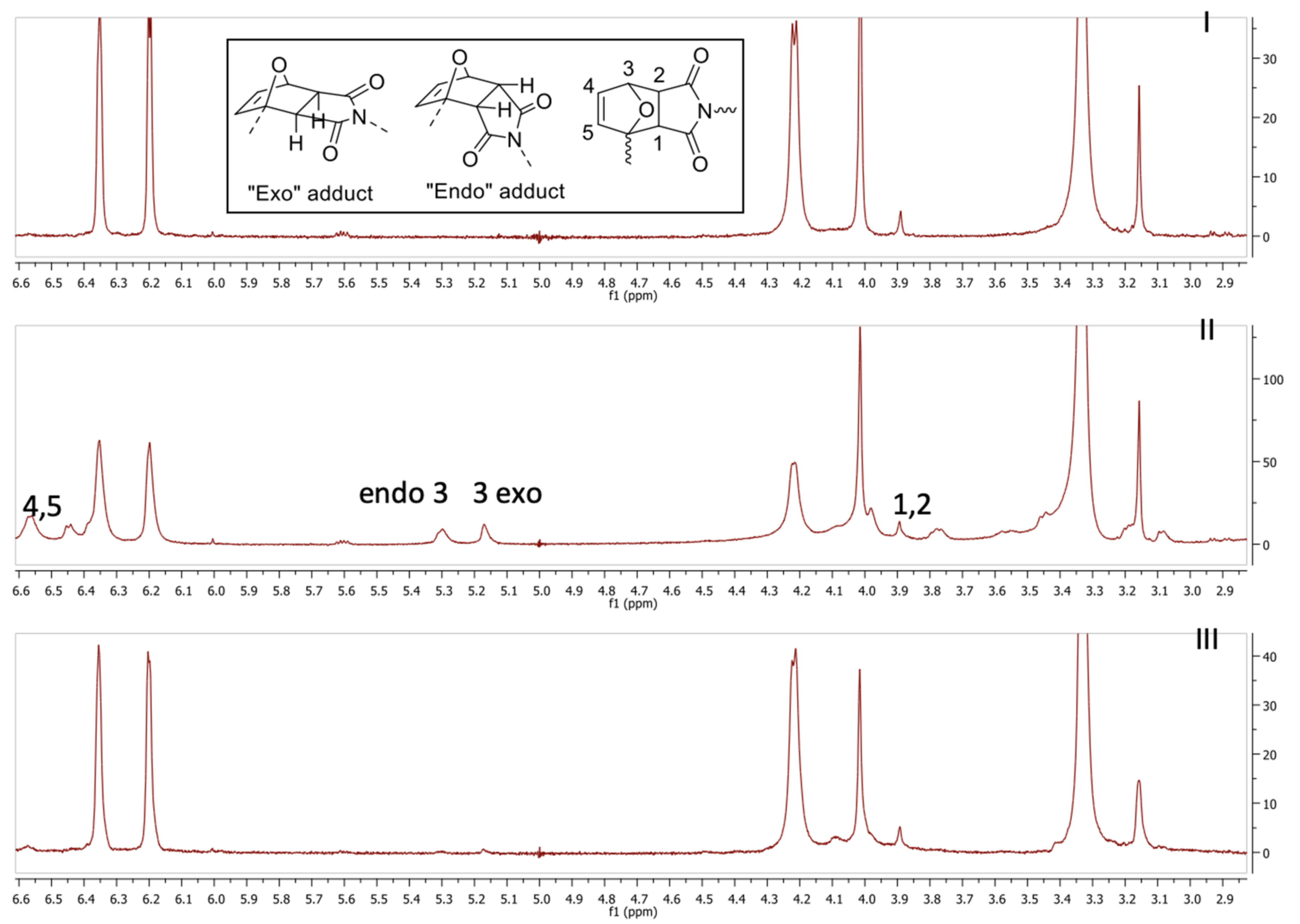
22]). In all these studies, this reversibility (as proved by FT-IR, but also by using model compounds) is also coupled with the thermal and mechanical one, as also shown in this paper (see below). However, in these published works, no light could be shed on the chemistry of crosslinking as the prepared samples, in all cases insoluble networks, did not allow any accurate spectroscopic analysis. In the present case, the relatively low molecular weight allowed the elucidation of the mechanism of crosslinking at the molecular level by solution NMR. First, a sample containing uncrosslinked polymers and bismaleimide in a 1:1 ratio of maleimide to furan groups in DMSO-d6 was prepared. Immediately after mixing, an NMR spectrum was recorded (spectrum I in
Figure 2). Next the sample was heated to 50 °C for 3 h; this should ensure complete formation of the crosslinks. Afterwards, another NMR spectrum was recorded, and the signals attributed to the DA adduct of furan and maleimide could clearly be detected (spectrum II in
Figure 2). There have been various reports describing [
12,
22] the assignments of these signals based on model compounds. Interestingly, there are two clearly distinct peaks for the endo and exo adducts visible. To the best of our knowledge, this is the first direct confirmation for the formation of both species since similar works on different systems describe an NMR spectrum of a crosslinked polymer network [
22], but only see a single peak which is attributed to both isomers. When the sample was subsequently heated to 150 °C for 5 min and a new spectrum was recorded. the adduct signals disappeared indicating that the RDA reaction had completely occurred (spectrum III in
Figure 2). This is further proof, in addition to the gelation process and its reverse (vide supra), of the reversibility of this crosslinking system in solution.
Being able to observe the two DA adducts in the NMR, an attempt was made to quantify their relative ratio as a function of temperature. This is quite interesting as such a shift in the ratio of these two adducts has been frequently invoked in the literature in order to clarify the thermal behavior of thermal reversible networks [
12,
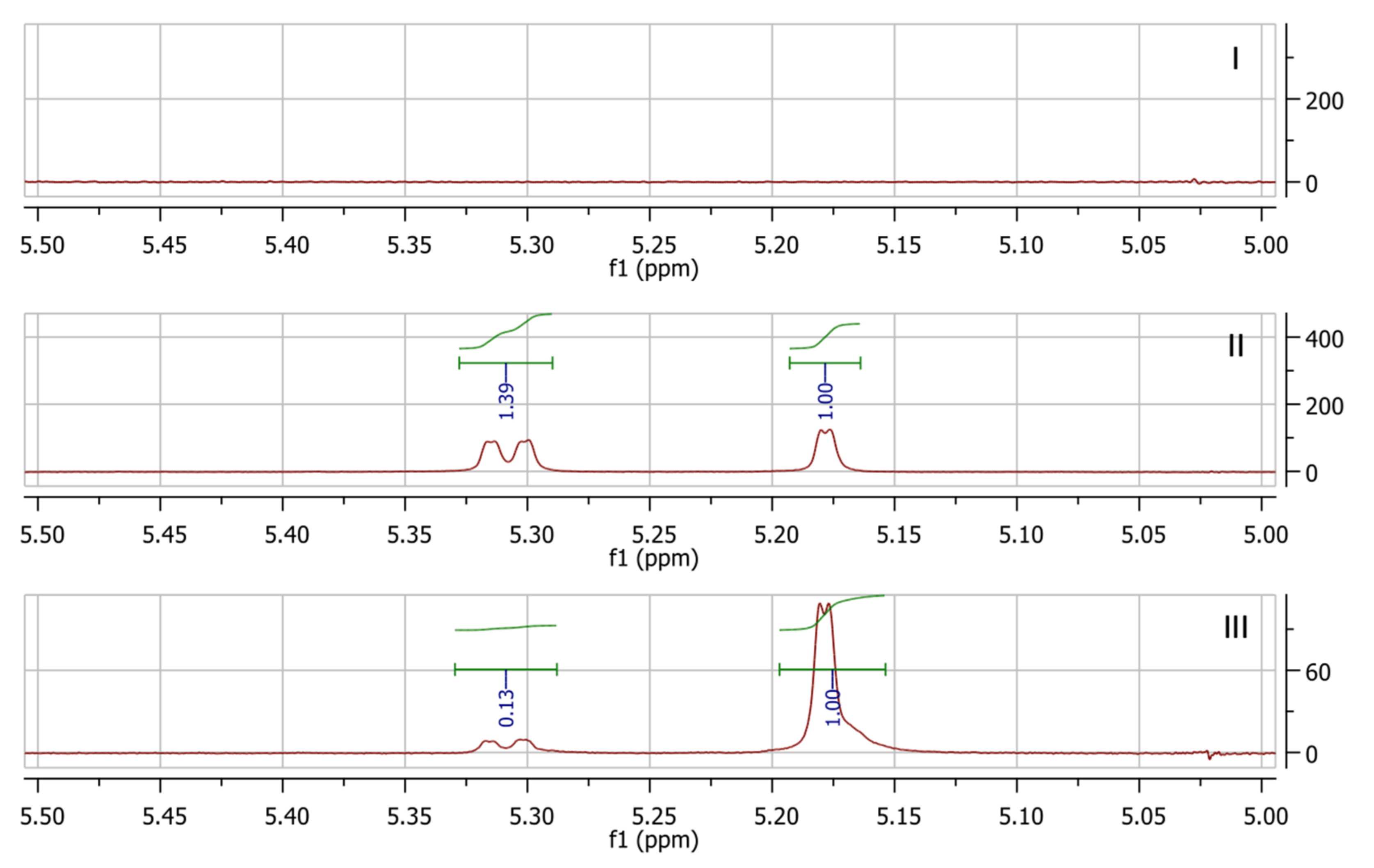
33], yet no clear proof has ever been reported for a crosslinked polymeric system. For the present system, in another experiment, the same solution containing uncrosslinked polymer and bismaleimide in a 1:1 maleimide:furan group ratio was created. A spectrum was recorded immediately after mixing (spectrum I in
Figure 3). Next, the sample was heated to 65 °C for 2 h, and a new spectrum was recorded (spectrum II in
Figure 3). Once again, the signals belonging to the DA adduct were found, and the ratio of endo:exo adducts was 58:42. It is well known that the DA reaction is an equilibrium reaction, and that the exo isomer is thermodynamically favored and the endo adduct is kinetically favored. When the sample was kept as 65 °C for 48 h and another spectrum was recorded (spectrum III in
Figure 3), the ratio of endo:exo isomers shifted significantly to 11:88. This constitutes, to the best of our knowledge, the first direct confirmation of the formation of two adducts in as well as their nature (thermodynamically and kinetically favored) for furan maleimide crosslinking reactions on a polymeric system. Indeed, other studies have shown a similar behavior, but only model compounds or singular adducts have been used [
22].
The formation of the two DA adducts and their relative ratio might be of pivotal importance for thermal reversibility of the system, as suggested for the related ones [
34].
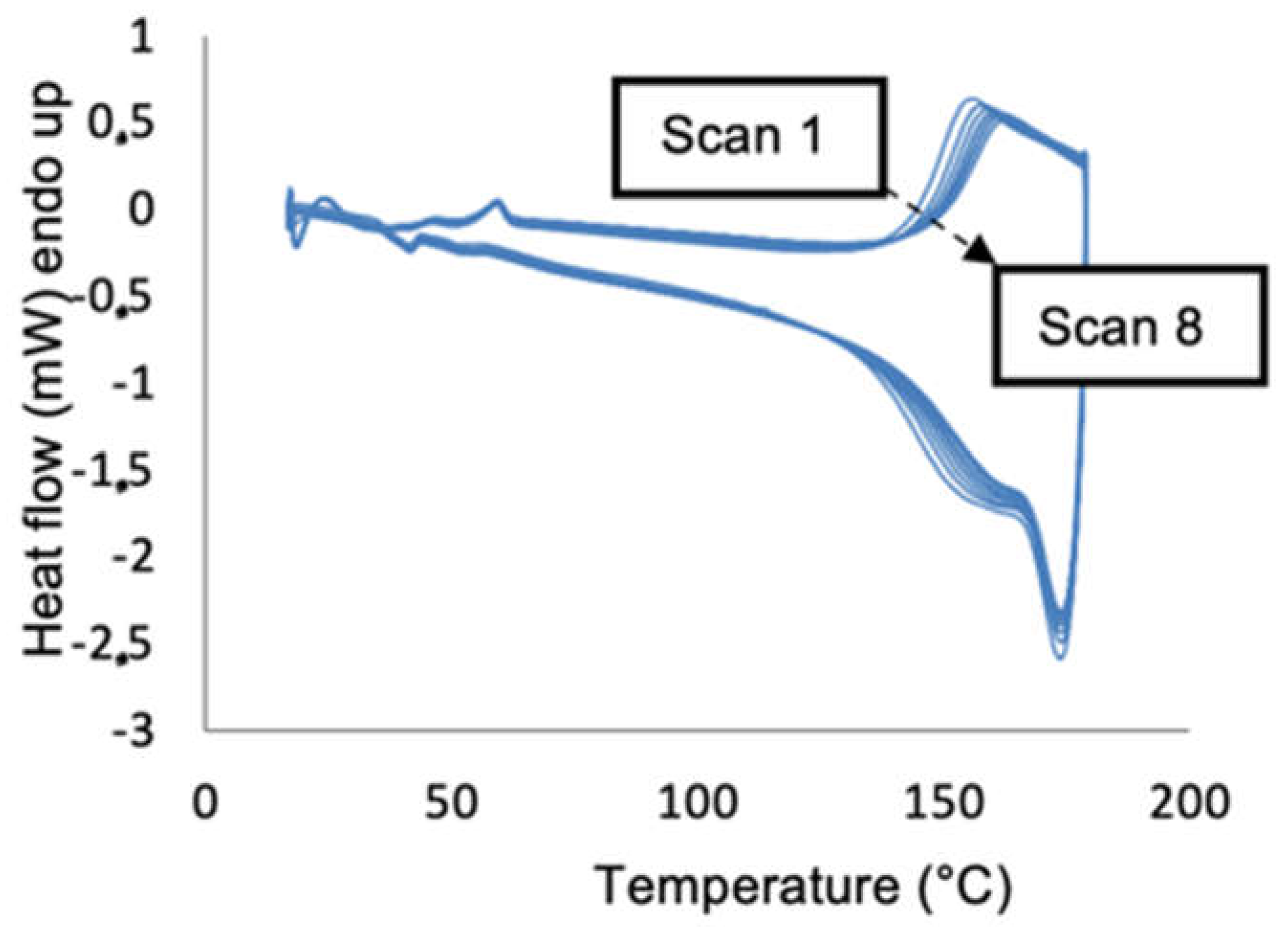
Thermal reversibility was tested by cyclical DSC measurements, according to which the material was heated to 180 °C and subsequently cooled to room temperature eight consecutive times (
Figure 4 for DPA-fur/tereph). The DA coupling is exothermic, while the RDA decoupling is an endothermic process. The integral of the corresponding peaks is an indication of the extent of the reaction.
The results suggest that the system shows thermoreversible coupling and decoupling: the heat capacity of the material did not change significantly over the course of all cycles, which is an indication of full reversibility [
12,
35]. The peak at 150 °C is related to the RDA decoupling reaction, while the DA coupling is not immediately apparent in this graph as it occurred gradually while cooling the sample. Over the course of all cycles, the maximum of the RDA peak at 150 °C shifted to higher values. A similar shift has been reported for related systems [
12,
22], for which this was firstly and hypothetically attributed to the RDA reaction being too slow with respect to the DSC timescale. However, this was later disproven, and the difference likely lies in a change in the ratio of endo and exo DA adducts. It has previously been established [
34] that the exo isomer is thermodynamically favored, which makes it likely that the kinetically favored endo adduct is initially formed in excess. Nevertheless, during the course of heating and cooling this equilibrium shifts towards the more favored exo product, which in turn should have a higher temperature of RDA decoupling. This is in striking agreement with the NMR experiments (vide supra) and thus provides direct conformation of the formation and importance of the adducts on the thermal behavior.
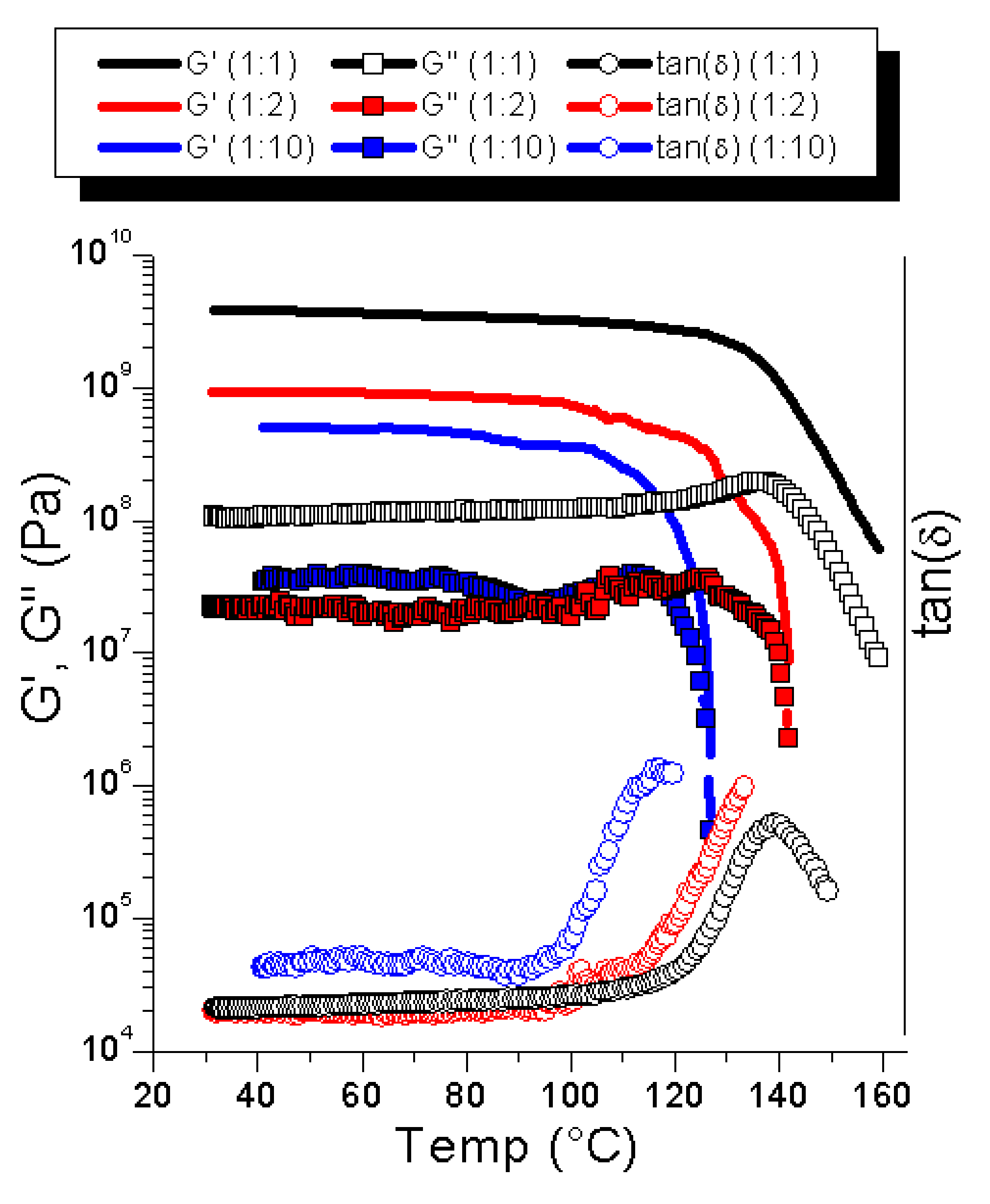
In order to assess the mechanical properties of the material, DMTA measurements were performed. The formation of solid, homogeneous bars is an indication of the reversibility of the compound, as without reversible crosslinking taking place, only a compressed powder would be obtained. In order to elucidate the effect of the crosslinking degree on the softening temperature (tan (δ) peak), various samples containing different maleimide:furan ratios were prepared.
Figure 5 shows G′ and G″ as well as the tan(δ) for samples with 1:1 1:2 and 1:10 mol:mol ratios of maleimide:furan groups.
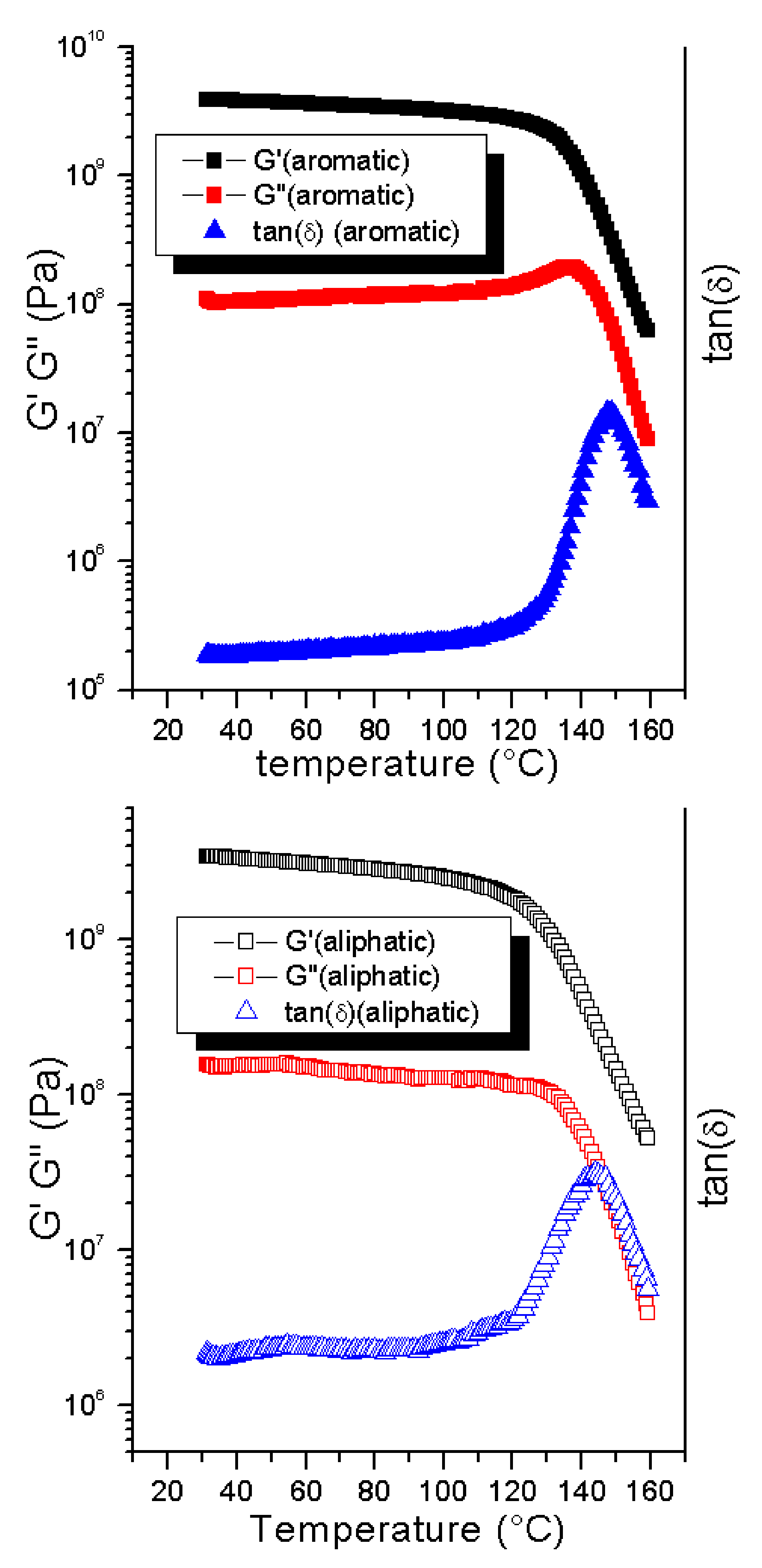
The softening temperatures were 137 °C, 135 °C, and 120 °C for the 1:1, 1:2, and 1:10 ratios of maleimide:furan, respectively. The effect of the MAL:FUR ratio on the softening point was less expressed than that reported for the related systems (
Figure 6).
A highly substituted (60%) polyketone-based system (PK50) displays a drop in softening temperature from 102 °C to 86 °C upon changing the MAL:FUR ratio from 1:1 to 1:2 (halving the amount of bismaleimide). An additional halving of the amount of bismaleimide led to an even larger decrease of the softening point (22 °C lower). A similar trend was observed when looking at the system obtained by the copolymerization of bis(hydroxymethyl)furane and succinic acid (PFS) [
36]: when halving the amount of bismaleimide, the softening point dropped from 38 °C to 9 °C. Since this value is below the T
g of the uncrosslinked material (15 °C) [
37], the authors attribute this to a disturbance in the chain packing due to the presence of a crosslinker. For DPA-fur, the effect of halving the amount of bismaleimide on the softening temperature was practically insignificant; only a drop of 2 °C was observed. The large difference in the softening point for the other systems probably stems from the low T
g of the uncrosslinked polymer. When the amount of crosslinker was reduced, the material behaved increasingly similar to a branched polymer structure than a three-dimensional network. The T
g of the material will become closer to that of the uncrosslinked polymer as less crosslinker is added. Interestingly, when reducing the amount of crosslinker even further to the ratio of 1:10 MAL:FUR, the softening point of DPA-fur also dropped below the T
g of the uncrosslinked polymer. The aromatic groups present in DPA-fur are likely involved in π-π stacking interactions, which are disturbed by the presence of small amounts of crosslinker. Apparently, the π-π-interactions have a larger influence on the softening point than the small amount of crosslinker present. Finally, the softening point observed for the 1:1 MAL:FUR DPA-fur network was much closer to the theoretical RDA temperature for furane and maleimide groups (e.g., 150 [
35]), which could be an indication of a more stable crosslink. Furthermore, as the material remains crosslinked for a larger range of temperatures (e.g., decrosslinking occurs at higher temperatures), this should provide a larger window of application for the resulting material than the PFS- and PK50-based materials.
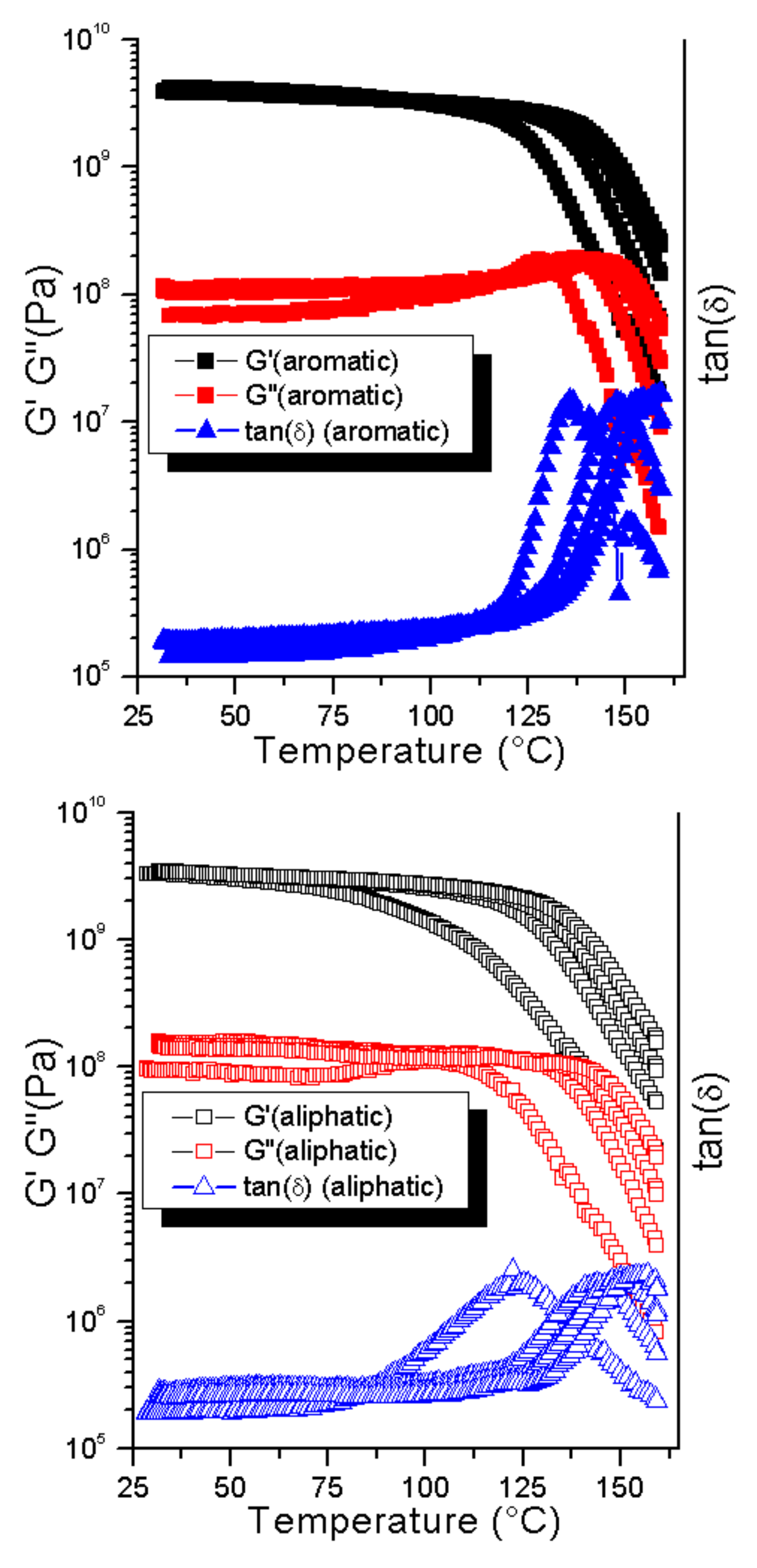
Another way of influencing the thermal behavior is the use of a different crosslinker [
36]. When the aromatic bismaleimide was substituted by an aliphatic one containing a C12 chain between the maleimide groups, a decrease in the softening point by 10 °C was observed (
Figure 7).
These results are comparable to the findings reported by Zeng et al. where a drop of approximately 10 °C was reported, as well for the 1:2 ratio of MAL:FUR when substituting the aromatic bismaleimide for an aliphatic one containing a C
2OC
2OC
2 ether bridge [
38]. This behavior likely comes from the increased flexibility of the aliphatic bismaleimide compared to its aromatic counterpart. This increased flexibility should result in a less rigid network. Furthermore, as suggested by the gelation experiments, the aliphatic crosslinker should be able to participate in backbiting coordination, limiting its availability for the formation of crosslinks. Independent of the exact mechanism, the observed results indicate that in the case of the present polymer, the amount and structure of the crosslinker might be conveniently used to manipulate the mechanical behavior of the end product.
In order to assess the mechanical reworkability and thermal recovery of the material, cyclical DMTA measurements were performed. The samples were heated to 160 °C and successively cooled back to room temperature (20 °C) for four cycles (
Figure 8).
During cooling, the material was expected to re-form the DA crosslinks that had been broken at high temperatures by the RDA reaction (which is dominant at these elevated temperatures). If the DA reaction is performed consistently, the material should display the same mechanical properties in all subsequent cycles. A relatively large change in properties was observed between the first and second cycle; this is consistent with the shift observed in DSC, and the explanation given of a kinetically favored orientation (endo vs. exo) still holds true. However, after each consecutive cycle, the softening point of the material increased. If the only equilibrium involved is the changing from an endo to exo isomer, the difference should not be as large as that observed. Most likely, a more permanent change occurs in the material under these circumstances. A well-known side reaction of the Diels–Alder adduct of furan and maleimide is the formation of an aromatic ring via the elimination of water [
35]. The mechanical properties of the material were not expected to change significantly in the low-temperature region as in both cases (reversible crosslinked and aromatized crosslinked) a covalently crosslinked network was obtained. However, as the aromatization leads to an irreversible network under these conditions, less crosslinks will be broken in each consecutive cycle, explaining the ever increasing softening point. This indicates that the observed system is only able to display full reversibility for a few thermal cycles.
4. Conclusions
In this work, diphenolic acid was successfully functionalized with furfurylamine, yielding a furan-bearing diol. This was incorporated into an aromatic polyester by reacting it with terephtaloyl chloride in an interfacial polycondensation reaction. The polymer obtained contains pendant furan groups, which are able to participate in reversible crosslinking, forming a thermally reversible network.
Reversibility was shown in solution by gelation experiments, where a gel was formed between 15 and 45 min when employing the aromatic crosslinker, depending on the ratio of maleimide to furan groups. Gelation with an aliphatic crosslinker took significantly longer: 143 to 368 min, depending on the maleimide to furan ratio. Decoupling (evident by the transition of gel to solution) occurred for all samples in approximately 3 min at 150 °C for all experiments.
Reversibility in the solid state was shown by the cyclical DSC measurements where the peak for RDA decoupling had a maximum at 154 °C for the first cycle. The maximum shifted slightly towards 160 °C for the eighth cycle, but all the other thermal properties of the material remained constant. The small shift in the maximum is attributed to differences in the ratio of endo and exo adducts formed. The NMR measurements showed a clear shift from a kinetic equilibrium to a thermodynamic one.
The material displays the recovery of mechanical properties, as evident from the cyclical DMTA measurements, and after an initial change, the polymer displays a high degree of recovery. However, the increase in the softening point after each cycle suggests that irreversible aromatization of the furanes and maleimides occurs.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

